Simple Summary
Medullary thyroid carcinoma (MTC) is driven by a small number of pathogenic genetic variants and tumours usually exhibit a correspondingly low tumour mutational burden. This reduces tumour visibility to the immune system and impacts the immune cell profile of the tumour microenvironment. In the last decade targeted pathway inhibitors have revolutionized the therapeutic landscape for patients with advanced disease, with increasing options for systemic therapy tailored to the molecular signature of the tumour. Therefore, understanding the molecular basis of disease, pathogenesis of immune evasion and mechanisms of escape of pathway inhibition is of paramount importance. Here, we summarize genetic and molecular drivers of MTC and their relevance to tumour immunogenicity, the cellular milieu of the tumour microenvironment, and response to targeted therapy.
Abstract
In this review, we explore the underlying molecular biology of medullary thyroid carcinoma (MTC) and its interplay with the host immune system. MTC is consistently driven by a small number of specific pathogenic variants, beyond which few additional genetic events are required for tumorigenesis. This explains the exceedingly low tumour mutational burden seen in most MTC, in contrast to other cancers. However, because of the low tumour mutational burden (TMB), there is a correspondingly low level of tumour-associated neoantigens that are presented to the host immune system. This reduces tumour visibility and vigour of the anti-tumour immune response and suggests the efficacy of immunotherapy in MTC is likely to be poor, acknowledging this inference is largely based on the extrapolation of data from other tumour types. The dominance of specific RET (REarranged during Transfection) pathogenic variants in MTC tumorigenesis rationalizes the observed efficacy of the targeted RET-specific tyrosine kinase inhibitors (TKIs) in comparison to multi-kinase inhibitors (MKIs). Therapeutic durability of pathway inhibitors is an ongoing research focus. It may be limited by the selection pressure TKI treatment creates, promoting survival of resistant tumour cell clones that can escape pathway inhibition through binding-site mutations, activation of alternate pathways, and modulation of the cellular and cytokine milieu of the tumour microenvironment (TME).
1. Introduction
Medullary thyroid carcinoma (MTC) is a rare neuroendocrine malignancy that arises from the parafollicular ‘C’ cells of the thyroid gland and accounts for 1–2% of all thyroid cancers [1]. It carries a poorer prognosis than differentiated thyroid cancer; however, there is significant variability in its clinical course; some patients living for decades with low volume nodal disease, whereas others die quickly from rapidly progressive metastases [2,3]. In the last decade, targeted pathway inhibitors have revolutionised the therapeutic landscape for patients with advanced disease, with increasing options for systemic therapy tailored to the molecular signature of the tumour. Therefore, understanding the molecular basis of disease, pathogenesis of immune evasion and mechanisms of escape of pathway inhibition is of paramount importance. In this review, we summarise genetic and molecular drivers of MTC and their relevance to tumour immunogenicity, the cellular milieu of the tumour microenvironment, and response to targeted therapy.
2. Materials and Methods
A comprehensive literature review utilising the PubMed database and Google Scholar search engine was performed. Keywords included “medullary thyroid cancer”, “medullary thyroid carcinoma”, “RET”, “tumour microenvironment”, “tyrosine kinase inhibitor”, “tyrosine kinase inhibitor resistance”, “tumour infiltrating lymphocytes” “immunotherapy”, and “targeted therapy”. The search was limited to articles published in English, and primarily focussed on articles published in the last 10 years, with the inclusion of selected relevant papers published outside of this timeframe. Following identification, papers were evaluated to ensure their relevance, appropriate methodology and clinical significance.
3. Genetic and Molecular Drivers of Disease
MTC can be broadly classified into hereditary and sporadic disease, with distinct associated molecular pathogenesis. Approximately 25% of MTC cases are ‘hereditary’ and associated with pathogenic germline variants in RET (REarranged during Transfection) protooncogene which cause MEN2 syndrome [4], including 5–7% of patients with apparently ‘sporadic’ disease in whom the mutation arises de novo [5]. Patients with pathogenic germline variants in RET have a lifetime risk of developing MTC that approaches 100% [6] and inheritance is autosomal dominant. The spectrum of clinical phenotypes includes Multiple Endocrine Neoplasia (MEN) type 2A and type 2B. Familial Medullary Thyroid Cancer (FMTC) syndrome is now recognized as a forme fruste of MEN2A [1]. Within these groups, there is both inter- and intra-family variation in clinical manifestations. Different RET mutations confer different levels of risk with respect to the age at which MTC typically develops and associated prognosis. For example, patients with the RET codon M918T variant are at the highest risk of aggressive disease and are recommended to undergo thyroidectomy within the first six months of life, whereas patients with RET codon C634 variants (responsible for approximately 75% of MEN2A) are at intermediate risk and should undergo thyroidectomy by the age of 5 y [1].
4. The RET Protooncogene and Hereditary Disease
RET encodes a transmembrane tyrosine kinase receptor (TKR), composed of an extracellular domain with a cysteine-rich region and four cadherin-like regions, a transmembrane domain, and an intracellular tyrosine kinase domain (Figure 1) [7]. Initially described by Takahashi et al. in the 1980s, it is located on chromosome 10q11.2 [4,8] and plays a crucial role in development of the genitourinary tract and nervous system [9,10,11] as a functional receptor for glial cell-derived neurotrophic factors (GDNFs).
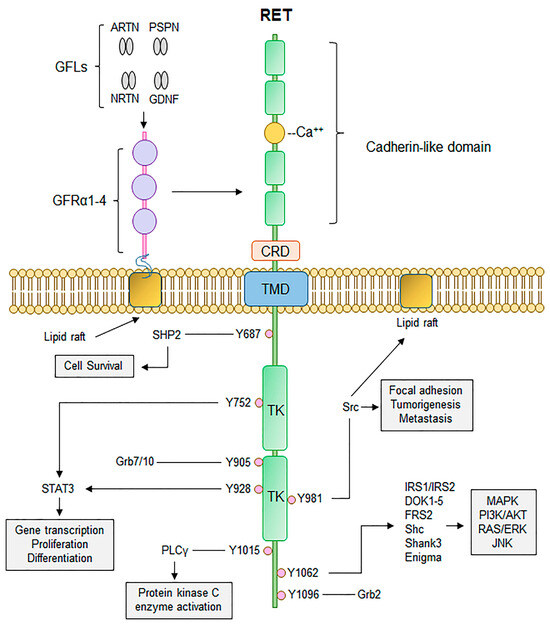
Figure 1.
Canonical RET signalling. RET activation occurs upon fulfilment of multiple steps. Binding of GDNF-family ligands (GFLs), to co-receptor GFRα1-4, concurrently with binding of calcium ions to the calcium binding domain, induces recruitment of RET, forming RET-GFRα complex. Formation of RET-GFRα complex brings two RET monomers in close proximity to induce homodimerization and cross phosphorylation of key RET tyrosine residues that recruit adaptor proteins important for propagation of RET signalling, such as PI3K/AKT, MAPK, and RAS/RAF/ERK. Thus, activation of RET signalling ultimately promotes cell proliferation, growth, and survival through activation of multiple downstream signalling cascades. CRD, cysteine-rich domain; TMD, transmembrane domain; TK, tyrosine kinase domain. Adapted from [12]. Published under a Creative Commons Attribution (CC BY) License.
In absence of ligand binding, the RET TKR exists as a single unphosphorylated TKR. With ligand binding, it undergoes dimerization and phosphorylation to activate several intracellular signalling pathways, including the p38 mitogen-activated protein kinase (MAPK) pathway, the phosphatidyl-inositol 3-kinase (PI3K) pathway and the extracellular signal-related kinase (ERK) pathway, which result in cellular proliferation [13]. Pathogenic variants cause constitutional activation and therefore drive downstream pathways independent of ligand binding to the extracellular domain.
The mechanism of oncogenic activation differs depending on the region of the RET TKR that is abnormal. Variants in the extracellular domain trigger ligand-independent dimerization and autophosphorylation, which in turn activate downstream intracellular signalling pathways. In comparison, if the variant occurs in the intracellular tyrosine kinase domain, autophosphorylation occurs without dimerization, which may drive activation of additional downstream pathways that are not typically controlled by the RET TKR when activated through the extracellular domain [14]. In RET fusion events, the transmembrane domain is absent and the kinase domain is constituently active [12].
Several distinct activating RET single nucleotide variants, with corollary phenotypic patterns [1], drive the majority of MTC cases; although, RET appears to be the only germline driver gene mutation recurrently involved in MTC [15]. The most common is the M918T variant, followed by variants in cysteine codons (C634, C620, C618, C630, C609 and C611). However, with improved recognition and genetic screening technology, over 100 mutation hotspots have been identified [1]. The type of RET variant may help to differentiate sporadic and hereditary MTC. For example, C630 variants suggest sporadic disease, whereas mutational hotspots of C609, C611, D631 and V804 suggest hereditary disease [16]. Furthermore, routine genetic screening of MTC patients with apparent sporadic disease results in reclassification of approximately 5–7% of cases as hereditary, mostly within the subgroup of MEN2A [17].
The specific driver event also informs assessment of disease biology and associated phenotypic manifestations [1], through activation of distinct intracellular signalling cascades with associated transcriptional implications and gene expression [18]. For example, neuronatin (NNAT), cell division cycle 14B (CDC14B) and protein tyrosine phosphatase receptor type T (PTPRT) are upregulated in patients with variants in the cysteine-rich region of the RET TKR extracellular domain (“MEN2A-like”) [14], whereas gamma-aminobutyric acid type A receptor subunit rho1 (GABRR1) and neurotrophic tyrosine receptor kinase 3 (NTRK3) are upregulated up to five-fold in patients with variants in the intracellular tyrosine kinase domain (“MEN2B-like”), which is associated with an aggressive biological phenotype [19]. Variants associated with MEN2B have also been demonstrated to suppress expression of genes required for recruitment and action of NK cells and T-cells in the tumour microenvironment (TME), such as chemokine C-X3-C motif ligand 1 (CX3CL1), with inflammatory cell infiltrates only seen in MEN2A/FMTC-associated tumours [20]. Interestingly, gene expression profiles of MTC driven by germline (hereditary) compared to somatic (sporadic) variants are not significantly different [14], suggesting activation of similar signalling pathways for a given RET mutation [21].
From an oncogenesis perspective, secondary genetic events are required before MTC develops in patients with pathogenic germline RET variants [22], analogous to the ‘two-hit’ hypothesis [23]. However, the second event (e.g., loss of the normal allele or duplication of the RET-mutant allele [24]) may require only a minor genetic alteration, particularly in association with an M918T mutation, as overall mutational burden in MTC is exceedingly low [15], and oncogenic progression of C cells from normal, to hyperplastic, to malignant evolves rapidly, and may occur in the first months of life [25].
5. Sporadic Disease
Although MTC is associated with a low mutational burden [15], most tumours are driven by a specific identifiable pathogenic driver variant [26]. In sporadic disease, somatic pathogenic RET variants drive approximately half of cases, with rat sarcoma virus (RAS) mutations driving 70% of RET wild-type tumours [27]. Interestingly, these two dominant driver mutations appear to be mutually exclusive. In addition to RET and RAS, uncommon variants in BRAF and NF1 have also been described as oncogenic drivers and there is an increased risk of MTC in NF1 syndrome [28]. There remains a small proportion of sporadic MTC cases with unknown driver events.
Additional pathogenic variants in tumour suppressor genes or DNA repair genes, as well as upregulation of genes that have a synergistic effect in oncogenesis may also contribute to disease progression, with mutations in tumour protein 53 (TP53), cluster of differentiation 117 (KIT), mutS homologue 6 (MSH6), mutL homologue 1 (MLH1), ataxia-telangiectasia mutated (ATM), von Hippel-Lindau (VHL), phosphatase and tensin homolog (PTEN), cyclin dependent kinase 2A (CDKN2A) and serine/threonine kinase 11 (STK11) seen in RET or RAS mutant tumours [16,29]. However, these are secondary genetic events that contribute to a more aggressive tumour phenotype rather than primary driver variants [30]. Similarly, epigenetic factors including microRNA overexpression (miR-183 and miR-375) may modulate the clinical phenotype of disease [31].
6. Molecular Subtyping
More recently, a proteome-based stratification of MTC into three molecular subtypes (metabolic, basal and mesenchymal) has been proposed, with distinct genetic and epigenetic profiles [28]. For example, tumours driven by the RET M918T variant were predominantly of the mesenchymal subtype, with prevalent upregulation of extracellular matrix pathways, frequent epigenetic DNA methylation and the poorest prognosis, whereas RAS-driven tumours were more likely to be of the metabolic subtype, with upregulation of pathways related to cellular metabolism, higher frequency of somatic copy number alterations (CHEK2, MUTYH, TP53, ATM, MLH1), strong activation of the MAPK and PI3K pathways, and associated with an intermediate prognosis [28]. In comparison, the basal subtype was the most genetically stable and carried the best prognosis. It retained a greater degree of neuroendocrine differentiation, with upregulation neuroendocrine markers including CEA, chromogranin A, synaptophysin and neural cell adhesion molecule 1 (NCAM1, CD56), more closely resembling normal C cells. Further studies are required to determine whether this classification system will prove clinically useful.
7. Interplay between the Immune System and Medullary Thyroid Cancer
An Overview of the Immune System and Carcinogenesis
The immune system plays an important role in suppressing cancer development [32]. It has been more than half a century since Burnet described the concept of immunological surveillance as fundamental to the maintenance of tissue homeostasis, occurring through lymphocyte-mediated recognition and elimination of genetically mutated somatic cells [33]. Despite initial scepticism, this hypothesis was supported by observing oncogenic viruses caused tumours with increased frequency in immunocompromised patients. Subsequently, observational studies from around the world have reported immunocompromised patients have higher standardised incidence ratios for the development several cancers with no known viral trigger, including colonic, lung, pancreatic, urothelial, endocrine and malignant melanoma [34].
The immune system can be described as a collection of physiological processes that enable recognition and elimination of foreign, or “non-self” antigens, and is broadly subdivided into innate and adaptive processes. Innate immunity involves recognition of the structural components of an ‘intruder’, known as pathogen-associated molecular patterns, through several germline-encoded pathogen recognition receptors [35]. Cellular mediators of the innate immune response include macrophages, neutrophils, and natural killer cells. Activation leads to antigen presentation, phagocytosis, and apoptosis, as well as expression of pro-inflammatory cytokines. These cytokines trigger an iterative augmentation of the innate immune response, as well as engagement of adaptive immune pathways. Adaptive immunity is mediated by clonal expansion of effector T and B cells, targeted to a specific antigen, initiated by innate immune signals [35].
Most tumour cells express antigens that can be recognised by the immune system. Despite this, cancer cells can adapt to evade detection through a process known as immunoediting [36]. Normally, the immune system is able to clear tumour, or hold it in a state of ‘equilibrium’ through innate (natural killer and dendritic cells) and adaptive (CD4 and CD8 T cells) mechanisms, until mutating tumour cells acquire the ability to evade detection or elimination [37] and therefore progress to a clinically significant pathology [38]. By manipulating the cytokine and chemokine milieu of the TME, cancer cells may suppress activation or efficacy of immune cells, resulting in ‘tolerance’ rather than clearance [7]. This escape from immune surveillance may be considered the ‘seventh hallmark’ of cancer [39], and similarly, the vigour of the immune response to genetically altered tissue has been demonstrated to be a prognostic factor for survival in melanoma [40], colorectal, breast and ovarian cancer [38]. Most published data describing the immune microenvironment in thyroid cancer pertain to differentiated thyroid cancer (papillary thyroid carcinoma and follicular thyroid carcinoma) and anaplastic thyroid cancer [41], and given fundamental differences in the underlying neuroendocrine biology of MTC, the degree to which these results can be extrapolated is uncertain.
8. Mechanisms of Immune Evasion
8.1. Immune Suppression Mediated by the Tumour Microenvironment
The TME is composed of extracellular matrix, lymphatics, mesenchymal and immune cells, and plays an important role in the suppression of the anti-tumour immune response [4]. The TME differs from physiological tissue, with relative tissue hypoxia, increased acidity due to lactate-producing metabolic processes, and increased reactive oxygen species [42]. Substrates for cellular metabolism are consumed by tumour cells beyond the ability of homeostatic regulation, and secondary by-products accumulate [43]. Infiltrating immune cells require nutrients and a physiological interstitial milieu to mount an effective immune response [44], and hence, the hostile nature of the TME impairs the immune cell effector function. Cancer cells may adapt to rely on aerobic glycolysis instead of oxidation phosphorylation and therefore deplete glucose in the TME required for T cell activation and effector function [45]. Similarly, elevated lactate levels inhibit T cell signalling [46] and reduce production of effector cytokines, including perforin and granzyme B [47].
The cytokine and chemokine milieu of the TME affects the recruitment and differentiation of key immune effector cells and may create either a tumour-inhibiting or tumour-promoting environment. For example, increased expression of immune suppressive cytokines, such as transforming growth factor β (TGF-β), vascular endothelial growth factor (VEGF) and interleukin 10 (IL10) may inhibit anti-tumour immune response [48]. In MTC, the specific RET driver variant may alter expression of genes encoding cytokines and chemokines involved with recruitment and stimulation of T cells and NK cells to the TME [20]. Furthermore, recruitment of immune suppressive cells, such as myeloid derived suppressor cells and M2 macrophages, may render effector T cells in the TME dysfunctional through production of immunosuppressive cytokines [49]. Pozdeyev et al. described the effect of myeloid infiltrate in MTC, in which CD163+ M2 macrophages were frequently present [30], producing cytokines and chemokines that promote angiogenesis, including vascular endothelial growth factor (VEGF) and prostaglandin E2 [50]. These macrophages also manifested a dysfunctional phenotype, lacking the Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) activation pathways normally triggered by pro-inflammatory cytokines, and contributing to tumour ‘tolerance’ [51]. Specific genetic events, such as alterations in the B-catenin/WNT pathway and loss of PTEN, may also impair intra-tumoral infiltration of functional antigen presenting cells, thereby diminishing the anti-tumour immune response [49]. Similarly, additional inhibitory proteins upregulated on the tumour cell surface, such as integrin associated protein (CD47), may affect efficacy of phagocytic cells and have been described to promote tumour progression and metastasis in MTC [52]. Ultimately, for a sustained anti-tumour immune response, activation and clonal expansion of key effector cells must be initiated and iteratively expanded [41], and if the TME disrupts this process through the balance of cytokines and chemokines, the cancer is able to escape immune control and progress to become clinically significant [42].
8.2. Immune Suppression through Surface Receptor Co-Stimulatory Inhibition
In addition to the cytokine milieu of the TME, cell-surface receptors play an important role in modulation of lymphocyte activation, through co-stimulatory and co-inhibitory ligand pairs. There is accumulating evidence that immune co-inhibitory receptors (CIRs) and their respective ligands on peri-tumoral lymphocytes interact within the TME, thereby facilitating immune evasion and escape [53]. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death receptor 1 (PD-1) have been demonstrated to be key regulators of T-cell anti-tumour immune response and are effective targets for immunotherapeutic agents [54,55].
8.2.1. Cytotoxic T-Lymphocyte-Associated Protein 4 (CTLA-4)
CTLA-4 binds to CD80 and CD86 and is a co-stimulatory signal involved in T-cell activation and survival. Expressed on the T cell surface, CTLA-4 outcompetes CD28 for ligand binding and therefore inhibits T-cell activation [56]. However, tumour cells may adapt to express CTLA-4, modulating the anti-tumour immune response [57]. In MTC, it has been suggested that tumour-associated CTLA-4 expression is exclusive to sporadic cases [58] and may contribute to the stage-adjusted worse prognosis seen when comparing sporadic and hereditary disease [59].
8.2.2. PD-1
The programmed death receptor (PD-1) [60] is expressed on activated T cells, B cells, monocytes, NK cells and DCs. Through the binding of its ligand PD-L1, it acts to inhibit the cytotoxic T cell response [61]. Tumours may harness this immunosuppressive mechanism through expression of PD-L1, adversely affecting prognosis in several cancers [62], including papillary thyroid cancer [63]. In MTC, these inhibitory pathways have not been extensively explored and their contribution to oncogenesis is unclear. Two small cohort studies have demonstrated very low PD-L1 expression on primary MTC tumours [30,64]. In a larger series of 201 consecutive primary MTCs, PD-L1 staining was observed in 14% of cases, with expression correlating with advanced TNM stage and prognosis [65].
In addition to the upregulation of PD-L1 on the surface of tumour cells, cancer cell-intrinsic expression of both PD-L1 and PD-1 may result in modulation of the PD-1/PD-L1 axis, with downstream activation of the mTOR signalling pathway [66]. In MTC, PD-1/PD-L1 co-expression may occur in up to 50% of PD-1 positive tumours [58,67], and correlate with advanced disease stage [67]. However, the clinical relevance of these findings remains uncertain.
The PD1/PD-L1 pathway can be disrupted by PD-1 inhibitors, such as pembrolizumab and nivolumab, which enhance tumour recognition by cytotoxic T cells [68,69], and have been shown to induce durable anti-tumour response for a variety of tumours, many of which were not considered to be particularly susceptible to immunotherapy [70,71]. However, predicting which patients will respond to therapy is challenging [72], and overall response rates to PD-1 inhibitors remain low (~20–25% of treated patients) [73]. In a small phase II study that included 7 patients with MTC, no pathological responses were seen with combination anti-PD1 and anti-CTLA4 treatment (nivolumab and ipilimumab), whereas in anaplastic thyroid cancer a partial response was seen in 3/10 of patients, including two with a complete response [74]. These findings require validation in larger cohorts; however, they suggest anti-PD1 immunotherapy has limited efficacy in MTC. This lack of efficacy may be affected by the low tumour mutational burden (TMB) seen in MTC and expression of additional regulatory molecules, such as polio virus receptor (CD155). Expressed in a variety of cancers including metastatic MTC, it interacts with CD8+ T cells to promote to immune evasion [30].
9. Emerging Co-Inhibitory Receptors
The T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) has recently been identified as a potential therapeutic target [75]. When expressed on T-cells, TIM-3 is an indicator of T cell exhaustion in both chronic viral infections and malignancy [76,77], and its overexpression on tumour cells has been described in several solid tumours, including lung, gastric, colon, hepatocellular and urological malignancies [78], with increased levels correlating with poor survival [78,79]. Expression of TIM-3 has been described in 48% of primary MTC tumours and was associated with extensive locoregional metastasis, advanced stage and disease recurrence [58]. However, TIM-3 was expressed solely on tumour cells and not on tumour-infiltrating lymphocytes (TILs). Corroborating this finding, an analysis of TILs in MTC tumours demonstrated sparse TIM-3 expression [30]. The exact mechanism of MTC tumour intrinsic TIM-3 remains poorly understood but is thought to involve the nuclear-factor kB (NF-kB) pathway [80]. NF- kB encompasses a family of transcription factors involved in the regulation of cytokines and their receptors, as well as cell adhesion molecules [81], and may play a role in carcinogenesis through inhibition of apoptosis and promotion of cell cycle progression [80].
CD276 (B7 Homolog 3, B7-H3) is a recently described immune checkpoint molecule, that suppresses T cell activation and proliferation, and is overexpressed in a variety of malignancies including MTC [82]. It has been shown to promote tumour cell immune evasion and T cell inhibition, by altering secretion of pro-inflammatory cytokines [7]. In the context of MTC, CD276 was found to be expressed three-fold higher in tumour tissue, although correlation with histopathological factors and prognosis was equivocal [7,82].
Tumour-Infiltrating Lymphocytes (TILs)
The presence of TILs is an indicator of anti-tumour immune activation and is associated with improved prognosis in several malignancies [83,84]. The International TILs Working Group (ITWG) published a standardised approach for assessment of TILs in breast cancer [85], which has since been applied to several other solid malignancies [83,84]. In brief, the ITWG approach reports TILs as a percentage of the surface area of the stromal component within the borders of invasive tumour, and excludes TILs outside this area, as well as in zones affected by crush artifact or necrosis from biopsy sites [85]. Using a standardised methodology for assessment of TILs is particular important in thyroid cancer, as T cell infiltration into the thyroid gland is a common histopathological finding, for example, in chronic lymphocytic (Hashimoto’s) thyroiditis [86]. In differentiated thyroid cancer, lymphocytic infiltration has been reported to correlate with improved survival [87] and lower rates of extrathyroidal extension [88], suggesting the immune response may be suppressing tumour growth. In MTC, data are limited. Scopsi et al. found lymphocytic infiltrate to be associated with a favourable prognosis; however, they commented that no true TILs were identified [89]. French et al. demonstrated that background lymphocytic infiltrate had no correlation with disease stage or prognosis, whereas true TILs were associated with advanced disease stage, locally invasive tumours and lymph node metastases [90]. Pozdeyev et al. reported organised immune cell infiltration (predominantly, CD8 T cells) in 49% of primary and 90% of metastatic MTC tumours [30]; however, this was not assessed using the ITWG approach, and the prognostic correlation requires further research.
The phenotypic subtype of TILs also influences the degree to which they recognise tumour antigens [91] and the response to immunotherapy. For example, not all TILs display signs of clonal expansion to tumour antigens and rather can be considered ‘bystander’ T cells (CD 39– CD8+ cells). A predominance of bystander cells may be associated with poor response to immunotherapy [92]. Furthermore, a high proportion of infiltrating regulatory T cells (Tregs—CD3 + CD4 + CD25 + FoxP3+), and a low CD8:Treg ratio have been demonstrated to correlate with tumour size and lymph node metastases in papillary thyroid cancer [90]. Hence, the absolute number of TILs alone may not necessarily imply an immunologically ‘hot’ [93] tumour or predict a favourable response to immunotherapy.
Tregs develop in the presence of TGF-β and temper the immune response to allow tolerance of self-antigens [94]. Within TME, they can impair immune clearance of tumour cells through secretion of immunosuppressive cytokines and expression of co-inhibitory cell surface molecules (e.g., CTLA-4) and have been demonstrated to be associated with tumour progression and reduced survival in several cancers [95]. They have also been shown to interfere with effector lymphocyte ionised calcium uptake, which disrupts the NF-kB signalling pathway required for T cell activation [96]. However, there are conflicting data in the literature regarding prognostic significance of Tregs; their role in the TME may differ according to tumour type, with improved prognosis reported with Treg infiltration in the setting of colorectal cancer [97] and squamous cell carcinoma of the head and neck [95]. This may be due to general immune infiltration by multiple T cell subsets, and some authors suggest that the CD8:Treg ratio may be a better indicator of balance between immune tolerance and activation within the TME [98]. Interestingly, Salama et al. noted that although infiltration of Tregs in the TME was associated with an improved prognosis in colorectal cancer, high Treg density in adjacent normal mucosa was associated with a worse prognosis [97]. In MTC, limited data suggest that Tregs in the TME have a negative prognostic impact [99]; however, this hypothesis requires further empiric research.
10. Efficacy of Immunotherapy
Utilisation of immune checkpoint inhibitors (ICIs) for advanced disease has improved survival in several solid malignancies, most notably melanoma [100], non-small cell lung cancer [101], renal-cell carcinoma [102] and colorectal cancers with mismatch repair deficiency [103]. However, ICI therapy is not effective in all patients. Tumour mutational burden (TMB), the number of somatic mutations seen per megabase of tumour genome, has been demonstrated to be a predictive biomarker for response to ICI therapy, with increased TMB (>10 mutations/Mb) associated with greater response rates [104,105]. In tumours with <10 mutations/Mb, the objective response rate to ICI therapy is only 6% [104]. The underlying mechanism of ICI therapy efficacy with increased TMB likely relates to the higher proportion tumour-specific neoantigens that develop as a consequence of increased TMB [106], which can then be displayed on major histocompatibility complex (MHC) molecules of the tumour cell and recognised by T cells [107]. In comparison to most other solid tumours, MTC is associated with a low TMB, with the majority of tumours harbouring <1 mutation/Mb [108]. Most MTC tumours are driven by RET mutations, with few other mutated somatic genes [109], suggesting the MTC tumorigenesis pathway does not depend on the same degree of accumulation of genetic driver events required in other solid tumours, such as the adenoma–carcinoma sequence of colorectal cancer [110], explaining the low observed TMB. Therefore, it is unlikely that MTC patients will benefit from ICI therapy.
Pathogenic variants in genes encoding the antigen processing and presentation apparatus also affect the response to immunotherapy. Regulation of MHC proteins on the cell surface facilitates antigen presentation and determines ‘visibility’ of the cell to the immune system. Epigenetic modulation of MHC-I expression is required in utero for the foetus to avoid maternal immune attack of paternal MHC-I alleles, and this evolutionarily-preserved mechanism can also be exploited by cancer cells, particularly neuroendocrine tumours, to evade immune surveillance [111], with downregulation of MHC class I surface proteins and loss of B2-microglobulin. The clinical relevance of changes in MHC protein expression in MTC requires further evaluation, with only limited data published to date [30,112].
11. Current Systemic Treatment Options in MTC
11.1. Targeted Therapy with Pathway Inhibition
Although MTC is associated with a low TMB, the majority of driver events are clinically actionable with currently approved pathway inhibition therapies [113,114]. TKIs are homologs of adenosine triphosphate (ATP) and competitively occupy the ATP binding sites of tyrosine kinase receptors, thus inhibiting activation of associated signalling pathways [115]. The RET TKR shares similarities with other tyrosine kinases and therefore can be targeted by both multitarget tyrosine kinase inhibitors (MKIs) and selective tyrosine kinase inhibitors. MKIs, including vandetanib and cabozantinib, act on several TKRs, including the VEGF receptor, platelet derived growth factor (PDGF) receptor, hepatocyte growth factor (c-MET) receptor, epidermal growth factor receptor (EGFR) and the RET TKR; although, the dominant contribution to therapeutic efficacy comes from VEGF receptor inhibition (Figure 2). Given the broad range of receptor targets, MKIs are also associated with significant off-target toxicities, particularly affecting the gastrointestinal tract and liver. Among patients treated with vandetanib and cabozantinib, 35% and 79% required a dose reduction, and 12% and 16% required permanent discontinuation, respectively [116,117].
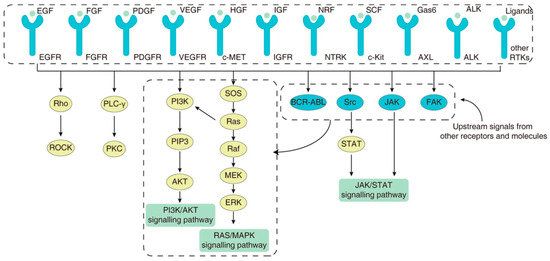
Figure 2.
The variety of surface tyrosine kinase receptors that play an important role in the maintenance of cellular homeostasis, through regulation of intracellular signalling pathways. The final effector pathways can be activated by several surface receptors, as well as by other intracellular activation pathways, highlighting the inherent difficulty in achieving sustained therapeutic efficacy through the blockade of a particular tyrosine kinase receptor. Adapted from [118]. Published under a Creative Commons Attribution (CC BY) License.
The need for better response rates and tolerability led to the development of selective RET inhibitors; selpercatinib [119] and pralsetinib [120]. Clinical trials have demonstrated lower rates of treatment discontinuation (2%) and reduced the severity of adverse effects associated with selpercatinib treatment compared to MKIs [121], with a significantly better objective response rate of 73% [119]. A recent phase 3 trial of selpercatinib in advanced RET-mutant MTC demonstrated a 12-month progression-free survival rate of 86.8% in comparison to 65.7% for the MKI control group, with 38.8% of patients requiring dose reduction due to toxicities vs. 77.3% in the MKI group [122]. The median progression-free survival had not been reached at the time of publication, and additional follow-up is required to define its ultimate durability.
11.2. Escape of Pathway Inhibition
Efficacy of TKIs may be limited by both ‘on-target’ and ‘off-target’ resistance mechanisms (Figure 2). On-target resistance refers to mutations that affect binding in the target kinase domain, whereas off-target resistance occurs through upregulation of bypass pathways [121,123].
12. On-Target Resistance
A large proportion of on-target mutations involve structural alteration of the receptor ATP-binding pocket (steric inhibition), which renders the drug inactive [124]. This may be an intrinsic or acquired mutation. For example, resistance to vandetanib occurs in MTCs with RETp.Val804Met ‘gatekeeper’ variants [121], in which a conformational change to spatial arrangement of the binding site prevents vandetanib from binding. In this situation, the amino acid substitution results in a greater hydrophobic force that impedes binding of the TKI to the receptor. Similarly, tumour-specific mutations in the target tyrosine-kinase receptor may confer intrinsic resistance to TKI treatment, such as EGFR mutations in patients with lung adenocarcinoma [125] and PDGFR in gastrointestinal stromal tumours [126].
In addition to gatekeeper variants, specific changes in exposed kinase residues of the receptor binding region (‘solvent front mutations’) may confer resistance. For example, the RET solvent front variant G810A results in the addition of a methyl group, which creates hydrophobic disruption of vandetanib binding, but still allows binging of nintedanib due to presence of a corresponding methyl group absent in vandetanib. Hence, RET G810A confers resistance to vandetanib, but not nintedanib [127]. Similarly, RET L881V-driven tumours are resistant to lenvatinib, cabozantinib and vandetanib due to absence of a phenyl ring required for binding [127], and presence of this phenyl ring on RET L730V confers resistance to nintedanib, due to a hydrophobic interaction [118]. Such solvent front mutations may be acquired during treatment because of selection pressure. For example, following an initial dramatic response to selpercatinib, emergence of RET G810R, G810S and G810C solvent front mutations result in the development of on-target resistance [128]. Furthermore, RET V804L/M and G810S mutations may confer pan-TKI resistance to the MKIs and RET-specific treatments [124]. Bypassing these mechanisms of resistance through the design of structurally different RET inhibitors is an ongoing area of research focus [129].
The binding affinity of TKIs may also be moderated by TKR mutations and hence also contribute to treatment resistance. For example, the RETS904F mutation has been demonstrated to increase the autophosphorylation of the mutant RET kinase, as well as the its ATP binding affinity, both of which reduce the efficacy of MKI therapy [130].
13. Off-Target Resistance
Off-target resistance can develop when proliferative pathways are activated by alternative mechanisms, such as MET [131] and KRAS that bypass the targeted kinase [121,130] (Figure 3). Because several receptor tyrosine kinases can activate the same downstream pathways (PI3K/AKT and RAS/MAPK), the therapeutic effect of blocking a particular tyrosine kinase receptor is reduced when tumour cells driver activation of the downstream pathways through alternative TKRs [132,133].
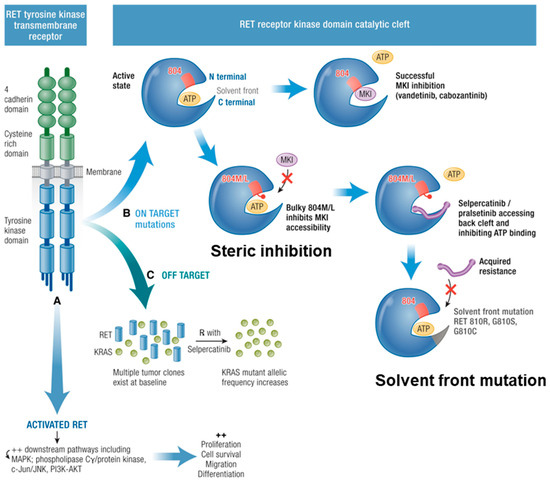
Figure 3.
Mechanisms of MKI resistance. RET tyrosine kinase transmembrane receptor has constitutive activation due to RET point mutations leading to downstream pathway activation. (A) Resistance mechanisms are described. (B) On target: the RET receptor kinase domain catalytic cleft is activated when ATP causes phosphorylation. MKIs (vandetanib, cabozantinib) can hinder the ATP binding when there is no V804M/L mutation with bulky hydrophobic side chains: steric inhibition. Acquired solvent front mutations RET G810R, G810S, G810C hinder this binding rendering selpercatinib ineffective. (C) Off Target mutations are shown with multiple tumour clones existing at baseline, reduction in RET, and subsequent increase in KRAS (or MET) allelic frequency. Adapted from [121]. Published under a Creative Commons Attribution (CC BY) License.
The selection pressure created by prolonged treatment allows selection of resistant clones, in which mutations in the TKR may result in constituent activation, limiting the efficacy of TKIs. In addition, resistance may develop through cytokine and chemokine modulation of the TME [134]. For example, the presence of inflammatory cytokines such as tumour necrosis factor-alpha may modulate the downstream pathways of the EGFR and hence reduce the efficacy of EGFR-targeted TKIs [135]. Similarly, chemokine signalling mediated through chemokine receptor 2 activates the downstream PI3K/AKT pathway which has been demonstrated to impact the efficacy of TKI therapy [136]. Furthermore, immunosuppressive M2 macrophages may reduce the efficacy of TKI therapy through the production of chemokines which can modulate both surface receptors such as MET [137] and downstream intracellular pathways [134].
Additional mechanisms of off-target resistance include the modification of the metabolic cellular pathways [138] and epigenetic modification, including DNA methylation [139], histone modifications [140] and mRNA modification [141]. Lastly, adaptive mechanisms to alter drug metabolism and limit intracellular drug concentration, including lysosomal sequestration and drug efflux, may also contribute to development of off-target resistance [118].
14. Conclusions and Future Directions
In this review, we highlight that MTC is consistently driven by a small number of specific pathogenic variants, beyond which few additional genetic events are required for tumorigenesis. This homogeneity of driver events explains the exceedingly low tumour mutational burden seen in MTC, in contrast to other cancers. However, as a result, there is a correspondingly low level of tumour-associated neoantigens presented to the host immune system. This reduces tumour visibility and the vigour of the anti-tumour immune response. In addition, it suggests the efficacy of immunotherapy in MTC is likely to be poor, acknowledging this inference is largely based on the extrapolation of data from other tumour types. Specific to MTC, the immune microenvironment has not been extensively described, with conflicting data published to date. Correlation of the cytokine and immune cell profile of the TME with the underlying molecular subtype, clinicopathological factors and prognosis, as well as description of changes that occur in the TME with TKI therapy remain important areas for future research.
The dominance of specific RET pathogenic variants in MTC tumorigenesis rationalises the observed superior efficacy of the targeted RET TKIs in comparison to MKIs. Therapeutic durability of RET-specific pathway inhibitors is also superior to that of the MKIs; however, the development of resistance to pathway inhibition remains an inherent limitation of TKI treatment. Resistance may develop through the selection pressure TKI treatment creates, promoting survival of resistant tumour cell clones that can escape pathway inhibition through binding-site mutations, activation of alternate pathways, and modulation of the cellular and cytokine milieu of the TME. The optimal therapeutic strategies to delay the emergency of resistance and the approach to management once resistance occurs remain important areas for future research.
Author Contributions
Conceptualization, A.J.P. and S.B.S.; methodology, A.J.P., H.S.-B., A.J.P. and H.S.-B.—original draft preparation, A.J.P.; writing—review and editing, A.J.P., R.C.-B., A.J.G. and S.B.S.; supervision, S.B.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma: The American Thyroid Association Guidelines Task Force on medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Roman, S.; Lin, R.; Sosa, J.A. Prognosis of medullary thyroid carcinoma: Demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2006, 107, 2134–2142. [Google Scholar] [CrossRef]
- Papachristos, A.J.; Nicholls, L.E.; Mechera, R.; Aniss, A.M.; Robinson, B.; Clifton-Bligh, R.; Gill, A.J.; Learoyd, D.; Sidhu, S.B.; Glover, A. Management of medullary thyroid cancer: Patterns of recurrence and outcomes of reoperative surgery. Oncologist 2023, 28, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Jaber, T.; Dadu, R.; Hu, M.I. Medullary thyroid carcinoma. Curr. Opin. Endocrinol. Diabetes Obes. 2021, 28, 540–546. [Google Scholar] [CrossRef]
- Elisei, R.; Romei, C.; Cosci, B.; Agate, L.; Bottici, V.; Molinaro, E.; Sculli, M.; Miccoli, P.; Basolo, F.; Grasso, L. RET genetic screening in patients with medullary thyroid cancer and their relatives: Experience with 807 individuals at one center. J. Clin. Endocrinol. Metab. 2007, 92, 4725–4729. [Google Scholar] [CrossRef]
- Sippel, R.S.; Kunnimalaiyaan, M.; Chen, H. Current management of medullary thyroid cancer. Oncologist 2008, 13, 539–547. [Google Scholar] [CrossRef]
- Hincza-Nowak, K.; Kowalik, A.; Walczyk, A.; Pałyga, I.; Gąsior-Perczak, D.; Płusa, A.; Kopczyński, J.; Chrapek, M.; Góźdź, S.; Kowalska, A. Immune Profiling of Medullary Thyroid Cancer—An Opportunity for Immunotherapy. Genes 2021, 12, 1534. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.G.; Inabnet, W.B.; Lo, C.Y. Endocrine Surgery; Springer: London, UK, 2009. [Google Scholar]
- Schuchardt, A.; D’Agati, V.; Larsson-Blomberg, L.; Costantini, F.; Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994, 367, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Pützer, B.M. Mechanisms of disease: Cancer targeting and the impact of oncogenic RET for medullary thyroid carcinoma therapy. Nat. Clin. Pract. Oncol. 2006, 3, 564–574. [Google Scholar] [CrossRef]
- Subbiah, V.; Yang, D.; Velcheti, V.; Drilon, A.; Meric-Bernstam, F. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J. Clin. Oncol. 2020, 38, 1209–1221. [Google Scholar] [CrossRef]
- Regua, A.T.; Najjar, M.; Lo, H.-W. RET signaling pathway and RET inhibitors in human cancer. Front. Oncol. 2022, 12, 932353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Oczko-Wojciechowska, M.; Swierniak, M.; Krajewska, J.; Kowalska, M.; Kowal, M.; Stokowy, T.; Wojtas, B.; Rusinek, D.; Pawlaczek, A.; Czarniecka, A. Differences in the transcriptome of medullary thyroid cancer regarding the status and type of RET gene mutations. Sci. Rep. 2017, 7, 42074. [Google Scholar] [CrossRef] [PubMed]
- Qu, N.; Shi, X.; Zhao, J.-J.; Guan, H.; Zhang, T.-T.; Wen, S.-S.; Liao, T.; Hu, J.-Q.; Liu, W.-Y.; Wang, Y.-L. Genomic and transcriptomic characterization of sporadic medullary thyroid carcinoma. Thyroid 2020, 30, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Minna, E.; Romeo, P.; Dugo, M.; De Cecco, L.; Aiello, A.; Pistore, F.; Carenzo, A.; Greco, A.; Borrello, M.G. Medullary thyroid carcinoma mutational spectrum update and signaling-type inference by transcriptional profiles: Literature meta-analysis and study of tumor samples. Cancers 2022, 14, 1951. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Cosci, B.; Renzini, G.; Bottici, V.; Molinaro, E.; Agate, L.; Passannanti, P.; Viola, D.; Biagini, A.; Basolo, F. RET genetic screening of sporadic medullary thyroid cancer (MTC) allows the preclinical diagnosis of unsuspected gene carriers and the identification of a relevant percentage of hidden familial MTC (FMTC). Clin. Endocrinol. 2011, 74, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.; Clayton, D.; Schuffenecker, I.; Lenoir, G.; Cote, G.; Gagel, R.F.; Van Amstel, H.K.P.; Lips, C.J.; Nishisho, I.; Takai, S.-I. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2: International RET Mutation Consortium analysis. JAMA 1996, 276, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- McGregor, L.M.; McCune, B.K.; Graff, J.R.; McDowell, P.R.; Romans, K.E.; Yancopoulos, G.D.; Ball, D.W.; Baylin, S.B.; Nelkin, B.D. Roles of trk family neurotrophin receptors in medullary thyroid carcinoma development and progression. Proc. Natl. Acad. Sci. USA 1999, 96, 4540–4545. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, D.; Koczan, D.; Ricken, P.; Rimpler, U.; Pahnke, J.; Li, Z.; Pützer, B.M. Transcriptome analysis in mouse tumors induced by Ret-MEN2/FMTC mutations reveals subtype-specific role in survival and interference with immune surveillance. Endocr.-Relat. Cancer 2009, 16, 211–224. [Google Scholar] [CrossRef]
- Ameur, N.; Lacroix, L.; Roucan, S.; Roux, V.; Broutin, S.; Talbot, M.; Dupuy, C.; Caillou, B.; Schlumberger, M.; Bidart, J.-M. Aggressive inherited and sporadic medullary thyroid carcinomas display similar oncogenic pathways. Endocr.-Relat. Cancer 2009, 16, 1261–1272. [Google Scholar] [CrossRef]
- Smith-Hicks, C.L.; Sizer, K.C.; Powers, J.F.; Tischler, A.S.; Costantini, F. C-cell hyperplasia, pheochromocytoma and sympathoadrenal malformation in a mouse model of multiple endocrine neoplasia type 2B. EMBO J. 2000, 19, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Hereditary cancer: Two hits revisited. J. Cancer Res. Clin. Oncol. 1996, 122, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Torres-Cruz, J.; Pack, S.D.; Koch, C.A.; Vortmeyer, A.O.; Mannan, P.; Lubensky, I.A.; Gagel, R.F.; Zhuang, Z. Amplification and overexpression of mutant RET in multiple endocrine neoplasia type 2-associated medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Leboulleux, S.; Travagli, J.; Caillou, B.; Laplanche, A.; Bidart, J.; Schlumberger, M.; Baudin, E. Medullary thyroid carcinoma as part of a multiple endocrine neoplasia type 2B syndrome: Influence of the stage on the clinical course. Cancer 2002, 94, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Pozdeyev, N.; Korch, C.; Marlow, L.A.; Smallridge, R.C.; Copland, J.A.; Henderson, Y.C.; Lai, S.Y.; Clayman, G.L.; Onoda, N. Comprehensive genetic characterization of human thyroid cancer cell lines: A validated panel for preclinical studies. Clin. Cancer Res. 2019, 25, 3141–3151. [Google Scholar] [CrossRef] [PubMed]
- Barletta, J.A.; Nosé, V.; Sadow, P.M. Genomics and epigenomics of medullary thyroid carcinoma: From sporadic disease to familial manifestations. Endocr. Pathol. 2021, 32, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Sun, Y.; Shen, C.; Zhang, Y.; Shi, R.; Zhang, F.; Liao, T.; Lv, G.; Zhu, Z.; Jiao, L. Integrated proteogenomic characterization of medullary thyroid carcinoma. Cell Discov. 2022, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Viswanathan, K.; Ahadi, M.S.; Ahmadi, S.; Alzumaili, B.; Bani, M.-A.; Baudin, E.; Behrman, D.B.; Capelletti, M.; Chau, N.G. Association of the genomic profile of medullary thyroid carcinoma with tumor characteristics and clinical outcomes in an international multicenter study. Thyroid 2024, 34, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Pozdeyev, N.; Erickson, T.A.; Zhang, L.; Ellison, K.; Rivard, C.J.; Sams, S.; Hirsch, F.R.; Haugen, B.R.; French, J.D. Comprehensive Immune Profiling of Medullary Thyroid Cancer. Thyroid 2020, 30, 1263–1279. [Google Scholar] [CrossRef]
- Abraham, D.; Jackson, N.; Gundara, J.S.; Zhao, J.; Gill, A.J.; Delbridge, L.; Robinson, B.G.; Sidhu, S.B. MicroRNA profiling of sporadic and hereditary medullary thyroid cancer identifies predictors of nodal metastasis, prognosis, and potential therapeutic targets. Clin. Cancer Res. 2011, 17, 4772–4781. [Google Scholar] [CrossRef]
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.; Redmond, W.L.; Seliger, B.; Marincola, F.M. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. Eur. J. Cancer 2017, 81, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F. The concept of immunological surveillance. Immunol. Asp. Neoplasia 1970, 13, 1–27. [Google Scholar]
- Birkeland, S.A.; Storm, H.H.; Lamm, L.U.; Barlow, L.; Blohmé, I.; Forsberg, B.; Eklund, B.; Fjeldborg, O.; Friedberg, M.; Frödin, L. Cancer risk after renal transplantation in the Nordic countries, 1964–1986. Int. J. Cancer 1995, 60, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, P.W.; Vaidya, S.A.; Cheng, G. The art of war: Innate and adaptive immune responses. Cell. Mol. Life Sci. 2003, 60, 2604–2621. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The concept of immune surveillance against tumors. The first theories. Oncotarget 2017, 8, 7175–7180. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nature Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Clemente, C.G.; Mihm, M.C., Jr.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Fallahi, P.; Galdiero, M.R.; Ruffilli, I.; Elia, G.; Ragusa, F.; Paparo, S.R.; Patrizio, A.; Mazzi, V.; Varricchi, G. Immune and inflammatory cells in thyroid cancer microenvironment. Int. J. Mol. Sci. 2019, 20, 4413. [Google Scholar] [CrossRef]
- Rao, D.; Verburg, F.; Renner, K.; Peeper, D.S.; Lacroix, R.; Blank, C.U. Metabolic profiles of regulatory T cells in the tumour microenvironment. Cancer Immunol. Immunother. 2021, 70, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 2020, 9, e55185. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Hervas-Stubbs, S.; Glennie, M.; Pardoll, D.M.; Chen, L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat. Rev. Cancer 2007, 7, 95–106. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Allavena, P.; Garlanda, C.; Borrello, M.G.; Sica, A.; Mantovani, A. Pathways connecting inflammation and cancer. Curr. Opin. Genet. Dev. 2008, 18, 3–10. [Google Scholar] [CrossRef]
- Modica, R.; Minotta, R.; Liccardi, A.; Cannavale, G.; Benevento, E.; Colao, A. Evaluation of Neutrophil-to-Lymphocyte Ratio (NLR), Platelet-to-Lymphocyte Ratio (PLR) and Systemic Immune-Inflammation Index (SII) as Potential Biomarkers in Patients with Sporadic Medullary Thyroid Cancer (MTC). J. Pers. Med. 2023, 13, 953. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Shang, Z.; Wang, W.; Gu, C.; Wei, Y.; Zhu, Y.; Yang, C.; Zhang, T.; Zhu, Y.; Zhu, Y.; et al. Immune Co-inhibitory Receptors CTLA-4, PD-1, TIGIT, LAG-3, and TIM-3 in Upper Tract Urothelial Carcinomas: A Large Cohort Study. J. Immunother. 2023, 46, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef]
- Kern, R.; Panis, C. CTLA-4 expression and its clinical significance in breast cancer. Arch. Immunol. Ther. Exp. 2021, 69, 16. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, C.-W.; Tan, L.-C.; Wen, S.-S.; Liao, T.; Zhang, Y.; Chen, T.-Z.; Ma, B.; Yu, P.-C.; Lu, Z.-W. Immune co-inhibitory receptors PD-1, CTLA-4, TIM-3, LAG-3, and TIGIT in medullary thyroid cancers: A large cohort study. J. Clin. Endocrinol. Metab. 2021, 106, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Saltiki, K.; Simeakis, G.; Anagnostou, E.; Zapanti, E.; Anastasiou, E.; Alevizaki, M. Different outcomes in sporadic versus familial medullary thyroid cancer. Head Neck 2019, 41, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. OncoTargets Ther. 2016, 9, 5023–5039. [Google Scholar]
- Chowdhury, S.; Veyhl, J.; Jessa, F.; Polyakova, O.; Alenzi, A.; MacMillan, C.; Ralhan, R.; Walfish, P.G. Programmed death-ligand 1 overexpression is a prognostic marker for aggressive papillary thyroid cancer and its variants. Oncotarget 2016, 7, 32318. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, M.; Rebecchini, C.; Saglietti, C.; Bulliard, J.-L.; Marino, L.; de Leval, L.; Sykiotis, G.P. Very low expression of PD-L1 in medullary thyroid carcinoma. Endocr.-Relat. Cancer 2017, 24, L35–L38. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yu, P.-C.; Lei, B.-W.; Li, C.-W.; Zhang, Y.; Tan, L.-C.; Shi, R.-L.; Wang, J.; Ma, B.; Xu, W.-B. Association between programmed death-ligand 1 expression and clinicopathological characteristics, structural recurrence, and biochemical recurrence/persistent disease in medullary thyroid carcinoma. Thyroid 2019, 29, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef]
- Bi, Y.; Ren, X.; Bai, X.; Meng, Y.; Luo, Y.; Cao, J.; Zhang, Y.; Liang, Z. PD-1/PD-L1 expressions in medullary thyroid carcinoma: Clinicopathologic and prognostic analysis of Chinese population. Eur. J. Surg. Oncol. 2019, 45, 353–358. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, rv324–rv328. [Google Scholar] [CrossRef]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: Meta-analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Lorch, J.H.; Barletta, J.A.; Nehs, M.; Uppaluri, R.; Alexander, E.K.; Haddad, R.I.; Hanna, G.J.; Margalit, D.N.; Tishler, R.B.; Schoenfeld, J.D. A phase II study of nivolumab (N) plus ipilimumab (I) in radioidine refractory differentiated thyroid cancer (RAIR DTC) with exploratory cohorts in anaplastic (ATC) and medullary thyroid cancer (MTC). J. Clin. Oncol. 2020, 38, 6513. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K. Tim-3 inhibits T helper type 1–mediated auto-and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Yuan, L.; Gao, Q.; Yuan, P.; Zhao, P.; Yuan, H.; Fan, H.; Li, T.; Qin, P.; Han, L. Circulating and tumor-infiltrating Tim-3 in patients with colorectal cancer. Oncotarget 2015, 6, 20592. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, P.; Liang, T.; Wang, L.; Hu, L. TIM-3 is a potential prognostic marker for patients with solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 31705–31713. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhang, X.; Xia, X.; Zhang, C.; Liang, X.; Gao, L.; Zhang, X.; Ma, C. Ectopic expression of TIM-3 in lung cancers: A potential independent prognostic factor for patients with NSCLC. Am. J. Clin. Pathol. 2012, 137, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Gallel, P.; Pallares, J.; Dolcet, X.; Llobet, D.; Eritja, N.; Santacana, M.; Yeramian, A.; Palomar-Asenjo, V.; Lagarda, H.; Mauricio, D.; et al. Nuclear factor-kappaB activation is associated with somatic and germ line RET mutations in medullary thyroid carcinoma. Hum. Pathol. 2008, 39, 994–1001. [Google Scholar] [CrossRef]
- Matias-Guiu, X.; De Lellis, R. Medullary thyroid carcinoma: A 25-year perspective. Endocr. Pathol. 2014, 25, 21–29. [Google Scholar] [CrossRef]
- Hincza-Nowak, K.; Kowalik, A.; Walczyk, A.; Palyga, I.; Gasior-Perczak, D.; Plusa, A.; Kopczynski, J.; Chrapek, M.; Gozdz, S.; Kowalska, A. CD276 as a Candidate Target for Immunotherapy in Medullary Thyroid Cancer. Int. J. Mol. Sci. 2023, 24, 10019. [Google Scholar] [CrossRef] [PubMed]
- Hendry, S.; Salgado, R.; Gevaert, T.; Russell, P.A.; John, T.; Thapa, B.; Christie, M.; Van De Vijver, K.; Estrada, M.V.; Gonzalez-Ericsson, P.I. Assessing tumor infiltrating lymphocytes in solid tumors: A practical review for pathologists and proposal for a standardized method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in melanoma, gastrointestinal tract carcinomas, non-small cell lung carcinoma and mesothelioma, endometrial and ovarian carcinomas, squamous cell carcinoma of the head and neck, genitourinary carcinomas, and primary brain tumors. Adv. Anat. Pathol. 2017, 24, 311. [Google Scholar] [PubMed]
- Fuchs, T.L.; Sioson, L.; Sheen, A.; Jafari-Nejad, K.; Renaud, C.J.; Andrici, J.; Ahadi, M.; Chou, A.; Gill, A.J. Assessment of tumor-infiltrating lymphocytes using International TILs Working Group (ITWG) system is a strong predictor of overall survival in colorectal carcinoma: A study of 1034 patients. Am. J. Surg. Pathol. 2020, 44, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Pénault-Llorca, F. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Rago, T.; Di Coscio, G.; Ugolini, C.; Scutari, M.; Basolo, F.; Latrofa, F.; Romani, R.; Berti, P.; Grasso, L.; Braverman, L. Clinical features of thyroid autoimmunity are associated with thyroiditis on histology and are not predictive of malignancy in 570 patients with indeterminate nodules on cytology who had a thyroidectomy. Clin. Endocrinol. 2007, 67, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, C.I.; Hall, P.; Dickman, P.W.; Zedenius, J. Clinically significant prognostic factors for differentiated thyroid carcinoma: A population-based, nested case–control study. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2006, 106, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, S.; Kawai, K.; Matsumoto, Y.; Mukuta, T.; Morita, T.; Hirai, K.; Matsuzuka, F.; Kakudoh, K.; Kuma, K.; Tamai, H. The correlation between papillary thyroid carcinoma and lymphocytic infiltration in the thyroid gland. J. Clin. Endocrinol. Metab. 1995, 80, 3421–3424. [Google Scholar]
- Scopsi, L.; Collini, P.; Sampietro, G.; Boracchi, P.; Pilotti, S. Prognostic impact of thyroid lymphocytic infiltration in patients with medullary thyroid carcinoma. Thyroid 1996, 6, 613–617. [Google Scholar] [CrossRef] [PubMed]
- French, J.D.; Weber, Z.J.; Fretwell, D.L.; Said, S.; Klopper, J.P.; Haugen, B.R. Tumor-associated lymphocytes and increased FoxP3+ regulatory T cell frequency correlate with more aggressive papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2010, 95, 2325–2333. [Google Scholar] [CrossRef]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019, 25, 89–94. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.-L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Menicali, E.; Guzzetti, M.; Morelli, S.; Moretti, S.; Puxeddu, E. Immune landscape of thyroid cancers: New insights. Front. Endocrinol. 2021, 11, 637826. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.-j.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [PubMed]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Poehnert, D.; Müller, J.; Scheumann, G.; Koch, M.; Lück, R. Regulatory T cells in peripheral blood, lymph node, and thyroid tissue in patients with medullary thyroid carcinoma. World J. Surg. 2010, 34, 1481–1487. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M. First-line nivolumab plus ipilimumab in advanced non–small-cell lung cancer (CheckMate 568): Outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J. Clin. Oncol. 2019, 37, 992. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Efremova, M.; Finotello, F.; Rieder, D.; Trajanoski, Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front. Immunol. 2017, 8, 1679. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Jiao, Y.; Sausen, M.; Leary, R.; Bettegowda, C.; Roberts, N.J.; Bhan, S.; Ho, A.S.; Khan, Z.; Bishop, J. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J. Clin. Endocrinol. Metab. 2013, 98, E364–E369. [Google Scholar] [CrossRef] [PubMed]
- Leslie, A.; Carey, F.; Pratt, N.; Steele, R. The colorectal adenoma–carcinoma sequence. Br. J. Surg. 2002, 89, 845–860. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.-C.; Kersbergen, A.; Lam, E.Y.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 2019, 36, 385–401.e388. [Google Scholar] [CrossRef]
- Ruan, X.; Yi, J.; Hu, L.; Zhi, J.; Zeng, Y.; Hou, X.; Huang, J.; Gu, P.; Hao, W.; Gao, M. Reduced MHC class II expression in medullary thyroid cancer identifies patients with poor prognosis. Endocr.-Relat. Cancer 2022, 29, 87–98. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.; Gaweł, D.; Godlewska, M. Novel inhibitor-based therapies for thyroid cancer—An update. Int. J. Mol. Sci. 2021, 22, 11829. [Google Scholar] [CrossRef] [PubMed]
- Prete, A.; Borges de Souza, P.; Censi, S.; Muzza, M.; Nucci, N.; Sponziello, M. Update on fundamental mechanisms of thyroid cancer. Front. Endocrinol. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Schlumberger, M.J.; Müller, S.P.; Schöffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639. [Google Scholar] [CrossRef]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: Molecular mechanisms and future perspective. Signal Transduct. Target. Ther. 2022, 7, 329. [Google Scholar] [CrossRef] [PubMed]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S. Efficacy of selpercatinib in RET-altered thyroid cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Jozaghi, Y.; Zafereo, M.; Williams, M.D.; Gule-Monroe, M.K.; Wang, J.; Grubbs, E.G.; Vaporciyan, A.; Hu, M.I.; Busaidy, N.; Dadu, R.; et al. Neoadjuvant selpercatinib for advanced medullary thyroid cancer. Head Neck 2021, 43, E7–E12. [Google Scholar] [CrossRef]
- Gild, M.L.; Clifton-Bligh, R.J.; Wirth, L.J.; Robinson, B.G. Medullary Thyroid Cancer: Updates and Challenges. Endocr. Rev. 2023, 44, 934–946. [Google Scholar] [CrossRef]
- Hadoux, J.; Elisei, R.; Brose, M.S.; Hoff, A.O.; Robinson, B.G.; Gao, M.; Jarzab, B.; Isaev, P.; Kopeckova, K.; Wadsley, J. Phase 3 trial of selpercatinib in advanced RET-mutant medullary thyroid cancer. N. Engl. J. Med. 2023, 389, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Gild, M.L.; Bullock, M.; Tsang, V.; Clifton-Bligh, R.J.; Robinson, B.G.; Wirth, L.J. Challenges and Strategies to Combat Resistance Mechanisms in Thyroid Cancer Therapeutics. Thyroid 2023, 33, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shen, T.; Mooers, B.H.; Hilberg, F.; Wu, J. Drug resistance profiles of mutations in the RET kinase domain. Br. J. Pharmacol. 2018, 175, 3504–3515. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Huang, Z.; Han, L.; Gong, Y.; Xie, C. Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer. Int. J. Oncol. 2021, 59, 90. [Google Scholar] [CrossRef] [PubMed]
- Gramza, A.W.; Corless, C.L.; Heinrich, M.C. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin. Cancer Res. 2009, 15, 7510–7518. [Google Scholar] [CrossRef] [PubMed]
- Terzyan, S.S.; Shen, T.; Liu, X.; Huang, Q.; Teng, P.; Zhou, M.; Hilberg, F.; Cai, J.; Mooers, B.H.; Wu, J. Structural basis of resistance of mutant RET protein-tyrosine kinase to its inhibitors nintedanib and vandetanib. J. Biol. Chem. 2019, 294, 10428–10437. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Tan, L.; Lin, J.J.; Wong, S.Q.; Hollizeck, S.; Ebata, K.; Tuch, B.B.; Yoda, S.; Gainor, J.F.; Sequist, L.V. RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. J. Thorac. Oncol. 2020, 15, 541–549. [Google Scholar] [CrossRef]
- Drilon, A.E.; Zhai, D.; Rogers, E.; Deng, W.; Zhang, X.; Ung, J.; Lee, D.; Rodon, L.; Graber, A.; Zimmerman, Z.F. The next-generation RET inhibitor TPX-0046 is active in drug-resistant and naïve RET-driven cancer models. J. Clin. Oncol. 2020, 38, 3616. [Google Scholar] [CrossRef]
- Nakaoku, T.; Kohno, T.; Araki, M.; Niho, S.; Chauhan, R.; Knowles, P.P.; Tsuchihara, K.; Matsumoto, S.; Shimada, Y.; Mimaki, S. A secondary RET mutation in the activation loop conferring resistance to vandetanib. Nat. Commun. 2018, 9, 625. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Vaquero, J.; Lobe, C.; Tahraoui, S.; Clapéron, A.; Mergey, M.; Merabtene, F.; Wendum, D.; Coulouarn, C.; Housset, C.; Desbois-Mouthon, C. The IGF2/IR/IGF1R pathway in tumor cells and myofibroblasts mediates resistance to EGFR inhibition in cholangiocarcinoma. Clin. Cancer Res. 2018, 24, 4282–4296. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Chiang, C.-L.; Hung, J.-Y.; Lee, M.-H.; Su, W.-C.; Wu, S.-Y.; Wei, Y.-F.; Lee, K.-Y.; Tseng, Y.-H.; Su, J. Resistance profiles of anaplastic lymphoma kinase tyrosine kinase inhibitors in advanced non–small-cell lung cancer: A multicenter study using targeted next-generation sequencing. Eur. J. Cancer 2021, 156, 1–11. [Google Scholar] [CrossRef]
- Zhou, S.-L.; Zhou, Z.-J.; Hu, Z.-Q.; Huang, X.-W.; Wang, Z.; Chen, E.-B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 2016, 150, 1646–1658.e1617. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Gong, K.; Ali, S.; Ali, N.; Shallwani, S.; Hatanpaa, K.J.; Pan, E.; Mickey, B.; Burma, S.; Wang, D.H. A TNF–JNK–Axl–ERK signaling axis mediates primary resistance to EGFR inhibition in glioblastoma. Nat. Neurosci. 2017, 20, 1074–1084. [Google Scholar] [CrossRef]
- Ou, B.; Cheng, X.; Xu, Z.; Chen, C.; Shen, X.; Zhao, J.; Lu, A. A positive feedback loop of β-catenin/CCR2 axis promotes regorafenib resistance in colorectal cancer. Cell Death Dis. 2019, 10, 643. [Google Scholar] [CrossRef]
- Dong, N.; Shi, X.; Wang, S.; Gao, Y.; Kuang, Z.; Xie, Q.; Li, Y.; Deng, H.; Wu, Y.; Li, M. M2 macrophages mediate sorafenib resistance by secreting HGF in a feed-forward manner in hepatocellular carcinoma. Br. J. Cancer 2019, 121, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Noel, B.M.; Ouellette, S.B.; Marholz, L.; Dickey, D.; Navis, C.; Yang, T.-Y.; Nguyen, V.; Parker, S.J.; Bernlohr, D.; Sachs, Z. Multiomic profiling of tyrosine kinase inhibitor-resistant K562 cells suggests metabolic reprogramming to promote cell survival. J. Proteome Res. 2019, 18, 1842–1856. [Google Scholar] [CrossRef]
- Niu, X.; Liu, F.; Zhou, Y.; Zhou, Z.; Zhou, D.; Wang, T.; Li, Z.; Ye, X.; Yu, Y.; Weng, X. Genome-wide DNA methylation analysis reveals GABBR2 as a novel epigenetic target for EGFR 19 deletion lung adenocarcinoma with induction erlotinib treatment. Clin. Cancer Res. 2017, 23, 5003–5014. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.F.; Chen, L.T.; Wang, H.D.; Liu, Y.H.; Shiah, S.G. Transcriptional suppression of Dicer by HOXB-AS3/EZH2 complex dictates sorafenib resistance and cancer stemness. Cancer Sci. 2022, 113, 1601–1612. [Google Scholar] [CrossRef]
- Liu, S.; Li, Q.; Li, G.; Zhang, Q.; Zhuo, L.; Han, X.; Zhang, M.; Chen, X.; Pan, T.; Yan, L. The mechanism of m6A methyltransferase METTL3-mediated autophagy in reversing gefitinib resistance in NSCLC cells by β-elemene. Cell Death Dis. 2020, 11, 969. [Google Scholar] [CrossRef]
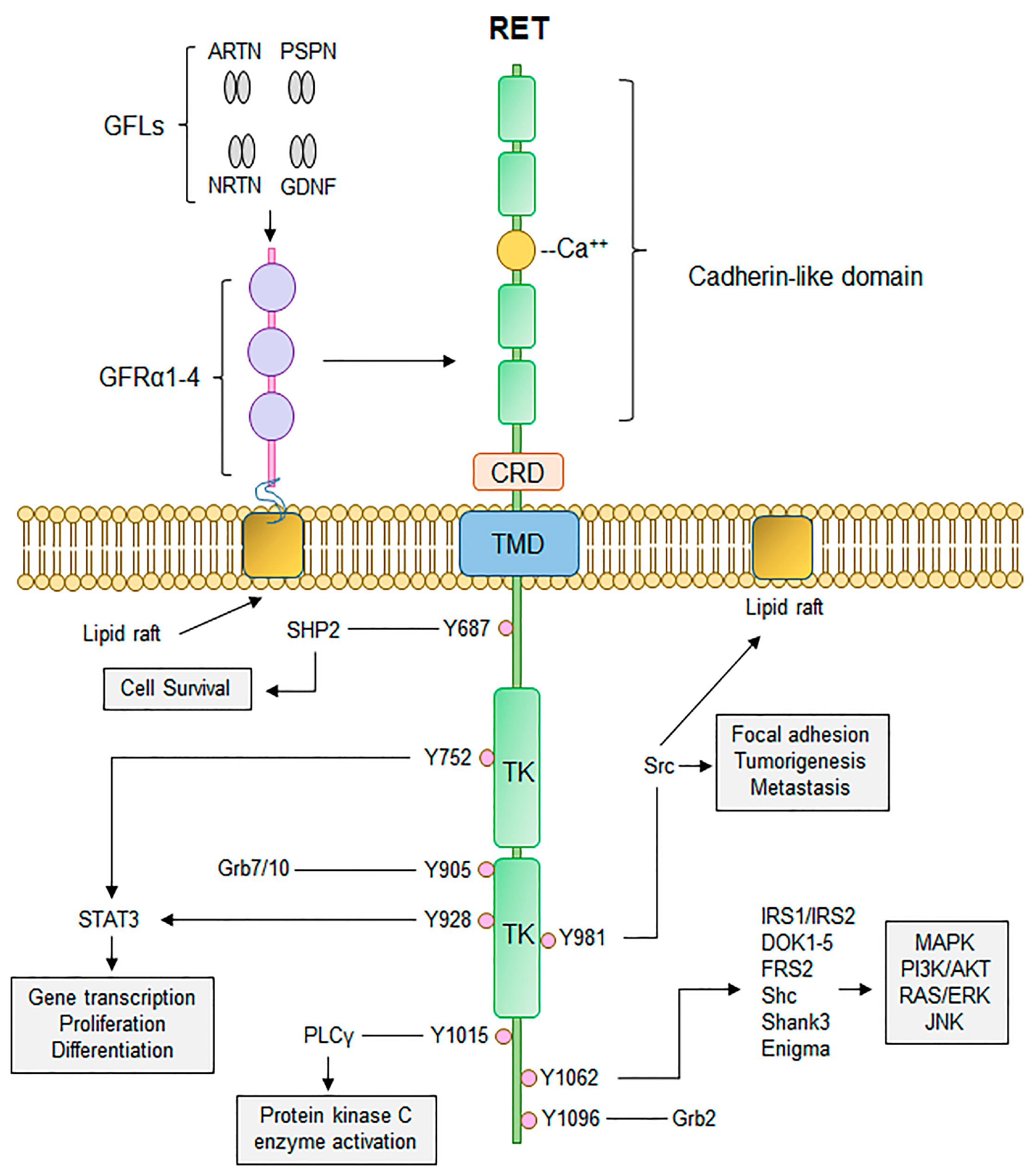
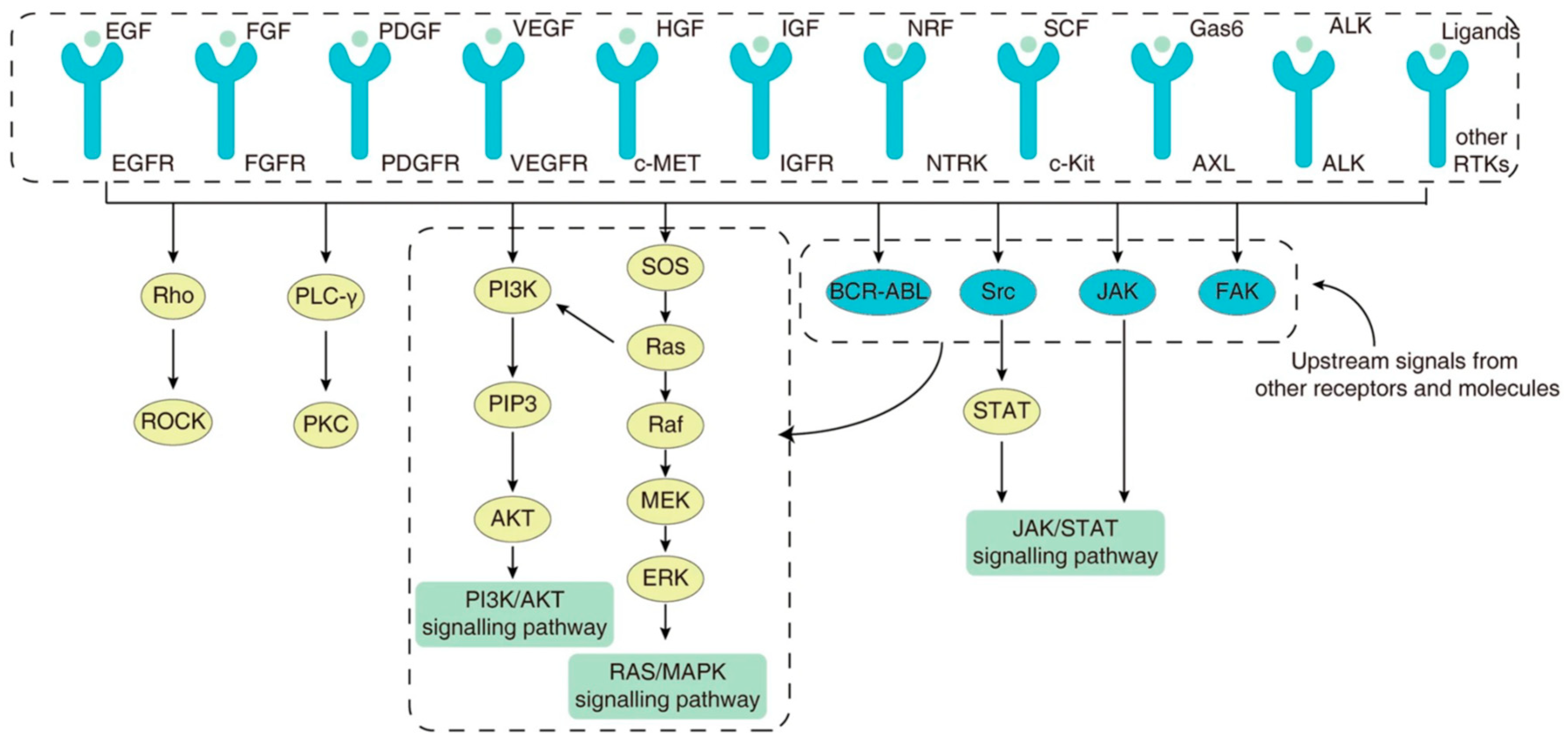
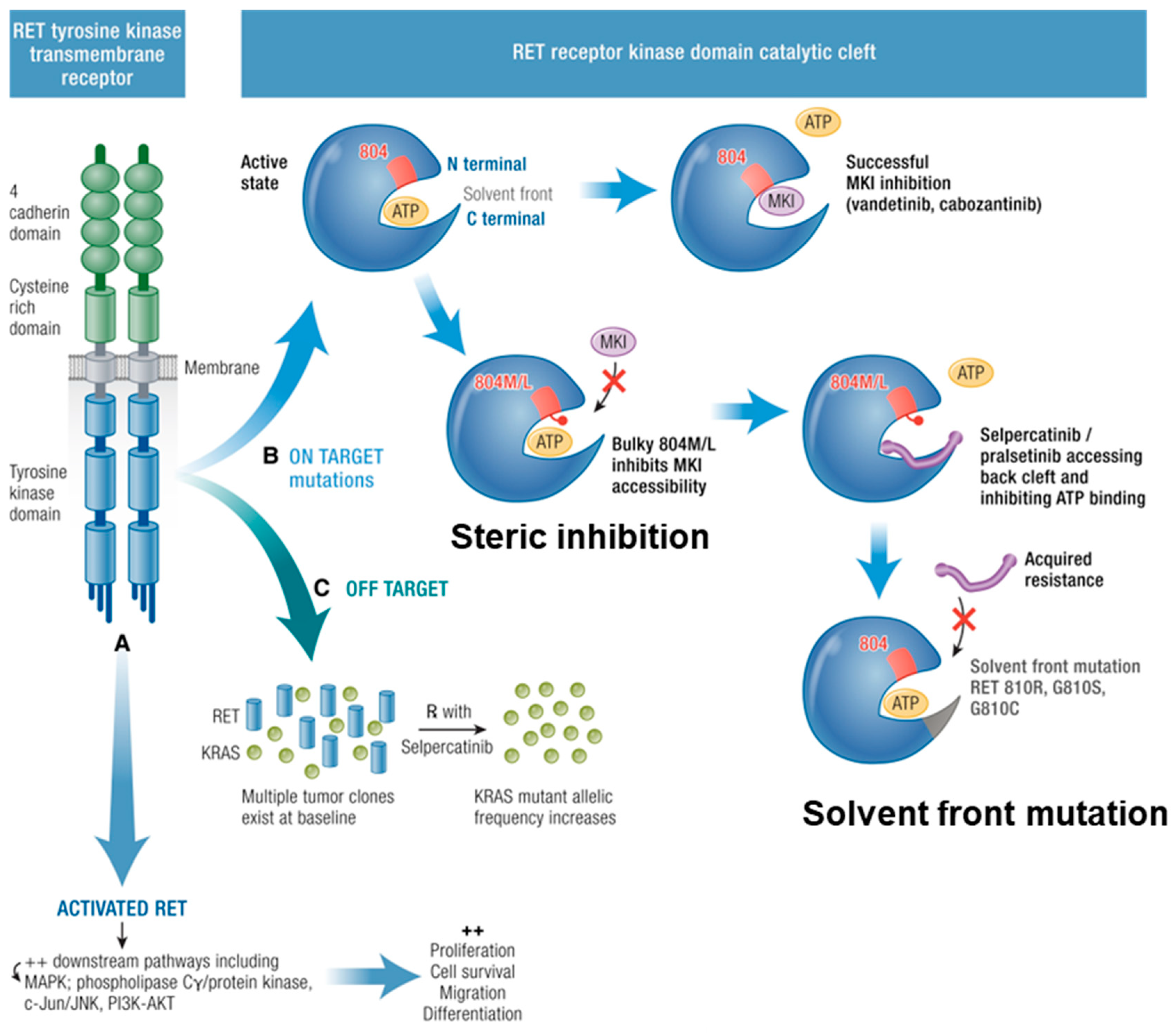
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).