
3D Chromatin Alteration by Disrupting β-Catenin/CBP Interaction Is Enriched with Insulin Signaling in Pancreatic Cancer

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Cell Lines and Reagents
2.2. Cell Proliferation Assays
2.3. Cell Migration Assays
2.4. Cell Apoptosis Analysis
2.5. In Situ Hi-C Profiling
2.6. RNA-Seq Profiling
2.7. CRISPR/Cas9-Mediated Genomic Deletion
2.8. RT-qPCR and 3C-qPCR
2.9. Western Blotting
2.10. shRNA Plasmid-Mediated Inhibition of RHBDD1 Expression
2.11. Hi-C Data Analysis
2.12. RNA-Seq Data Analysis
2.13. Enrichment of Signaling Pathways
3. Results
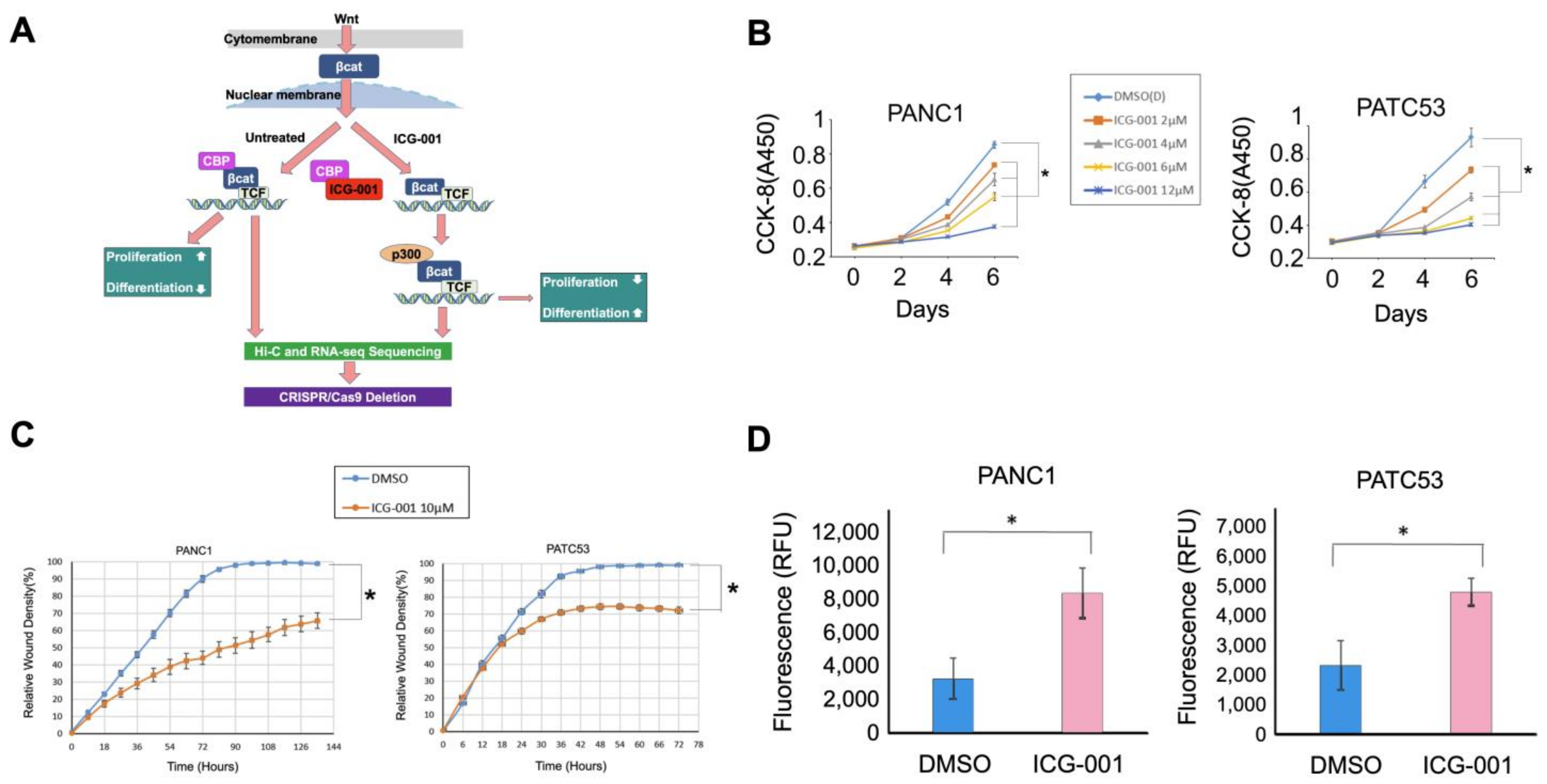
3.1. Pancreatic Cancer Cells Are Inhibited by Wnt Signaling Inhibitor ICG-001
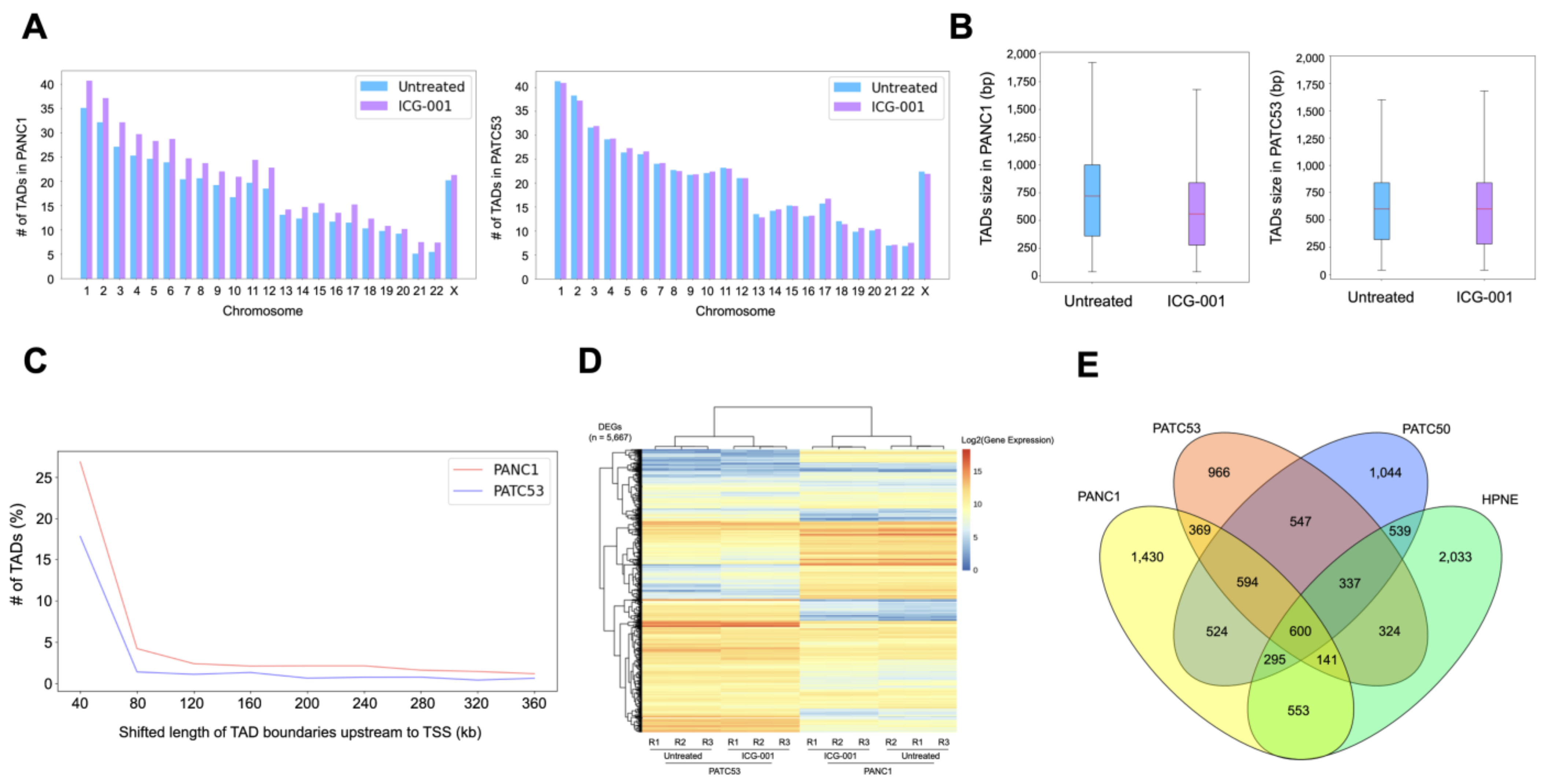
3.2. The 3D Chromatin Architecture of Pancreatic Cancer Cells Is Altered by ICG-001
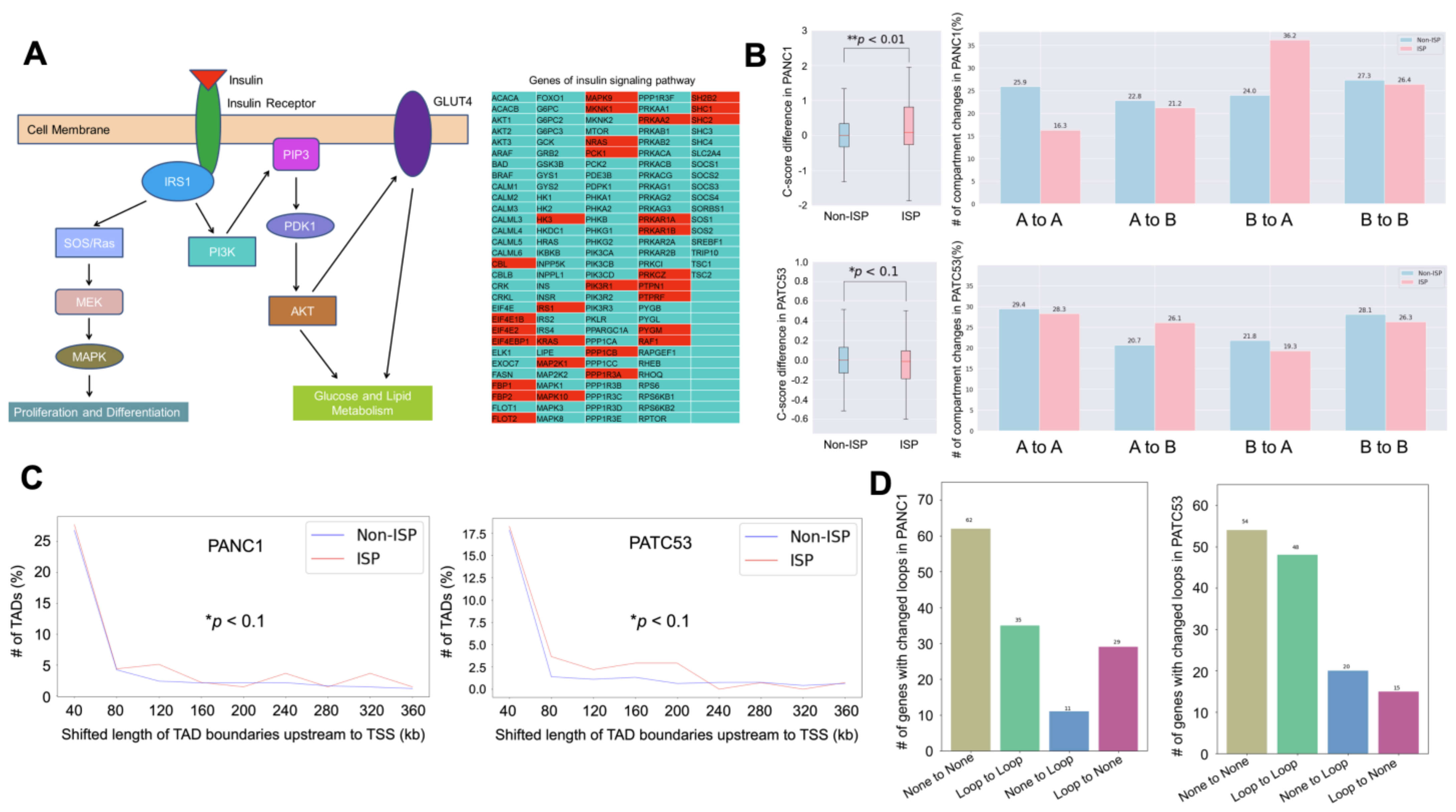
3.3. Insulin Signaling Pathway Is Highly Enriched in the Altered Chromatin Structure
3.4. Looping Events Associated with ISP Genes Are Weakened by ICG-001
3.5. Deletion of IRS1 Distal–Promoter Looping Impedes Pancreatic Cancer Cell Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Diaferia, G.R.; Balestrieri, C.; Prosperini, E.; Nicoli, P.; Spaggiari, P.; Zerbi, A.; Natoli, G. Dissection of transcriptional and cis-regulatory control of differentiation in human pancreatic cancer. EMBO J. 2016, 35, 595–617. [Google Scholar] [CrossRef]
- Mao, Y.; Shen, J.; Lu, Y.; Lin, K.; Wang, H.; Li, Y.; Chang, P.; Walker, M.G.; Li, D. RNA sequencing analyses reveal novel differentially expressed genes and pathways in pancreatic cancer. Oncotarget 2017, 8, 42537–42547. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.-S.; Hwang, C.-I.; Somerville, T.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, D.L.; Wang, Y.; Gaddis, M.; Zhou, Y.; Wang, J.; Witt, H.; Lin, S.; Farnham, P.J.; Jin, V.X.; Frietze, S.E. Three-dimensional analysis reveals altered chromatin interaction by enhancer inhibitors harbors TCF7L2-regulated cancer gene signature. J. Cell Biochem. 2019, 120, 3056–3070. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, D.L.; Boyd, J.R.; Stein, G.S.; Jin, V.X.; Frietze, S. Disruption of Broad Epigenetic Domains in PDAC Cells by HAT Inhibitors. Epigenomes 2019, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.P., IV; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef] [PubMed]
- White, B.D.; Chien, A.J.; Dawson, D.W. Dysregulation of Wnt/b-catenin signaling in gastrointestinal cancers. Gastroenterology 2011, 142, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Park, J.I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond beta-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.; Oh, S.W.; Kim, H.Y.; et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [PubMed]
- Arensman, M.D.; Telesca, D.; Lay, A.R.; Kershaw, K.M.; Wu, N.; Donahue, T.R.; Dawson, D.W. The CREB-Binding Protein Inhibitor ICG-001 Suppresses Pancreatic Cancer Growth. Mol. Cancer Ther. 2014, 13, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Manegold, P.; Lai, K.K.Y.; Wu, Y.; Teo, J.-L.; Lenz, H.-J.; Genyk, Y.S.; Pandol, S.J.; Wu, K.; Lin, D.P.; Chen, Y.; et al. Differentiation Therapy Targeting the β-Catenin/CBP Interaction in Pancreatic Cancer. Cancers 2018, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Taking the road less traveled—The therapeutic potential of CBP/β-catenin antagonists. Expert. Opin. Ther. Targets 2021, 25, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Gaddis, M.; Gerrard, D.; Frietze, S.; Farnham, P.J. Altering cancer transcriptomes using epigenomic inhibitors. Epigenetics Chromatin 2015, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Barutcu, A.R.; Lajoie, B.R.; McCord, R.P.; Tye, C.E.; Hong, D.; Messier, T.L.; Browne, G.; van Wijnen, A.J.; Lian, J.B.; Stein, J.L.; et al. Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells. Genome Biol. 2015, 16, 214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Gerrard, D.L.; Wang, J.; Li, T.; Yang, Y.; Fritz, A.J.; Rajendran, M.; Fu, X.; Stein, G.; Schiff, R.; et al. Temporal dynamic reorganization of 3D chromatin architecture in hormone-induced breast cancer and endocrine resistance. Nat. Commun. 2019, 10, 1522. [Google Scholar] [CrossRef] [PubMed]
- Rhie, S.K.; Perez, A.A.; Lay, F.D.; Schreiner, S.; Shi, J.; Polin, J.; Farnham, P.J. A high-resolution 3D epigenomic map reveals insights into the creation of the prostate cancer transcriptome. Nat. Commun. 2019, 10, 4154. [Google Scholar] [CrossRef] [PubMed]
- Yusufova, N.; Kloetgen, A.; Teater, M.; Osunsade, A.; Camarillo, J.M.; Chin, C.R.; Doane, A.S.; Venters, B.J.; Portillo-Ledesma, S.; Conway, J.; et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature 2021, 589, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Kloetgen, A.; Thandapani, P.; Ntziachristos, P.; Ghebrechristos, Y.; Nomikou, S.; Lazaris, C.; Chen, X.; Hu, H.; Bakogianni, S.; Wang, J.; et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat. Genet. 2020, 52, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Yang, J.; Wang, C.; Yang, G.; Wang, H.; Chen, Y.; Xu, R.; Fan, X.; You, L.; Zhang, T.; et al. High-resolution Hi-C maps highlight multiscale 3D epigenome reprogramming during pancreatic cancer metastasis. J. Hematol. Oncol. 2021, 14, 120. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Choppavarapu, L.; Fang, K.; Naeini, A.S.; Nosirov, B.; Li, J.; Yang, K.; He, Z.; Zhou, Y.; Schiff, R.; et al. The 3D genomic landscape of differential response to EGFR/HER2 inhibition in endocrine-resistant breast cancer cells. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194631. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cheng, X.; Yang, Y.; Li, T.; Li, J.; Huang, T.H.-M.; Wang, J.; Lin, S.; Jin, V.X. Modeling and analysis of Hi-C data by HiSIF identifies characteristic promoter-distal loops. Genome Med. 2020, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.-J.; Vert, J.-P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef]
- Zheng, X.; Zheng, Y. CscoreTool: Fast Hi-C compartment analysis at high resolution. Bioinformatics 2018, 34, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Teo, J.L.; Ma, H.; Nguyen, C.; Lam, C.; Kahn, M. Specific inhibition of CBP/beta-catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 12171–12176. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ono, M.; Chimge, N.-O.; Chosa, K.; Nguyen, C.; Melendez, E.; Lou, C.-H.; Lim, P.; Termini, J.; Lai, K.K.Y.; et al. Differential Kat3 Usage Orchestrates the Integration of Cellular Metabolism with Differentiation. Cancers 2021, 13, 5884. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhang, R.; Suzuki, R.; Li, S.-Q.; Roife, D.; Truty, M.J.; Chatterjee, D.; Thomas, R.M.; Cardwell, J.; Wang, Y.; et al. Two-dimensional culture of human pancreatic adenocarcinoma cells results in an irreversible transition from epithelial to mesenchymal phenotype. Lab. Investig. 2015, 95, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.T.; Lim, G.E.; Skovsø, S.; Yang, Y.H.C.; Albrecht, T.; Alejandro, E.U.; Hoesli, C.A.; Piret, J.M.; Warnock, G.L.; Johnson, J.D. Effects of insulin on human pancreatic cancer progression modeled in vitro. BMC Cancer 2014, 14, 814. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Guo, Y.; Du, J.; Gu, J.; Kong, L.; Tao, B.; Li, J.; Fu, D. The Intricate Crosstalk Between Insulin and Pancreatic Ductal Adenocarcinoma: A Review From Clinical to Molecular. Front. Cell Dev. Biol. 2022, 10, 844028. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Xu, Q.; Zhao, Y.; Stevens, J.V.; Young, S.H.; Sinnett-Smith, J.; Rozengurt, E. Insulin Receptor and GPCR Crosstalk Stimulates YAP via PI3K and PKD in Pancreatic Cancer Cells. Mol. Cancer Res. 2017, 15, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic-Arsic, M.; Kalideris, E.; Siveke, J.T. The role of insulin and IGF system in pancreatic cancer. J. Mol. Endocrinol. 2013, 50, R67–R74. [Google Scholar] [CrossRef] [PubMed]
- Yee, D. Insulin-like growth factor receptor inhibitors: Baby or the bathwater? J. Natl. Cancer Inst. 2012, 104, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Mutgan, A.C.; Besikcioglu, H.E.; Wang, S.; Friess, H.; Ceyhan, G.O.; Demir, I.E. Insulin/IGF-driven cancer cell-stroma crosstalk as a novel therapeutic target in pancreatic cancer. Mol. Cancer 2018, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Singh, J.; Lawres, L.; Dorans, K.J.; Garcia, C.; Burkhardt, D.B.; Robbins, R.; Bhutkar, A.; Cardone, R.; Zhao, X.; et al. Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma. Cell 2020, 181, 832–847.e18. [Google Scholar] [CrossRef] [PubMed]
- Poloz, Y.; Stambolic, V. Obesity and Cancer, a Case for Insulin Signaling. Cell Death Dis. 2015, 6, e2037. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.J.; Roddam, A.W.; Beral, V. Pancreatic cancer in type 1 and young-onset diabetes: Systematic review and meta-analysis. Br. J. Cancer 2007, 96, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Batabyal, P.; Hoorn, S.V.; Christophi, C.; Nikfarjam, M. Association of diabetes mellitus and pancreatic adenocarcinoma: A meta-analysis of 88 studies. Ann. Surg. Oncol. 2014, 21, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Tien, S.C.; Hsieh, P.K.; Jeng, Y.M.; Chang, M.C.; Chang, Y.T.; Chen, Y.J.; Chen, Y.J.; Eva, Y.H.; Lee, W.H. High Glucose Triggers Nucleotide Imbalance through O-GlcNAcylation of Key Enzymes and Induces KRAS Mutation in Pancreatic Cells. Cell Metab. 2019, 29, 1334–1349.e10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, W.; Zhou, J.; Chen, J.; Wang, X.; Cai, M.; Li, F.; Xu, W.; Carlsson, P.-O.; Sun, Z. Lipotoxicity reduces β cell survival through islet stellate cell activation regulated by lipid metabolism-related molecules. Exp. Cell Res. 2019, 380, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cao, S.; Li, Y.; Sun, B.; Chen, D.; Wang, D.; Zhou, Y. Pancreatic stellate cells facilitate pancreatic cancer cell viability and invasion. Oncol. Lett. 2019, 17, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Marzoq, A.J.; Mustafa, S.A.; Heidrich, L.; Hoheisel, J.D.; Alhamdani, M.S.S. Impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells. Sci. Rep. 2019, 9, 5303. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; He, Z.; Li, T.; Choppavarapu, L.; Hu, X.; Cao, R.; Leone, G.W.; Kahn, M.; Jin, V.X. 3D Chromatin Alteration by Disrupting β-Catenin/CBP Interaction Is Enriched with Insulin Signaling in Pancreatic Cancer. Cancers 2024, 16, 2202. https://doi.org/10.3390/cancers16122202
Zhou Y, He Z, Li T, Choppavarapu L, Hu X, Cao R, Leone GW, Kahn M, Jin VX. 3D Chromatin Alteration by Disrupting β-Catenin/CBP Interaction Is Enriched with Insulin Signaling in Pancreatic Cancer. Cancers. 2024; 16(12):2202. https://doi.org/10.3390/cancers16122202
Chicago/Turabian StyleZhou, Yufan, Zhijing He, Tian Li, Lavanya Choppavarapu, Xiaohui Hu, Ruifeng Cao, Gustavo W. Leone, Michael Kahn, and Victor X. Jin. 2024. "3D Chromatin Alteration by Disrupting β-Catenin/CBP Interaction Is Enriched with Insulin Signaling in Pancreatic Cancer" Cancers 16, no. 12: 2202. https://doi.org/10.3390/cancers16122202
APA StyleZhou, Y., He, Z., Li, T., Choppavarapu, L., Hu, X., Cao, R., Leone, G. W., Kahn, M., & Jin, V. X. (2024). 3D Chromatin Alteration by Disrupting β-Catenin/CBP Interaction Is Enriched with Insulin Signaling in Pancreatic Cancer. Cancers, 16(12), 2202. https://doi.org/10.3390/cancers16122202