
Unsupervised Analysis Reveals the Involvement of Key Immune Response Genes and the Matrisome in Resistance to BRAF and MEK Inhibitors in Melanoma


Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
3. Results
3.1. The Clustering of Tumors and Patients Reveals No Clustering by Pre-Treatment or Post-Treatment, or by Any Other Criteria
3.2. Analysis Using the Melanoma-Specific Gene Set also Returned No Significant Clustering
3.3. Unsupervised Differential Gene Expression Analysis
3.4. Gene Set Enrichment Analysis
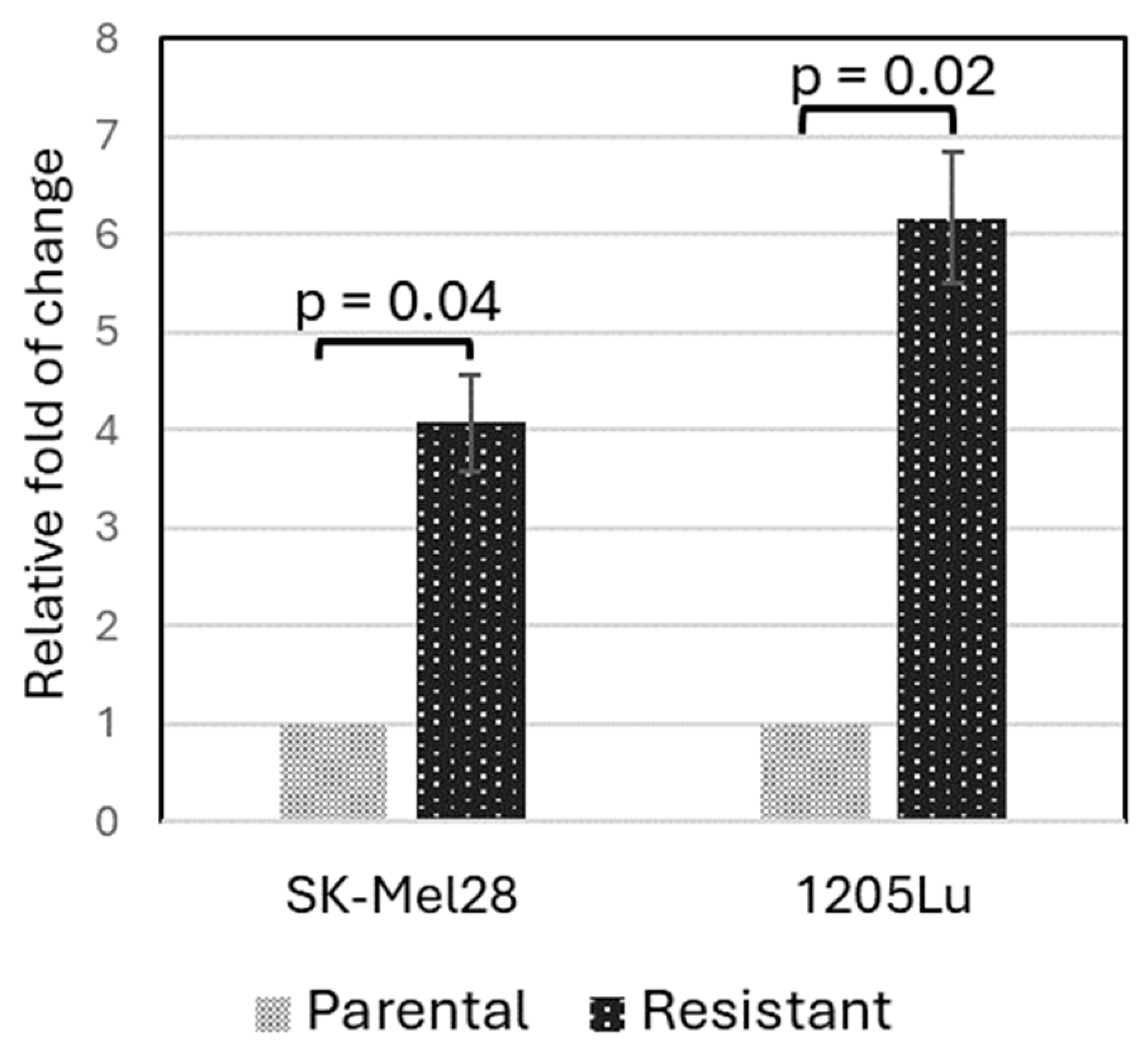
3.5. Validation of PLXNC1 Over-Expression in BRAFi-Resistant Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Larkin, J.; Del Vecchio, M.; Ascierto, P.A.; Krajsova, I.; Schachter, J.; Neyns, B.; Espinosa, E.; Garbe, C.; Sileni, V.C.; Gogas, H.; et al. Vemurafenib in patients with BRAF(V600) mutated metastatic melanoma: An open-label, multicentre, safety study. Lancet Oncol. 2014, 15, 436–444. [Google Scholar] [CrossRef]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef]
- Yun, S.; Vincelette, N.D.; Green, M.R.; Wahner Hendrickson, A.E.; Abraham, I. Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: A systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer Med. 2016, 5, 1481–1491. [Google Scholar] [CrossRef]
- Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Groot, J.W.B.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsova, I.; Liszkay, G.; et al. COLUMBUS 5-Year Update: A Randomized, Open-Label, Phase III Trial of Encorafenib Plus Binimetinib Versus Vemurafenib or Encorafenib in Patients with BRAF V600-Mutant Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 4178–4188. [Google Scholar] [CrossRef]
- Choi, J.; Landrette, S.F.; Wang, T.; Evans, P.; Bacchiocchi, A.; Bjornson, R.; Cheng, E.; Stiegler, A.L.; Gathiaka, S.; Acevedo, O.; et al. Identification of PLX4032-resistance mechanisms and implications for novel RAF inhibitors. Pigment Cell Melanoma Res. 2014, 27, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [PubMed]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef]
- Gopal, Y.N.; Rizos, H.; Chen, G.; Deng, W.; Frederick, D.T.; Cooper, Z.A.; Scolyer, R.A.; Pupo, G.; Komurov, K.; Sehgal, V.; et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1alpha and oxidative phosphorylation in melanoma. Cancer Res. 2014, 74, 7037–7047. [Google Scholar] [CrossRef] [PubMed]
- Rock, R.; Mayrhofer, J.E.; Torres-Quesada, O.; Enzler, F.; Raffeiner, A.; Raffeiner, P.; Feichtner, A.; Huber, R.G.; Koide, S.; Taylor, S.S.; et al. BRAF inhibitors promote intermediate BRAF(V600E) conformations and binary interactions with activated RAS. Sci. Adv. 2019, 5, eaav8463. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Bin Lim, S.; Chua, M.L.K.; Yeong, J.P.S.; Tan, S.J.; Lim, W.T.; Lim, C.T. Pan-cancer analysis connects tumor matrisome to immune response. NPJ Precis. Oncol. 2019, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Socovich, A.M.; Naba, A. The cancer matrisome: From comprehensive characterization to biomarker discovery. Semin. Cell Dev. Biol. 2019, 89, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Belotti, Y.; Lim, S.B.; Iyer, N.G.; Lim, W.T.; Lim, C.T. Prognostic Matrisomal Gene Panel and Its Association with Immune Cell Infiltration in Head and Neck Carcinomas. Cancers 2021, 13, 5761. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package Version 1(2), 726. Available online: https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf (accessed on 27 March 2024).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Korotkevich, G. Fast Gene Set Enrichment Analysis. bioRxiv 2021. [CrossRef]
- Zecena, H.; Tveit, D.; Wang, Z.; Farhat, A.; Panchal, P.; Liu, J.; Singh, S.J.; Sanghera, A.; Bainiwal, A.; Teo, S.Y.; et al. Systems biology analysis of mitogen activated protein kinase inhibitor resistance in malignant melanoma. BMC Syst. Biol. 2018, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Singh, A.; Yang, Z.; Garcia, A.; Kong, Y.; Meyskens, F.L., Jr. MiTF links Erk1/2 kinase and p21 CIP1/WAF1 activation after UVC radiation in normal human melanocytes and melanoma cells. Mol. Cancer 2010, 9, 214. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A. A description of the Molecular Signatures Database (MSigDB) Web site. Methods Mol. Biol. 2014, 1150, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Dias-Santagata, D.; Bergethon, K.; Iafrate, A.J.; Settleman, J.; Engelman, J.A. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci. Signal. 2010, 3, ra84. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Wang, D.; Lin, R.; Lv, Q.; Wang, W. IFI44 is an immune evasion biomarker for SARS-CoV-2 and Staphylococcus aureus infection in patients with RA. Front. Immunol. 2022, 13, 1013322. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nogales, A.; Martinez-Sobrido, L.; Topham, D.J. Interferon-Induced Protein 44 Interacts with Cellular FK506-Binding Protein 5, Negatively Regulates Host Antiviral Responses, and Supports Virus Replication. mBio 2019, 10, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Hooijkaas, A.; Gadiot, J.; Morrow, M.; Stewart, R.; Schumacher, T.; Blank, C.U. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology 2012, 1, 609–617. [Google Scholar] [CrossRef]
- Samaniego, R.; Dominguez-Soto, A.; Ratnam, M.; Matsuyama, T.; Sanchez-Mateos, P.; Corbi, A.L.; Puig-Kroger, A. Folate Receptor beta (FRbeta) Expression in Tissue-Resident and Tumor-Associated Macrophages Associates with and Depends on the Expression of PU.1. Cells 2020, 9, 1445. [Google Scholar] [CrossRef]
- Zhang, P.; Zhao, Y.; Xia, X.; Mei, S.; Huang, Y.; Zhu, Y.; Yu, S.; Chen, X. Expression of OLR1 gene on tumor-associated macrophages of head and neck squamous cell carcinoma, and its correlation with clinical outcome. Oncoimmunology 2023, 12, 2203073. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Huang, C.; Zhao, H.; Zhou, J.; Hu, M.; Chen, Q.; Ge, B.; Huang, Q. PLXNC1: A Novel Potential Immune-Related Target for Stomach Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 662707. [Google Scholar] [CrossRef] [PubMed]
- Ragaini, S.; Wagner, S.; Marconi, G.; Parisi, S.; Sartor, C.; Nanni, J.; Cristiano, G.; Talami, A.; Olivi, M.; Ocadlikova, D.; et al. An IDO1-related immune gene signature predicts overall survival in acute myeloid leukemia. Blood Adv. 2022, 6, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Hasegawa, H.; Hyodo, T.; Ito, S.; Asano, E.; Yuang, H.; Funasaka, K.; Shimokata, K.; Hasegawa, Y.; Hamaguchi, M.; et al. ARHGAP18, a GTPase-activating protein for RhoA, controls cell shape, spreading, and motility. Mol. Biol. Cell 2011, 22, 3840–3852. [Google Scholar] [CrossRef] [PubMed]
- Pena-Oyarzun, D.; Rodriguez-Pena, M.; Burgos-Bravo, F.; Vergara, A.; Kretschmar, C.; Sotomayor-Flores, C.; Ramirez-Sarmiento, C.A.; De Smedt, H.; Reyes, M.; Perez, W.; et al. PKD2/polycystin-2 induces autophagy by forming a complex with BECN1. Autophagy 2021, 17, 1714–1728. [Google Scholar] [CrossRef] [PubMed]
- Ramsdale, R.; Jorissen, R.N.; Li, F.Z.; Al-Obaidi, S.; Ward, T.; Sheppard, K.E.; Bukczynska, P.E.; Young, R.J.; Boyle, S.E.; Shackleton, M.; et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci. Signal. 2015, 8, ra82. [Google Scholar] [CrossRef]
- Konig, K.; Marth, L.; Roissant, J.; Granja, T.; Jennewein, C.; Devanathan, V.; Schneider, M.; Kohler, D.; Zarbock, A.; Rosenberger, P. The plexin C1 receptor promotes acute inflammation. Eur. J. Immunol. 2014, 44, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Granja, T.; Kohler, D.; Mirakaj, V.; Nelson, E.; Konig, K.; Rosenberger, P. Crucial role of Plexin C1 for pulmonary inflammation and survival during lung injury. Mucosal Immunol. 2014, 7, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Lazova, R.; Gould Rothberg, B.E.; Rimm, D.; Scott, G. The semaphorin 7A receptor Plexin C1 is lost during melanoma metastasis. Am. J. Dermatopathol. 2009, 31, 177–181. [Google Scholar] [CrossRef]
- Kumasaka, M.Y.; Yajima, I.; Iida, M.; Takahashi, H.; Inoue, Y.; Fukushima, S.; Ihn, H.; Takeda, K.; Naito, Y.; Yoshikawa, T.; et al. Correlated expression levels of endothelin receptor B and Plexin C1 in melanoma. Am. J. Cancer Res. 2015, 5, 1117–1123. [Google Scholar] [PubMed]
- Chen, J.; Liu, H.; Chen, J.; Sun, B.; Wu, J.; Du, C. PLXNC1 Enhances Carcinogenesis Through Transcriptional Activation of IL6ST in Gastric Cancer. Front. Oncol. 2020, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- NazimTurhal, S.; Dogan, M.; Esendagli, G.; Artac, M.; Korkmaz, L.; Coskun, H.S.; Goker, E.; PerranYumuk, F.; Bilgetekin, I.; Kose, F.; et al. The Relationship Between Plexin C1 Overexpression and Survival in Hepatocellular Carcinoma: A Turkish Oncology Group (TOG) Study. J. Gastrointest. Cancer 2022, 53, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.; Coumbe, J.E.M.; Coumbe, B.G.T.; Thomas, J.; Willsmore, Z.; Dimitrievska, M.; Yasuzawa-Parker, M.; Hoyle, M.; Ingar, S.; Geh, J.L.C.; et al. BRAF inhibitors and their immunological effects in malignant melanoma. Expert. Rev. Clin. Immunol. 2022, 18, 347–362. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Dataset | None or EDT * | Vemurafenib | Dabrafenib | All |
|---|---|---|---|---|
| GSE50509 | 28 | 8 | 25 | 61 |
| GSE61992 | 14 | 0 | 10 ** | 24 |
| Total | 42 | 8 | 35 | 85 |
| GSE50509 | GSE61992 | |||||||
|---|---|---|---|---|---|---|---|---|
| SYMBOL | logFC | AveExpr | p. Value | logFC | AveExpr | p. Value | Band | Gene Description |
| ARHGAP18 | 0.61 | 6.47 | 0.027 | 0.70 | 5.36 | 0.025 | 6q22.33e | Rho GTPase-activating protein 18 |
| FOLR2 | −0.69 | 7.32 | 0.012 | −0.64 | 5.71 | 0.017 | 11q13.4a | folate receptor beta |
| IFI44 | 0.77 | 8.22 | 0.039 | 1.36 | 5.98 | 0.020 | 1p31.1e | Interferon-induced protein 44 |
| LEF1 | 0.69 | 9.78 | 0.005 | 0.68 | 8.62 | 0.039 | 4q25b | lymphoid enhancer binding factor 1 |
| OLR1 | 0.79 | 6.56 | 0.011 | 0.81 | 6.34 | 0.013 | 12p13.2c | oxidized low-density lipoprotein receptor 1 |
| PKD2 | 0.80 | 7.96 | 0.004 | 0.75 | 4.95 | 0.029 | 4q22.1b | polycystin 2, transient receptor potential cation channel |
| PLXNC1 | 0.79 | 6.37 | 0.023 | 0.60 | 3.96 | 0.034 | 12q22c | Plexin C1 |
| SAMD9 | 0.84 | 6.91 | 0.017 | 1.16 | 4.95 | 0.031 | 7q21.2b | sterile alpha motif domain-containing 9 |
| SLAMF9 | −0.82 | 5.88 | 0.027 | 0.62 | 9.82 | 0.039 | 1q23.2c | SLAM family member 9 |
| TP53TG1 | −0.77 | 6.69 | 0.001 | 0.65 | 5.45 | 0.048 | 7q21.12 | TP53 target 1 |
| CEBPA | −0.72 | 7.63 | 0.026 | 0.93 | 8.39 | 0.011 | 19q13.11b | CCAAT enhancer-binding protein alpha |
| CKB | −0.87 | 7.06 | 0.020 | 0.93 | 4.39 | 0.003 | 14q32.33a | creatine kinase B |
| Dataset | Signature | Pathway | p val | padj | ES | NES | Size | Leading Edge Genes |
|---|---|---|---|---|---|---|---|---|
| GSE50509 | C1 | chr19q13 | 0.0000 | 0.0000 | −0.820 | −3.386 | 11 | TMEM145, PAFAH1B3, BLVRB, LIN7B, CNFN, LILRA2, LRFN3, CEBPA, ZNF296, NCCRP1 |
| C2:CP | NABA_MATRISOME | 0.0010 | 0.0021 | −0.714 | −2.380 | 15 | F12, CCL18, POSTN, SFRP4, SCUBE2, CXCL8, PDGFD, SULF1, PRG4, EMCN, PLXNC1, SPP1, SPARCL1, ANXA1 PLXNC1, SPP1, SPARCL1, ANXA1 | |
| C2:CP | NABA_MATRISOME_ASSOCIATED | 0.0142 | 0.0142 | −0.563 | −1.829 | 11 | F12, CCL18, SFRP4, SCUBE2, CXCL8, PDGFD, SULF1, EMCN, PLXNC1 | |
| C3:“MIR:MIRDB” | MIR106B_5P | 0.0002 | 0.0336 | 0.451 | 2.338 | 31 | KLF9, SOS1, BBX, IL1RAP, SSH1, KAT2B, PKD2, JAK1, NPAT, REV3L, ANO6, TMX3, PAFAH1B1, RBM12B, PDE3B, BMPR2, CALD1, SPTY2D1, ARID4B, PLEKHA3 | |
| C3:“MIR:MIRDB” | MIR20A_5P | 0.0002 | 0.0336 | 0.451 | 2.338 | 31 | KLF9, SOS1, BBX, IL1RAP, SSH1, KAT2B, PKD2, JAK1, NPAT, REV3L, ANO6, TMX3, PAFAH1B1, RBM12B, PDE3B, BMPR2, CALD1, SPTY2D1, ARID4B, PLEKHA3 | |
| C3:“MIR:MIRDB” | MIR106A_5P | 0.0003 | 0.0484 | 0.457 | 2.327 | 29 | KLF9, SOS1, BBX, IL1RAP, SSH1, KAT2B, PKD2, JAK1, NPAT, REV3L, ANO6, TMX3, RBM12B, PDE3B, BMPR2, CALD1, SPTY2D1, ARID4B, PLEKHA3 | |
| C3:“TFT:TFT_Legacy” | TAATTA_CHX10_01 | 0.0006 | 0.0493 | 0.423 | 2.144 | 31 | TSPAN7, SORBS2, TRIM24, CDH19, PRRX1, PPFIBP1, AMD1, MEF2C, OSBPL8, SECISBP2L, IFI16, BMPR2, CALD1, BAZ1A, CAB39, ZC3H7A, STXBP3, MYO1B, CPEB4, PELI2, EPB41L3 | |
| C5:“GO:BP” | GOBP_CIRCULATORY_SYSTEM_DEVELOPMENT | 0.0000 | 0.0145 | 0.470 | 2.484 | 35 | DUSP6, SOS1, SORBS2, PRRX1, MATR3, ARID2, EP300, KAT2B, PKD2, MEF2C, JAK1, SPRY2, LEF1, ROCK1, HTR2B, PDE3B, BMPR2, C1GALT1, ITGAV, CALD1 | |
| C5:“GO:BP” | GOBP_HEART_DEVELOPMENT | 0.0001 | 0.0145 | 0.573 | 2.445 | 18 | DUSP6, SOS1, SORBS2, MATR3, ARID2, EP300, KAT2B, PKD2, MEF2C, ROCK1, HTR2B, BMPR2 | |
| C5:“GO:BP” | GOBP_DEVELOPMENTAL_GROWTH | 0.0002 | 0.0145 | 0.567 | 2.418 | 18 | DUSP6, SOS1, SORBS2, IQGAP1, ARID5B, ARID2, EP300, MEF2C, SPRY2, MTM1, PAFAH1B1, BMPR2 | |
| C5:“GO:BP” | GOBP_ANIMAL_ORGAN_MORPHOGENESIS | 0.0001 | 0.0145 | 0.511 | 2.404 | 24 | CSGALNACT1, SOS1, SNX10, PRRX1, ARID5B, ARID2, EP300, PKD2, MEF2C, SPRY2, LEF1, HTR2B, PAFAH1B1, BMPR2 | |
| C5:“GO:BP” | GOBP_REGULATION_OF_PHOSPHORUS_METABOLIC_PROCESS | 0.0002 | 0.0145 | 0.448 | 2.324 | 33 | DUSP6, SOS1, IQGAP1, ACSL3, EP300, KAT2B, PKD2, MEF2C, SPRY2, TNIK, PTPN13, OSBPL8, GNAQ, ROCK1, HTR2B, BMPR2, BST1, CAB39, CD44, SASH1 | |
| C5:“GO:BP” | GOBP_VASCULATURE_DEVELOPMENT | 0.0004 | 0.0266 | 0.473 | 2.254 | 25 | SOS1, PRRX1, ARID2, PKD2, MEF2C, JAK1, SPRY2, LEF1, ROCK1, PDE3B, BMPR2, C1GALT1, ITGAV, CALD1, SASH1, HECTD1, ANXA1 | |
| C5:“GO:BP” | GOBP_TISSUE_DEVELOPMENT | 0.0005 | 0.0281 | 0.358 | 2.211 | 55 | MCOLN3, CSGALNACT1, SOS1, SORBS2, IQGAP1, SNX10, PRRX1, ARID5B, ARID2, EP300, PKD2, MEF2C, GSTM3, SPRY2, LEF1, UGCG, MTM1, ROCK1, ANO6, HTR2B, PAFAH1B1, BMPR2, C1GALT1, ITGAV | |
| C5:“GO:BP” | GOBP_DEPHOSPHORYLATION | 0.0008 | 0.0369 | 0.578 | 2.196 | 13 | NT5C3A, DUSP6, IQGAP1, SSH1, MEF2C, PTPN13, MTM1, ROCK1 | |
| C5:“GO:BP” | GOBP_POSITIVE_REGULATION_OF_PHOSPHORUS_METABOLIC_PROCESS | 0.0010 | 0.0404 | 0.496 | 2.126 | 19 | IQGAP1, ACSL3, PKD2, MEF2C, SPRY2, TNIK, OSBPL8, ROCK1, HTR2B, BMPR2, CAB39, CD44, SASH1 | |
| C5:“GO:BP” | GOBP_POSITIVE_REGULATION_OF_TRANSFERASE_ACTIVITY | 0.0011 | 0.0404 | 0.574 | 2.116 | 12 | IQGAP1, PKD2, SPRY2, OSBPL8, HTR2B, BMPR2, CAB39, SASH1, HNRNPA2B1, ARRDC4, TOM1L1 | |
| C5:“GO:BP” | GOBP_TUBE_MORPHOGENESIS | 0.0013 | 0.0456 | 0.451 | 2.183 | 26 | SOS1, PRRX1, ARID2, PKD2, MEF2C, JAK1, SPRY2, LEF1, ROCK1, PDE3B, BMPR2, C1GALT1, ITGAV, CALD1, SASH1, HECTD1, ANXA1 | |
| C5:“GO:BP” | GOBP_EMBRYO_DEVELOPMENT | 0.0016 | 0.0490 | 0.409 | 2.088 | 30 | SOS1, TBC1D23, PRRX1, ARID2, EP300, PKD2, MEF2C, SPRY2, LEF1, NPAT, ROCK1, HTR2B, PAFAH1B1, BMPR2, ITGAV | |
| C5:HPO | HP_DOWNSLANTED_PALPEBRAL_FISSURES | 0.0002 | 0.0343 | 0.706 | 2.192 | 10 | TSPAN7, CSGALNACT1, MPDZ, SOS1, PRRX1, ARID2, EP300 | |
| C8 | FAN_OVARY_CL1_GPRC5A_TNFRS12A_HIGH_SELECTABLE_FOLLICLE_STROMAL_CELL | 0.0003 | 0.0338 | 0.553 | 2.183 | 15 | ARID5B, AMD1, GSTM3, IFI16, UBL3, PLOD2, CALD1, SGCE, HNRNPA2B1, CTNNAL1, RHOBTB3, DDX21, EIF3J | |
| C8 | MANNO_MIDBRAIN_NEUROTYPES_HPERIC | 0.0013 | 0.0342 | 0.418 | 2.157 | 31 | DUSP6, SOS1, BBX, IQGAP1, MATR3, ARID2, SSH1, EP300, SPATA13, KAT2B, JAK1, SH3KBP1, ST3GAL5, NPAT, VPS26A, SNRK, OSBPL8, REV3L, BTBD1, ROCK1, TYW3, YTHDC2, IFI16, PAFAH1B1, ANKRD49, RAD21, SLC2A3, TBC1D4, BAZ1A, CAB39, TRAPPC8, CD44, ARID4B, HNRNPA2B1, ITPR1, ANXA1, TMEM123, BCLAF1, U2SURP, MYCBP2, SAMD9, PDS5A, FNBP4, ACAP2, ACTR2 | |
| C8 | BUSSLINGER_GASTRIC_IMMUNE_CELLS | 0.0009 | 0.0342 | 0.320 | 2.092 | 73 | DUSP6, SOS1, BBX, IQGAP1, MATR3, ARID2, SSH1, EP300, SPATA13, KAT2B, JAK1, SH3KBP1, ST3GAL5, NPAT, VPS26A, SNRK, OSBPL8, REV3L, BTBD1, ROCK1” | |
| C8 | FAN_EMBRYONIC_CTX_BRAIN_ENDOTHELIAL_2 | 0.0017 | 0.0342 | 0.531 | 2.045 | 14 | GNG12, JAK1, ST3GAL5, SNRK, CALD1, SASH1, SGCE, MYO1B, ITPR1, FRMD6, PHACTR2, SPARCL1 | |
| C8 | RUBENSTEIN_SKELETAL_MUSCLE_SMOOTH_MUSCLE_CELLS | 0.0016 | 0.0342 | 0.514 | 2.030 | 15 | BBX, SORBS2, ARID5B, MEF2C, TCEA1, TAX1BP1, SNRK, ROCK1, CALD1, SGCE, SPARCL1, NSA2, PCNP, PRSS23 | |
| C8 | MURARO_PANCREAS_MESENCHYMAL_STROMAL_CELL | 0.0021 | 0.0362 | 0.402 | 2.072 | 31 | DUSP6, KLF9, PRRX1, PPFIBP1, ARID5B, FAM114A1, SSH1, JAK1, SH3KBP1, ANO6, IFI16, PLOD2, ITGAV, SLC2A3, CALD1, BAZ1A, SASH1, ANXA1, FRMD6, DDX21 | |
| GSE61992 | C2:CGP | CHICAS_RB1_TARGETS_SENESCENT | 0.0000 | 0.0127 | 0.425 | 2.743 | 34 | PITPNC1, IFI6, HIPK2, TREM1, APCDD1L, VEGFA, TXNIP, IL6, CA12, GBP3, GK, FEZ1, CTSS, ODC1, CXCL5, PSMB8, EIF4EBP1, HLA-B, PRR11, CXCL10, IFITM1 |
| C2:CGP | PEREZ_TP53_AND_TP63_TARGETS | 0.0002 | 0.0406 | 0.569 | 2.459 | 14 | MYLIP, NEFL, CKB, GPT2, SEMA4D, RBP7, PPP1R16B, ADAP2, MAFB, RASGRP1 | |
| C2:CGP | YOSHIMURA_MAPK8_TARGETS_UP | 0.0002 | 0.0406 | 0.298 | 2.329 | 52 | NEFL, RGS22, ENPP6, CD247, ZNF226, LGALS9, FOXM1, PTPN3, KCNK1, CSNK1D, NELL2, FEZ1, ITGB3, CD74, CTSS, INPP5D, MAFB, PSMB8, TAP2, PLEK, DUSP4, HLA-DQA1, LYZ, KLRB1, CCL5, RGS18, GZMB, NMU, PFKFB3, CD52 | |
| C2:CGP | BLANCO_MELO_COVID19_SARS_COV_2_POS_PATIENT_LUNG_TISSUE_UP | 0.0003 | 0.0471 | 0.411 | 2.318 | 26 | OAS2, IFI6, TREM1, IFIT1, OLR1, TNF, OAS3, WAS, TYROBP, CD37, HSH2D, CXCL10, PLEK, IFITM1, SAMD9, PLAC8, RGS18, RTP4 | |
| C2:CP | NABA_MATRISOME_ASSOCIATED | 0.0003 | 0.0015 | 0.413 | 2.348 | 43 | SCUBE3, PLOD1, SEMA4D, VEGFA, IL6, LGALS9, ADAMTSL2, TNF, CTSS, CXCL5, PLXDC2, CTSO, CXCL10, CTSW, P4HA2, ADAMTS9, ANGPTL7, SERPINI1, PLXNA2, GPC3, FLT3LG, CCL5 | |
| C2:CP | NABA_MATRISOME | 0.0010 | 0.0025 | 0.374 | 2.120 | 50 | SCUBE3, PLOD1, SEMA4D, VEGFA, IL6, LGALS9, ADAMTSL2, NELL2, TNF, CTSS, CXCL5, PLXDC2, CTSO, CXCL10, CTSW, P4HA2, ADAMTS9, ANGPTL7, SERPINI1, PLXNA2, GPC3, FLT3LG, CCL5, PLXNC1, HAPLN1 | |
| C3:“MIR:MIRDB” | MIR548P | 0.0001 | 0.0142 | 0.611 | 2.678 | 14 | DSP, RRAS2, PITPNC1, VEGFA, CEBPA, CSNK1D, IPPK, CXCL5, PLXDC2, MAFB | |
| C3:“MIR:MIR_Legacy” | MIR548P | 0.0000 | 0.0103 | 0.611 | 2.677 | 14 | DSP, RRAS2, PITPNC1, VEGFA, CEBPA, CSNK1D, IPPK, CXCL5, PLXDC2, MAFB | |
| C3:“TFT:GTRD” | ZNF513_TARGET_GENES | 0.0002 | 0.0311 | 0.493 | 2.500 | 19 | SCUBE3, CKB, PLOD1, SEMA4D, JPH1, TXNIP, CD6, NPAS1, HKDC1, HLA-F, ERC2, BOC, ADAMTS9, PKD2 | |
| C5:“GO:BP” | GOBP_HOMEOSTATIC_PROCESS | 8.00 × 10−6 | 0.0056 | 0.301 | 2.545 | 73 | MYLIP, TRIM6, SLC24A3, CKB, CSF1R, RPE65, IFI6, HIPK2, VEGFA, JPH1, IFIT1, IL6, CEBPA, LGALS9, PTPN3, CA12, NELL2, HCLS1, TNF, ITGB3, GPR65, CD74, INPP5D, HKDC1, NOD2, MAFB, P2RY8, STAT1, NTSR1, HSH2D, CXCL10, CHRNA1, CD55, METRNL, PKD2, LYZ, SLC40A1, LDLR, PLAC8, CCL5, HK2, NMU | |
| C5:“GO:CC” | GOCC_INTRINSIC_COMPONENT_OF_PLASMA_MEMBRANE | 0.0006 | 0.0502 | 0.239 | 2.214 | 83 | SLC6A15, SLC24A3, KIR2DL3, TSPAN13, KCNF1, IL2RB, CSF1R, SEMA4D, IL6, ADORA2B, TSPAN32, KCNK1, IL18RAP, OLR1, CD4, TNF, TLR1, ITGB3, GPR65, MPZL1, CD6, ITGAL, NOD2, PCDHB13, P2RY8, HLA-B, NTSR1, TYROBP, CD37, KLRD1, CHRNA1, GPNMB, BOC, PKD2, HLA-DQA1, SLC7A7, PLXNA2, GPC3, SLC40A1, LDLR, LRRC8C, PLXNC1 | |
| C6 | STK33_NOMO_UP | 0.0008 | 0.0528 | 0.425 | 2.140 | 21 | SLC24A3, FCHO2, RNASEL, SEMA4D, TREM1, HAVCR2, PTPN3, OLR1, GBP3, TLR1, EOMES | |
| C7:VAX | NAKAYA_PBMC_FLUARIX_FLUVIRIN_AGE_18_50YO_7DY_DN | 0.0002 | 0.0149 | 0.573 | 2.473 | 14 | VEGFA, HAVCR2, SIK1, CD6, HLA-F, MAFB, NR4A2, CD55, METRNL, ITPRIP, SAMSN1, PFKFB3 | |
| C8 | TRAVAGLINI_LUNG_EREG_DENDRITIC_CELL | 7.99 × 10−5 | 0.0155 | 0.334 | 2.513 | 48 | HLA-DOA, CSF1R, HIPK2, SEMA4D, TREM1, VEGFA, HAVCR2, LST1, OLR1, GK, CD4, HCLS1, TLR1, CD74, PLXDC2, HLA-DMB, MAFB, TYROBP, RNASE6, NR4A2, ARHGAP18, PLEK, METRNL, DUSP4, HLA-DQA1, DNAJC15, LYZ | |
| C8 | TRAVAGLINI_LUNG_CILIATED_CELL | 0.0002 | 0.0155 | 0.393 | 2.460 | 31 | DSP, RGS22, CKB, OAS2, IFI6, HES2, GBP3, NELL2, CD4, PPME1, CTSS, KLHL6, ARHGAP18, DNER, P4HA2, RARRES1, DUSP4, ACAP1, GPC3, PLAC8, RTP4 | |
| C8 | TRAVAGLINI_LUNG_TREM2_DENDRITIC_CELL | 0.0002 | 0.0155 | 0.293 | 2.334 | 57 | HLA-DOA, FCHO2, CSF1R, IFI6, TREM1, HAVCR2, CEBPA, LGALS9, OLR1, GK, CD4, FBP1, CD74, CTSS, PLXDC2, HLA-DMB, MAFB, EIF4EBP1, STAT1, TYROBP, RNASE6, ARHGAP18, CXCL10, RARRES1, GPNMB, LY86, HLA-DQA1, SLC7A7, LYZ, SLC40A1 | |
| C8 | AIZARANI_LIVER_C25_KUPFFER_CELLS_4 | 0.0005 | 0.0265 | 0.375 | 2.350 | 31 | HLA-DOA, CSF1R, VEGFA, LGALS9, LST1, HCLS1, CTSS, HLA-DMB, MAFB, KLHL6, TYROBP, RNASE6, NR4A2, PLEK, CD55, METRNL, HLA-DQA1, LYZ, GZMB, SAMSN1, ITGB2 | |
| C8 | TRAVAGLINI_LUNG_NATURAL_KILLER_CELL | 0.0007 | 0.0276 | 0.448 | 2.357 | 22 | CD247, IL2RB, HAVCR2, AOAH, GPR65, PYHIN1, ITGAL, HSH2D, GNLY, CTSW, KLRD1, IFITM1, KLRB1, PLAC8, CCL5, GZMB, ITGB2 | |
| C8 | TRAVAGLINI_LUNG_MACROPHAGE_CELL | 0.0009 | 0.0315 | 0.440 | 2.264 | 21 | IFI6, TREM1, IL6, OLR1, GK, FBP1, CD74, CTSS, PLXDC2, TYROBP, GPNMB, HLA-DQA1, SLC7A7, LYZ | |
| C8 | TRAVAGLINI_LUNG_LIPOFIBROBLAST_CELL | 0.0013 | 0.0356 | 0.507 | 2.236 | 14 | PITPNC1, VEGFA, PVT1, CD74, NR4A2, RARRES1, GPNMB, IFITM1, LONRF2, LDLR, HK2 | |
| C8 | HAY_BONE_MARROW_NK_CELLS | 0.0015 | 0.0356 | 0.300 | 2.220 | 46 | KIR2DL3, PITPNC1, CD247, IL2RB, HAVCR2, HCST, AOAH, TBC1D10C, TSPAN32, IL18RAP, FEZ1, GPR65, PYHIN1, ITGAL, CERCAM, EOMES, HLA-F, HSH2D, GNLY, DOK2, CTSW, KLRD1, IFITM1 | |
| C8 | AIZARANI_LIVER_C2_KUPFFER_CELLS_1 | 0.0015 | 0.0356 | 0.361 | 2.148 | 28 | HLA-DOA, CSF1R, LGALS9, LST1, HCLS1, CTSS, PLXDC2, HLA-DMB, MAFB, KLHL6, TYROBP, RNASE6, CD37, PLEK, LY86, HLA-DQA1, LYZ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu-Smith, F.; Lin, J. Unsupervised Analysis Reveals the Involvement of Key Immune Response Genes and the Matrisome in Resistance to BRAF and MEK Inhibitors in Melanoma. Cancers 2024, 16, 2313. https://doi.org/10.3390/cancers16132313
Liu-Smith F, Lin J. Unsupervised Analysis Reveals the Involvement of Key Immune Response Genes and the Matrisome in Resistance to BRAF and MEK Inhibitors in Melanoma. Cancers. 2024; 16(13):2313. https://doi.org/10.3390/cancers16132313
Chicago/Turabian StyleLiu-Smith, Feng, and Jianjian Lin. 2024. "Unsupervised Analysis Reveals the Involvement of Key Immune Response Genes and the Matrisome in Resistance to BRAF and MEK Inhibitors in Melanoma" Cancers 16, no. 13: 2313. https://doi.org/10.3390/cancers16132313
APA StyleLiu-Smith, F., & Lin, J. (2024). Unsupervised Analysis Reveals the Involvement of Key Immune Response Genes and the Matrisome in Resistance to BRAF and MEK Inhibitors in Melanoma. Cancers, 16(13), 2313. https://doi.org/10.3390/cancers16132313