
Immune Microenvironment in Childhood Cancers: Characteristics and Therapeutic Challenges

Abstract
Simple Summary
Abstract
1. Introduction
2. Profiling the Immune Microenvironment of Pediatric Tumors
3. Influence of the Immune Microenvironment on Pediatric Tumor Biology
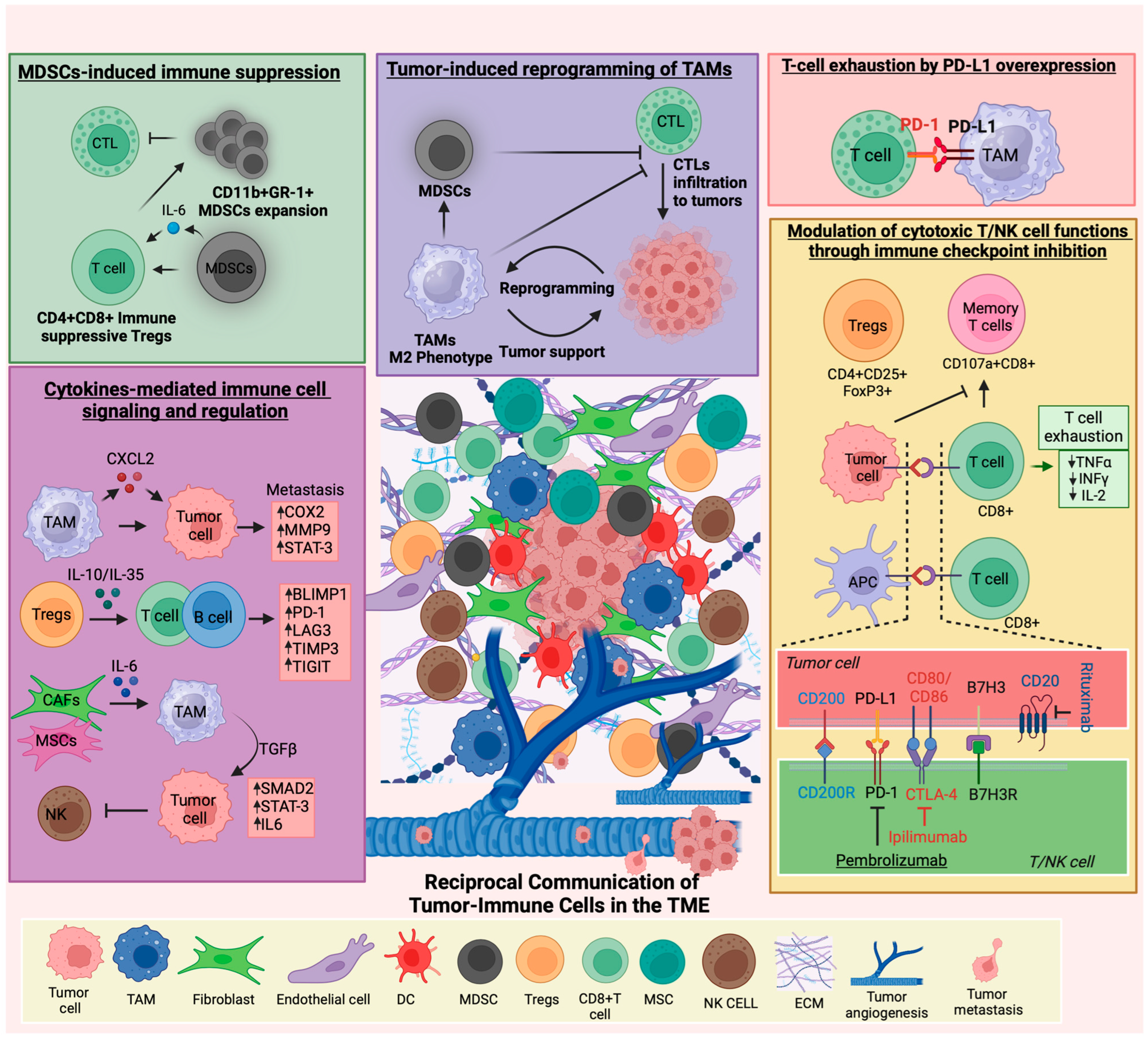
3.1. Immune Cell–Tumor Cell Interactions and Their Role in Tumor Progression
3.2. Metastasis and Immune Microenvironment
3.3. Immune Cell-Derived Cytokines
3.4. Immunotherapy Response and Predictors in Pediatric Tumors
4. Conclusions
Funding
Conflicts of Interest
Abbreviations
| WHO | World Health Organization |
| CNS | Central nervous system |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| JAK | Janus kinases |
| STAT | Signal transducer and activator of transcription |
| PI3K | Phosphoinositide 3-kinases |
| ALK | Anaplastic lymphoma kinase |
| NF1 | Neurofibromatosis type 1 |
| PTEN | Phosphatase and tensin homolog |
| FLT3 | FMS-like tyrosine kinase 3 |
| PIK3CA | Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha or p110α |
| PIK3R1 | Phosphoinositide-3-kinase regulatory subunit 1 |
| CDKN2A | Cyclin-dependent kinase Inhibitor 2A |
| TME | Tumor microenvironment |
| TILs | Tumor-infiltrating lymphocytes |
| Tregs | Regulatory T cells |
| NK cells | Natural killer cells |
| DCs | Dendritic cells |
| MDSCs | Myeloid-derived suppressor cells |
| ECM | Extracellular matrix |
| mAbs | Monoclonal antibodies |
| CAR | Chimeric antigen receptor |
| TAMs | Tumor-associated macrophages |
| TMB | Tumor mutational burden |
| LGG | Low-grade gliomas |
| HGG | High-grade gliomas |
| IFNγ | Interferon-gamma |
| CSF1R | Colony stimulating factor 1 receptor |
| CTLs | Cytotoxic T lymphocytes |
| PMN | Polymorphonuclear |
| PD-L1 | Programmed death ligand-1 |
| IL6 | Interleukin 6 |
| ES | Ewing sarcoma |
| TNF-α | Tumor necrosis factor-alpha |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| PD-1 | Programmed cell death protein 1 |
| MRD | Minimal residual disease |
| HSCT | Hematopoietic stem cell transplantation |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| SCT | Stem-cell transplantation |
| COG | Children’s oncology group |
| ADCC | Antibody-dependent cellular cytotoxicity |
References
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A.; IICC-3 Contributors. International incidence of childhood cancer, 2001–2010: A population-based registry study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Bonaventure, A.; Harewood, R.; Stiller, C.A.; Gatta, G.; Clavel, J.; Stefan, D.C.; Carreira, H.; Spika, D.; Marcos-Gragera, R.; Peris-Bonet, R.; et al. Worldwide comparison of survival from childhood leukaemia for 1995–2009, by subtype, age, and sex (CONCORD-2): A population-based study of individual data for 89 828 children from 198 registries in 53 countries. Lancet Haematol. 2017, 4, e202–e217. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.A.; King, J.B.; Lupo, P.J.; Durbin, E.B.; Tai, E.; Mills, K.; Van Dyne, E.; Buchanan Lunsford, N.; Henley, S.J.; Wilson, R.J. Counts, incidence rates, and trends of pediatric cancer in the United States, 2003–2019. J. Natl. Cancer Inst. 2023, 115, 1337–1354. [Google Scholar] [CrossRef] [PubMed]
- Curtin, S.C.; Anderson, R.N. Declines in Cancer Death Rates Among Youth: United States, 2001–2021. NCHS Data Brief 2023, 484, 1–8. [Google Scholar]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 30 April 2024).
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Jedraszek, K.; Malczewska, M.; Parysek-Wojcik, K.; Lejman, M. Resistance Mechanisms in Pediatric B-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2022, 23, 3067. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Ziegler, D.S.; Trahair, T.N.; Marshall, G.M.; Haber, M.; Norris, M.D. Too many targets, not enough patients: Rethinking neuroblastoma clinical trials. Nat. Rev. Cancer 2018, 18, 389–400. [Google Scholar] [CrossRef]
- Marchandet, L.; Lallier, M.; Charrier, C.; Baud’huin, M.; Ory, B.; Lamoureux, F. Mechanisms of Resistance to Conventional Therapies for Osteosarcoma. Cancers 2021, 13, 683. [Google Scholar] [CrossRef]
- Curran, E.K.; Godfrey, J.; Kline, J. Mechanisms of Immune Tolerance in Leukemia and Lymphoma. Trends Immunol. 2017, 38, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Bailur, J.K.; McCachren, S.S.; Pendleton, K.; Vasquez, J.C.; Lim, H.S.; Duffy, A.; Doxie, D.B.; Kaushal, A.; Foster, C.; DeRyckere, D.; et al. Risk-associated alterations in marrow T cells in pediatric leukemia. JCI Insight 2020, 5, e140179. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Molinari, V.; Jones, D.T.W.; Izquierdo, E.; Brouwer-Visser, J.; Giangaspero, F.; Haberler, C.; Pietsch, T.; Jacques, T.S.; et al. Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 2018, 33, 829–842 e825. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro Oncol. 2019, 21, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.D.; Franz, C.J.; Brennan, L.; Brouwer-Visser, J.; Tam, R.; Korski, K.; Koeppen, H.; Ziai, J.; Babitzki, G.; Ranchere-Vince, D.; et al. Immune contexture of paediatric cancers. Eur. J. Cancer 2022, 170, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Hendricks, W.P.D.; Nagulapally, A.B.; Kraveka, J.M.; Ferguson, W.S.; Brown, V.I.; Eslin, D.E.; Mitchell, D.; Cornelius, A.; Roberts, W.; et al. Genomic and Transcriptomic Analysis of Relapsed and Refractory Childhood Solid Tumors Reveals a Diverse Molecular Landscape and Mechanisms of Immune Evasion. Cancer Res. 2021, 81, 5818–5832. [Google Scholar] [CrossRef] [PubMed]
- Perzolli, A.; Koedijk, J.B.; Zwaan, C.M.; Heidenreich, O. Targeting the innate immune system in pediatric and adult AML. Leukemia 2024, 38, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Mizia-Malarz, A.; Sobol-Milejska, G. NK Cells as Possible Prognostic Factor in Childhood Acute Lymphoblastic Leukemia. Dis. Markers 2019, 2019, 3596983. [Google Scholar] [CrossRef]
- Okoye, C.; Tran, M.; Soladoye, E.; Akahara, D.E.; Emeasoba, C.M.; Ojinna, B.T.; Anasonye, E.; Obadare, O.O.; Diala, C.S.; Salaudeen, B.H.; et al. A Review of 10-Year Survivability of Immunotherapy in the Management of Colon Cancer. Cureus 2023, 15, e43189. [Google Scholar] [CrossRef]
- Myers, R.M.; Li, Y.; Barz Leahy, A.; Barrett, D.M.; Teachey, D.T.; Callahan, C.; Fasano, C.C.; Rheingold, S.R.; DiNofia, A.; Wray, L.; et al. Humanized CD19-Targeted Chimeric Antigen Receptor (CAR) T Cells in CAR-Naive and CAR-Exposed Children and Young Adults With Relapsed or Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2021, 39, 3044–3055. [Google Scholar] [CrossRef] [PubMed]
- Mayoh, C.; Gifford, A.J.; Terry, R.; Lau, L.M.S.; Wong, M.; Rao, P.; Shai-Hee, T.; Saletta, F.; Khuong-Quang, D.A.; Qin, V.; et al. A novel transcriptional signature identifies T-cell infiltration in high-risk paediatric cancer. Genome Med. 2023, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; De Luca, C.D.; Locatelli, F.; Velardi, E. Thymic Function and T-Cell Receptor Repertoire Diversity: Implications for Patient Response to Checkpoint Blockade Immunotherapy. Front. Immunol. 2021, 12, 752042. [Google Scholar] [CrossRef]
- Rajendran, S.; Hu, Y.; Canella, A.; Peterson, C.; Gross, A.; Cam, M.; Nazzaro, M.; Haffey, A.; Serin-Harmanci, A.; Distefano, R.; et al. Single-cell RNA sequencing reveals immunosuppressive myeloid cell diversity during malignant progression in a murine model of glioma. Cell Rep. 2023, 42, 112197. [Google Scholar] [CrossRef]
- Valind, A.; Verhoeven, B.M.; Enoksson, J.; Karlsson, J.; Christensson, G.; Manas, A.; Aaltonen, K.; Jansson, C.; Bexell, D.; Baryawno, N.; et al. Macrophage infiltration promotes regrowth in MYCN-amplified neuroblastoma after chemotherapy. Oncoimmunology 2023, 12, 2184130. [Google Scholar] [CrossRef]
- Cuadrado-Vilanova, M.; Liu, J.; Paco, S.; Aschero, R.; Burgueno, V.; Sirab, N.; Pascual-Pasto, G.; Correa, G.; Balaguer-Lluna, L.; Castillo-Ecija, H.; et al. Identification of immunosuppressive factors in retinoblastoma cell secretomes and aqueous humor from patients. J. Pathol. 2022, 257, 327–339. [Google Scholar] [CrossRef]
- Chen, S.; Chen, X.; Qiu, J.; Chen, P.; Han, X.; Wu, Y.; Zhuang, J.; Yang, M.; Wu, C.; Wu, N.; et al. Exosomes derived from retinoblastoma cells enhance tumour deterioration by infiltrating the microenvironment. Oncol. Rep. 2021, 45, 278–290. [Google Scholar] [CrossRef]
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.M.; Marie, B.; Larousserie, F.; Entz-Werle, N.; de Pinieux, G.; et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: An immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 trial. Oncoimmunology 2017, 6, e1331193. [Google Scholar] [CrossRef]
- Poli, E.; Cattelan, M.; Zanetti, I.; Scagnellato, A.; Giordano, G.; Zin, A.; Bisogno, G.; Bonvini, P. Autoantibody profiling of alveolar rhabdomyosarcoma patients unveils tumor-associated antigens with diagnostic and prognostic significance. Oncoimmunology 2021, 10, 1954765. [Google Scholar] [CrossRef] [PubMed]
- Miari, K.E.; Guzman, M.L.; Wheadon, H.; Williams, M.T.S. Macrophages in Acute Myeloid Leukaemia: Significant Players in Therapy Resistance and Patient Outcomes. Front. Cell Dev. Biol. 2021, 9, 692800. [Google Scholar] [CrossRef]
- Barros, M.H.; Hassan, R.; Niedobitek, G. Tumor-associated macrophages in pediatric classical Hodgkin lymphoma: Association with Epstein-Barr virus, lymphocyte subsets, and prognostic impact. Clin. Cancer Res. 2012, 18, 3762–3771. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Ligon, J.A.; Choi, W.; Cojocaru, G.; Fu, W.; Hsiue, E.H.; Oke, T.F.; Siegel, N.; Fong, M.H.; Ladle, B.; Pratilas, C.A.; et al. Pathways of immune exclusion in metastatic osteosarcoma are associated with inferior patient outcomes. J. Immunother. Cancer 2021, 9, e001772. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L.; El-Shanshory, M.R.; Abdou, S.H.; Attia, M.S.; Sobhy, S.M.; Zidan, M.F.; Zidan, A.A. Chemotherapy alters the increased numbers of myeloid-derived suppressor and regulatory T cells in children with acute lymphoblastic leukemia. Immunopharmacol. Immunotoxicol. 2018, 40, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Chen, Y.Y.; He, Y.Y.; Wang, J.Y.; Yang, J.P.; Zhong, S.L.; Jiang, N.; Zhou, P.; Jiang, H.; Zhou, J. Expansion and activation of granulocytic, myeloid-derived suppressor cells in childhood precursor B cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2017, 102, 449–458. [Google Scholar] [CrossRef]
- Grazioli, P.; Orlando, A.; Giordano, N.; Noce, C.; Peruzzi, G.; Abdollahzadeh, B.; Screpanti, I.; Campese, A.F. Notch-Signaling Deregulation Induces Myeloid-Derived Suppressor Cells in T-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2022, 13, 809261. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef]
- Kacher, J.; Manches, O.; Aspord, C.; Sartelet, H.; Chaperot, L. Impaired Antitumor Immune Response in MYCN-amplified Neuroblastoma Is Associated with Lack of CCL2 Secretion and Poor Dendritic Cell Recruitment. Cancer Res. Commun. 2022, 2, 577–589. [Google Scholar] [CrossRef]
- Cillo, A.R.; Mukherjee, E.; Bailey, N.G.; Onkar, S.; Daley, J.; Salgado, C.; Li, X.; Liu, D.; Ranganathan, S.; Burgess, M.; et al. Ewing Sarcoma and Osteosarcoma Have Distinct Immune Signatures and Intercellular Communication Networks. Clin. Cancer Res. 2022, 28, 4968–4982. [Google Scholar] [CrossRef]
- de Vasconcellos, J.F.; Laranjeira, A.B.; Zanchin, N.I.; Otubo, R.; Vaz, T.H.; Cardoso, A.A.; Brandalise, S.R.; Yunes, J.A. Increased CCL2 and IL-8 in the bone marrow microenvironment in acute lymphoblastic leukemia. Pediatr. Blood Cancer 2011, 56, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, B.M.; Mei, S.; Olsen, T.K.; Gustafsson, K.; Valind, A.; Lindstrom, A.; Gisselsson, D.; Fard, S.S.; Hagerling, C.; Kharchenko, P.V.; et al. The immune cell atlas of human neuroblastoma. Cell Rep. Med. 2022, 3, 100657. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Zhu, J.; Gu, S.; Yin, M.; Ma, J.; Pan, C.; Tang, J.; Zhang, P.; Liu, Y.; Bai, X.F.; et al. CD200 is overexpressed in neuroblastoma and regulates tumor immune microenvironment. Cancer Immunol. Immunother. 2020, 69, 2333–2343. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, E.Z.; Madney, Y.; Eldin, D.N.; Shafik, N.F. Overexpression of CD200 and CD123 is a major influential factor in the clinical course of pediatric acute myeloid leukemia. Exp. Mol. Pathol. 2021, 118, 104597. [Google Scholar] [CrossRef] [PubMed]
- Aref, S.; Azmy, E.; El-Bakry, K.; Ibrahim, L.; Abdel Aziz, S. Prognostic impact of CD200 and CD56 expression in pediatric B-cell acute lymphoblastic leukemia patients. Pediatr. Hematol. Oncol. 2017, 34, 275–285. [Google Scholar] [CrossRef]
- Coles, S.J.; Hills, R.K.; Wang, E.C.; Burnett, A.K.; Man, S.; Darley, R.L.; Tonks, A. Increased CD200 expression in acute myeloid leukemia is linked with an increased frequency of FoxP3+ regulatory T cells. Leukemia 2012, 26, 2146–2148. [Google Scholar] [CrossRef] [PubMed]
- Herbrich, S.; Baran, N.; Cai, T.; Weng, C.; Aitken, M.J.L.; Post, S.M.; Henderson, J.; Shi, C.; Richard-Carpentier, G.; Sauvageau, G.; et al. Overexpression of CD200 is a Stem Cell-Specific Mechanism of Immune Evasion in AML. J. Immunother. Cancer 2021, 9, e002968. [Google Scholar] [CrossRef] [PubMed]
- Coles, S.J.; Hills, R.K.; Wang, E.C.; Burnett, A.K.; Man, S.; Darley, R.L.; Tonks, A. Expression of CD200 on AML blasts directly suppresses memory T-cell function. Leukemia 2012, 26, 2148–2151. [Google Scholar] [CrossRef] [PubMed]
- Blaeschke, F.; Willier, S.; Stenger, D.; Lepenies, M.; Horstmann, M.A.; Escherich, G.; Zimmermann, M.; Rojas Ringeling, F.; Canzar, S.; Kaeuferle, T.; et al. Leukemia-induced dysfunctional TIM-3(+)CD4(+) bone marrow T cells increase risk of relapse in pediatric B-precursor ALL patients. Leukemia 2020, 34, 2607–2620. [Google Scholar] [CrossRef]
- Do, P.; Beckwith, K.A.; Cheney, C.; Tran, M.; Beaver, L.; Griffin, B.G.; Mo, X.; Liu, Y.; Lapalombella, R.; Hertlein, E.; et al. Leukemic B Cell CTLA-4 Suppresses Costimulation of T Cells. J. Immunol. 2019, 202, 2806–2816. [Google Scholar] [CrossRef]
- Simone, R.; Tenca, C.; Fais, F.; Luciani, M.; De Rossi, G.; Pesce, G.; Bagnasco, M.; Saverino, D. A soluble form of CTLA-4 is present in paediatric patients with acute lymphoblastic leukaemia and correlates with CD1d+ expression. PLoS ONE 2012, 7, e44654. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.X.; Xiao, H.R.; Wang, G.B.; Chen, X.W.; Li, C.G.; Mai, H.R.; Yuan, X.L.; Liu, G.S.; Wen, F.Q. Preliminary investigation on the abnormal mechanism of CD4(+)FOXP3(+)CD25(high) regulatory T cells in pediatric B-cell acute lymphoblastic leukemia. Exp. Ther. Med. 2018, 16, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, P.; Maas, M.L.; Gustafson, M.P.; Dickman, P.; Adams, R.H.; Watanabe, M.; Eshun, F.; Williams, J.; Seidel, M.J.; Dietz, A.B. Increased CTLA-4(+) T cells and an increased ratio of monocytes with loss of class II (CD14(+) HLA-DR(lo/neg)) found in aggressive pediatric sarcoma patients. J. Immunother. Cancer 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef]
- Marangoni, F.; Zhakyp, A.; Corsini, M.; Geels, S.N.; Carrizosa, E.; Thelen, M.; Mani, V.; Prussmann, J.N.; Warner, R.D.; Ozga, A.J.; et al. Expansion of tumor-associated Treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell 2021, 184, 3998–4015.e19. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Sho, M.; Sawai, T.; Kanehiro, H.; Maeda, K.; Yoshida, M.; Tsukada, R.; Nomura, M.; Okuyama, H. Potential role of the PD-L1 expression and tumor-infiltrating lymphocytes on neuroblastoma. Pediatr. Surg. Int. 2020, 36, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Siebert, N.; Zumpe, M.; Juttner, M.; Troschke-Meurer, S.; Lode, H.N. PD-1 blockade augments anti-neuroblastoma immune response induced by anti-GD(2) antibody ch14.18/CHO. Oncoimmunology 2017, 6, e1343775. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Kang, H.J.; Yalon-Oren, M.; Marshall, L.V.; Vezina, C.; Pappo, A.; Laetsch, T.W.; Petrilli, A.S.; Ebinger, M.; Toporski, J.; et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): Interim analysis of an open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Sundara, Y.T.; Kostine, M.; Cleven, A.H.; Bovee, J.V.; Schilham, M.W.; Cleton-Jansen, A.M. Increased PD-L1 and T-cell infiltration in the presence of HLA class I expression in metastatic high-grade osteosarcoma: A rationale for T-cell-based immunotherapy. Cancer Immunol. Immunother. 2017, 66, 119–128. [Google Scholar] [CrossRef]
- Su, Y.; Luo, B.; Lu, Y.; Wang, D.; Yan, J.; Zheng, J.; Xiao, J.; Wang, Y.; Xue, Z.; Yin, J.; et al. Anlotinib Induces a T Cell-Inflamed Tumor Microenvironment by Facilitating Vessel Normalization and Enhances the Efficacy of PD-1 Checkpoint Blockade in Neuroblastoma. Clin. Cancer Res. 2022, 28, 793–809. [Google Scholar] [CrossRef]
- Miracco, C.; Toti, P.; Gelmi, M.C.; Aversa, S.; Baldino, G.; Galluzzi, P.; De Francesco, S.; Petrelli, F.; Sorrentino, E.; Belmonte, G.; et al. Retinoblastoma Is Characterized by a Cold, CD8+ Cell Poor, PD-L1- Microenvironment, Which Turns Into Hot, CD8+ Cell Rich, PD-L1+ After Chemotherapy. Investig. Ophthalmol. Vis. Sci. 2021, 62, 6. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, F.; Yu, P.; Wang, J.; Liu, Z.; Bao, Q.; Zhang, W.; Wen, J. Sorafenib inhibits doxorubicin-induced PD-L1 upregulation to improve immunosuppressive microenvironment in Osteosarcoma. J. Cancer Res. Clin. Oncol. 2022, 149, 5127–5138. [Google Scholar] [CrossRef] [PubMed]
- Hofmeyer, K.A.; Ray, A.; Zang, X. The contrasting role of B7-H3. Proc. Natl. Acad. Sci. USA 2008, 105, 10277–10278. [Google Scholar] [CrossRef] [PubMed]
- Haydar, D.; Houke, H.; Chiang, J.; Yi, Z.; Ode, Z.; Caldwell, K.; Zhu, X.; Mercer, K.S.; Stripay, J.L.; Shaw, T.I.; et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro Oncol. 2021, 23, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, E.I.; Du, H.; Shou, P.; Song, F.; Suzuki, K.; Ahn, S.; Li, G.; Ferrone, S.; Su, L.; Savoldo, B.; et al. Preclinical Evaluation of B7-H3-specific Chimeric Antigen Receptor T Cells for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 3141–3153. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Poolen, G.C.; van Vliet, L.C.; Schipper, J.G.; Broekhuizen, R.; Monnikhof, M.; Van Hecke, W.; Vermeulen, J.F.; Bovenschen, N. Pediatric medulloblastoma express immune checkpoint B7-H3. Clin. Transl. Oncol. 2022, 24, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Hont, A.B.; Dumont, B.; Sutton, K.S.; Anderson, J.; Kentsis, A.; Drost, J.; Hong, A.L.; Verschuur, A. The tumor microenvironment and immune targeting therapy in pediatric renal tumors. Pediatr. Blood Cancer 2023, 70 (Suppl. 2), e30110. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, T.; Miyachi, M.; Sugimoto, Y.; Yagyu, S.; Kikuchi, K.; Tsuchiya, K.; Iehara, T.; Hosoi, H. Reduced B7-H3 expression by PAX3-FOXO1 knockdown inhibits cellular motility and promotes myogenic differentiation in alveolar rhabdomyosarcoma. Sci. Rep. 2021, 11, 18802. [Google Scholar] [CrossRef]
- Dillekas, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef]
- Tian, K.; Du, G.; Wang, X.; Wu, X.; Li, L.; Liu, W.; Wu, R. MMP-9 secreted by M2-type macrophages promotes Wilms’ tumour metastasis through the PI3K/AKT pathway. Mol. Biol. Rep. 2022, 49, 3469–3480. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Guo, W.; Ren, T.; Huang, Y.; Wang, S.; Liu, K.; Zheng, B.; Yang, K.; Zhang, H.; Liang, X. Tumor-associated macrophages promote lung metastasis and induce epithelial-mesenchymal transition in osteosarcoma by activating the COX-2/STAT3 axis. Cancer Lett. 2019, 440–441, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, M.; Wei, R.; Wu, J. Identification and Functional Analysis of EPOR(+) Tumor-Associated Macrophages in Human Osteosarcoma Lung Metastasis. J. Immunol. Res. 2020, 2020, 9374240. [Google Scholar] [CrossRef] [PubMed]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.; Burger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.; Lankester, A.C.; et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: A rationale for treatment with macrophage activating agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, O.; Yoshida, M.; Koma, Y.; Yanai, T.; Hasegawa, D.; Kosaka, Y.; Nishimura, N.; Yokozaki, H. Collaboration of cancer-associated fibroblasts and tumour-associated macrophages for neuroblastoma development. J. Pathol. 2016, 240, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Asgharzadeh, S.; Salo, J.A.; Ji, L.; Oberthuer, A.; Fischer, M.; Berthold, F.; Hadjidaniel, M.; Liu, C.W.; Metelitsa, L.S.; Pique-Regi, R.; et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J. Clin. Oncol. 2012, 30, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowska, S.; Beury, D.W.; Gopalan, V.; Tycko, A.K.; Qin, H.; Clements, M.E.; Drake, J.; Nwanze, C.; Murgai, M.; Rae, Z.; et al. Genetically engineered myeloid cells rebalance the core immune suppression program in metastasis. Cell 2021, 184, 2033–2052.e21. [Google Scholar] [CrossRef] [PubMed]
- Kos, K.; Aslam, M.A.; van de Ven, R.; Wellenstein, M.D.; Pieters, W.; van Weverwijk, A.; Duits, D.E.M.; van Pul, K.; Hau, C.S.; Vrijland, K.; et al. Tumor-educated T(regs) drive organ-specific metastasis in breast cancer by impairing NK cells in the lymph node niche. Cell Rep. 2022, 38, 110447. [Google Scholar] [CrossRef] [PubMed]
- Brinkrolf, P.; Landmeier, S.; Altvater, B.; Chen, C.; Pscherer, S.; Rosemann, A.; Ranft, A.; Dirksen, U.; Juergens, H.; Rossig, C. A high proportion of bone marrow T cells with regulatory phenotype (CD4+CD25hiFoxP3+) in Ewing sarcoma patients is associated with metastatic disease. Int. J. Cancer 2009, 125, 879–886. [Google Scholar] [CrossRef]
- Fritzsching, B.; Fellenberg, J.; Moskovszky, L.; Sapi, Z.; Krenacs, T.; Machado, I.; Poeschl, J.; Lehner, B.; Szendroi, M.; Bosch, A.L.; et al. CD8(+)/FOXP3(+)-ratio in osteosarcoma microenvironment separates survivors from non-survivors: A multicenter validated retrospective study. Oncoimmunology 2015, 4, e990800. [Google Scholar] [CrossRef]
- Almeida, R.D.S.; Ramos, A.M.L.; Luna, C.F.; Pedrosa, F.; Donadi, E.A.; Lucena-Silva, N. Cytokines and soluble HLA-G levels in bone marrow stroma and their association with the survival rate of patients exhibiting childhood T-cell acute lymphoblastic leukemia. Cytokine 2018, 102, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Kaya, Z.; Yuce, D.; Kirkiz, S.; Kocak, U.; Ozmen, F. Prognostic role of serum cytokines and soluble HLA-G levels in children with leukemia who undergo allogeneic stem cell transplantation. Cytokine 2022, 153, 155869. [Google Scholar] [CrossRef] [PubMed]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef]
- Rutishauser, R.L.; Martins, G.A.; Kalachikov, S.; Chandele, A.; Parish, I.A.; Meffre, E.; Jacob, J.; Calame, K.; Kaech, S.M. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 2009, 31, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Z.; Guo, X.; Liao, R.; Yang, K.; Ye, L.; You, Z. Involvement of IL-10 and TGF-beta in HLA-E-mediated neuroblastoma migration and invasion. Oncotarget 2016, 7, 44340–44349. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Wang, S.; Fang, T.; Qiu, X.; Wang, X.; Zhou, X.; Morse, M.A.; Hobeika, A.; Wu, W.; Yang, H.; et al. Changes in Peripheral Blood Regulatory T Cells and IL-6 and IL-10 Levels Predict Response of Pediatric Medulloblastoma and Germ Cell Tumors With Residual or Disseminated Disease to Craniospinal Irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, L.; Wittig, S.; Gruhn, B. Interleukin-10-592 polymorphism: Impact on relapse and survival after allogeneic hematopoietic stem cell transplantation in children with hematological malignancies. J. Cancer Res. Clin. Oncol. 2022, 148, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, G.; Taçyildiz, N.; Babacan, E.; Ünal, E.; İkincioğullari, A.; Eğin, Y.; Göksel, D.; Ertem, U.; Dağdemir, A.; Cin, Ş.; et al. Serum Interleukin-10 in Childhood Malignant Lymphomas (Preliminary Report). Pediatr. Res. 1999, 45, 775. [Google Scholar] [CrossRef]
- Stevens, A.M.; Horton, T.M.; Glasser, C.L.; Gerbing, R.B.; Aplenc, R.; Alonzo, T.A.; Redell, M.S. IL-10 and TNFalpha are associated with decreased survival in low-risk pediatric acute myeloid leukemia; a children’s oncology group report. Pediatr. Hematol. Oncol. 2023, 40, 147–158. [Google Scholar] [CrossRef]
- Zhang, X.L.; Komada, Y.; Chipeta, J.; Li, Q.S.; Inaba, H.; Azuma, E.; Yamamoto, H.; Sakurai, M. Intracellular cytokine profile of T cells from children with acute lymphoblastic leukemia. Cancer Immunol. Immunother. 2000, 49, 165–172. [Google Scholar] [CrossRef]
- Whitehead, T.P.; Wiemels, J.L.; Zhou, M.; Kang, A.Y.; McCoy, L.S.; Wang, R.; Fitch, B.; Petrick, L.M.; Yano, Y.; Imani, P.; et al. Cytokine Levels at Birth in Children Who Developed Acute Lymphoblastic Leukemia. Cancer Epidemiol. Biomark. Prev. 2021, 30, 1526–1535. [Google Scholar] [CrossRef]
- Magalhaes-Gama, F.; Kerr, M.W.A.; de Araujo, N.D.; Ibiapina, H.N.S.; Neves, J.C.F.; Hanna, F.S.A.; Xabregas, L.A.; Carvalho, M.; Alves, E.B.; Tarrago, A.M.; et al. Imbalance of Chemokines and Cytokines in the Bone Marrow Microenvironment of Children with B-Cell Acute Lymphoblastic Leukemia. J. Oncol. 2021, 2021, 5530650. [Google Scholar] [CrossRef]
- Knorr, F.; Damm-Welk, C.; Ruf, S.; Singh, V.K.; Zimmermann, M.; Reiter, A.; Woessmann, W. Blood cytokine concentrations in pediatric patients with anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica 2018, 103, 477–485. [Google Scholar] [CrossRef]
- Bte Syed Sulaiman, N.; Kuick, C.H.; Chang, K.T.E.; Wan, K.R.; Looi, W.S.; Low, D.C.Y.; Seow, W.T.; Low, S.Y.Y. Cytokines in Pediatric Pilocytic Astrocytomas: A Clinico-Pathological Study. NeuroSci 2021, 2, 95–108. [Google Scholar] [CrossRef]
- Tapia, L.I.; Olivares, M.; Torres, J.P.; De la Maza, V.; Valenzuela, R.; Contardo, V.; Tordecilla, J.; Alvarez, A.M.; Varas, M.; Zubieta, M.; et al. Cytokine and chemokine profiles in episodes of persistent high-risk febrile neutropenia in children with cancer. Cytokine 2021, 148, 155619. [Google Scholar] [CrossRef]
- Narendran, G.; Tomfohr, L.; Schulte, F. Inflammatory cytokines and depression in children with cancer: A review of the literature. Pediatr. Hematol. Oncol. 2018, 35, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Singh, A.; Yadav, G.; Kushwaha, R.; Ali, W.; Verma, S.P.; Singh, U.S. Serum Tumor Necrosis Factor-Alpha Levels in Acute Leukemia and Its Prognostic Significance. Cureus 2022, 14, e24835. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chu, X.; Gao, L.; Ling, J.; Xiao, P.; Lu, J.; Wang, Y.; He, H.; Li, J.; Hu, Y.; et al. High Expression of Interleukin-3 Receptor Alpha Chain (CD123) Predicts Favorable Outcome in Pediatric B-Cell Acute Lymphoblastic Leukemia Lacking Prognosis-Defining Genomic Aberrations. Front. Oncol. 2021, 11, 614420. [Google Scholar] [CrossRef]
- Wittwer, N.L.; Brumatti, G.; Marchant, C.; Sandow, J.J.; Pudney, M.K.; Dottore, M.; D’Andrea, R.J.; Lopez, A.F.; Ekert, P.G.; Ramshaw, H.S. High CD123 levels enhance proliferation in response to IL-3, but reduce chemotaxis by downregulating CXCR4 expression. Blood Adv. 2017, 1, 1067–1079. [Google Scholar] [CrossRef]
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 2011, 43, 932–939. [Google Scholar] [CrossRef]
- Oliveira, M.L.; Veloso, A.; Garcia, E.G.; Iyer, S.; Pereira, C.; Barreto, V.M.; Langenau, D.M.; Barata, J.T. Mutant IL7R collaborates with MYC to induce T-cell acute lymphoblastic leukemia. Leukemia 2022, 36, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kong, L.; Kim, S.; Lee, S.; Oh, S.; Jo, S.; Jang, I.; Kim, T.D. The Role of IL-7 and IL-7R in Cancer Pathophysiology and Immunotherapy. Int. J. Mol. Sci. 2022, 23, 10412. [Google Scholar] [CrossRef] [PubMed]
- Peace, D.J.; Cheever, M.A. Toxicity and therapeutic efficacy of high-dose interleukin 2. In vivo infusion of antibody to NK-1.1 attenuates toxicity without compromising efficacy against murine leukemia. J. Exp. Med. 1989, 169, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.S.; Hallner, A.; Brune, M.; Nilsson, S.; Thoren, F.B.; Martner, A.; Hellstrand, K. Immunotherapy with HDC/IL-2 may be clinically efficacious in acute myeloid leukemia of normal karyotype. Hum. Vaccin. Immunother. 2020, 16, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Petit, A.; Ducassou, S.; Leblanc, T.; Pasquet, M.; Rousseau, A.; Ragu, C.; Cachanado, M.; Nelken, B.; Bertrand, Y.; Michel, G.; et al. Maintenance Therapy With Interleukin-2 for Childhood AML: Results of ELAM02 Phase III Randomized Trial. Hemasphere 2018, 2, e159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hresko, M.E.; Picton, L.K.; Su, L.; Hollander, M.J.; Nunez-Cruz, S.; Zhang, Z.; Assenmacher, C.A.; Sockolosky, J.T.; Garcia, K.C.; et al. A human orthogonal IL-2 and IL-2Rbeta system enhances CAR T cell expansion and antitumor activity in a murine model of leukemia. Sci. Transl. Med. 2021, 13, eabg6986. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.M.; Miller, J.M.; Munoz, J.O.; Gaikwad, A.S.; Redell, M.S. Interleukin-6 levels predict event-free survival in pediatric AML and suggest a mechanism of chemotherapy resistance. Blood Adv. 2017, 1, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Inoue, K.; Ogawa, H.; Yamagami, T.; Soma, T.; Miyake, S.; Hirata, M.; Kishimoto, T. The expression of IL-6 and its related genes in acute leukemia. Leuk. Lymphoma 1996, 21, 49–52. [Google Scholar] [CrossRef]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef]
- Bruserud, O.; Tore Gjertsen, B.; Brustugun, O.T.; Bassoe, C.F.; Nesthus, I.; Espen Akselsen, P.; Buhring, H.J.; Pawelec, G. Effects of interleukin 10 on blast cells derived from patients with acute myelogenous leukemia. Leukemia 1995, 9, 1910–1920. [Google Scholar]
- Chang, J.S.; Zhou, M.; Buffler, P.A.; Chokkalingam, A.P.; Metayer, C.; Wiemels, J.L. Profound deficit of IL10 at birth in children who develop childhood acute lymphoblastic leukemia. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- Fitch, B.A.; Zhou, M.; Situ, J.; Surianarayanan, S.; Reeves, M.Q.; Hermiston, M.L.; Wiemels, J.L.; Kogan, S.C. Decreased IL-10 accelerates B-cell leukemia/lymphoma in a mouse model of pediatric lymphoid leukemia. Blood Adv. 2022, 6, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Su, H.; Yan, M.; Zhang, L.; Tang, J.; Li, Q.; Gu, X.; Gong, Q. Interleukin-33 Promotes Cell Survival via p38 MAPK-Mediated Interleukin-6 Gene Expression and Release in Pediatric AML. Front. Immunol. 2020, 11, 595053. [Google Scholar] [CrossRef] [PubMed]
- Naef, P.; Radpour, R.; Jaeger-Ruckstuhl, C.A.; Bodmer, N.; Baerlocher, G.M.; Doehner, H.; Doehner, K.; Riether, C.; Ochsenbein, A.F. IL-33-ST2 signaling promotes stemness in subtypes of myeloid leukemia cells through the Wnt and Notch pathways. Sci. Signal. 2023, 16, eadd7705. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.; Edwards, D.K.t.; Eide, C.A.; Newell, L.; Traer, E.; Medeiros, B.C.; Pollyea, D.A.; Deininger, M.W.; Collins, R.H.; Tyner, J.W.; et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep. 2017, 18, 3204–3218. [Google Scholar] [CrossRef] [PubMed]
- Fitter, S.; Bradey, A.L.; Kok, C.H.; Noll, J.E.; Wilczek, V.J.; Venn, N.C.; Law, T.; Paisitkriangkrai, S.; Story, C.; Saunders, L.; et al. CKLF and IL1B transcript levels at diagnosis are predictive of relapse in children with pre-B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2021, 193, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Arriero, M.D.M.; Villatoro, A. Interleukin-1beta as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev. 2017, 31, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Fultang, L.; Gamble, L.D.; Gneo, L.; Berry, A.M.; Egan, S.A.; De Bie, F.; Yogev, O.; Eden, G.L.; Booth, S.; Brownhill, S.; et al. Macrophage-Derived IL1beta and TNFalpha Regulate Arginine Metabolism in Neuroblastoma. Cancer Res. 2019, 79, 611–624. [Google Scholar] [CrossRef]
- Ladenstein, R.; Potschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Yaniv, I.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): A multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1617–1629. [Google Scholar] [CrossRef]
- Pession, A.; Prete, A.; Locatelli, F.; Pierinelli, S.; Pession, A.L.; Maccario, R.; Magrini, E.; De Bernardi, B.; Paolucci, P.; Paolucci, G. Immunotherapy with low-dose recombinant interleukin 2 after high-dose chemotherapy and autologous stem cell transplantation in neuroblastoma. Br. J. Cancer 1998, 78, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Cicek, F.; Troschke-Meurer, S.; Ceylan, K.; Jahns, L.J.; Zumpe, M.; Siebert, N.; Ehlert, K.; Lode, H.N. Impact of IL-2 on Treatment Tolerance in Patients With High-Risk Neuroblastoma Treated With Dinutuximab Beta-Based Immunotherapy. Front. Pediatr. 2020, 8, 582820. [Google Scholar] [CrossRef] [PubMed]
- Raeber, M.E.; Sahin, D.; Karakus, U.; Boyman, O. A systematic review of interleukin-2-based immunotherapies in clinical trials for cancer and autoimmune diseases. EBioMedicine 2023, 90, 104539. [Google Scholar] [CrossRef] [PubMed]
- Ara, T.; Song, L.; Shimada, H.; Keshelava, N.; Russell, H.V.; Metelitsa, L.S.; Groshen, S.G.; Seeger, R.C.; DeClerck, Y.A. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 2009, 69, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Louault, K.; Porras, T.; Lee, M.H.; Muthugounder, S.; Kennedy, R.J.; Blavier, L.; Sarte, E.; Fernandez, G.E.; Yang, F.; Pawel, B.R.; et al. Fibroblasts and macrophages cooperate to create a pro-tumorigenic and immune resistant environment via activation of TGF-beta/IL-6 pathway in neuroblastoma. Oncoimmunology 2022, 11, 2146860. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J.; et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef]
- Holst, C.B.; Christensen, I.J.; Skjoth-Rasmussen, J.; Hamerlik, P.; Poulsen, H.S.; Johansen, J.S. Systemic Immune Modulation in Gliomas: Prognostic Value of Plasma IL-6, YKL-40, and Genetic Variation in YKL-40. Front. Oncol. 2020, 10, 478. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Tanaka, K.; Itonaga, I.; Iwasaki, T.; Tsumura, H. Interaction between human osteosarcoma and mesenchymal stem cells via an interleukin-8 signaling loop in the tumor microenvironment. Cell Commun. Signal. 2018, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Baglio, S.R.; Schrijver, C.D.; Massaro, C.; Lagerweij, T.; Martin, C.G.; Giorgio, C.; Brandolini, L.; Gavioli, E.M.; Allegretti, M.; Pegtel, D.M. Combined IL-6 and IL-8 inhibition to overcome mesenchymal stem cell (MSC)-induced resistance to antimetastatic drugs in osteosarcoma. J. Clin. Oncol. 2022, 40, 10037. [Google Scholar] [CrossRef]
- Cheng, M.; Cai, W.; Huang, W.; Chen, Y.; Wu, Z.; Luo, P.; Yan, W. Histone deacetylase 6 regulated expression of IL-8 is involved in the doxorubicin (Dox) resistance of osteosarcoma cells via modulating ABCB1 transcription. Eur. J. Pharmacol. 2018, 840, 1–8. [Google Scholar] [CrossRef]
- Xiao, H.; Chen, L.; Luo, G.; Son, H.; Prectoni, J.H.; Zheng, W. Effect of the cytokine levels in serum on osteosarcoma. Tumour Biol. 2014, 35, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.S.; Jian, T.Y.; Sung, S.Y.; Hsieh, C.L.; Huang, M.H.; Fang, C.L.; Wong, T.T.; Lin, Y.L. Enrichment of Tumor-Infiltrating B Cells in Group 4 Medulloblastoma in Children. Int. J. Mol. Sci. 2022, 23, 5287. [Google Scholar] [CrossRef] [PubMed]
- Huettner, C.; Czub, S.; Kerkau, S.; Roggendorf, W.; Tonn, J.C. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res. 1997, 17, 3217–3224. [Google Scholar] [PubMed]
- Sreenivasan, L.; Wang, H.; Yap, S.Q.; Leclair, P.; Tam, A.; Lim, C.J. Autocrine IL-6/STAT3 signaling aids development of acquired drug resistance in Group 3 medulloblastoma. Cell Death Dis. 2020, 11, 1035. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Jayasekara, W.S.N.; Picard, D.; Chen, J.; Watkins, D.N.; Cain, J.E.; Remke, M.; Gough, D.J. A Sexually Dimorphic Role for STAT3 in Sonic Hedgehog Medulloblastoma. Cancers 2019, 11, 1702. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Nong, L.; Wei, Y.; Qin, S.; Zhou, Y.; Tang, Y. Association of interleukin-12 polymorphisms and serum IL-12p40 levels with osteosarcoma risk. DNA Cell Biol. 2013, 32, 605–610. [Google Scholar] [CrossRef]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; Vom Berg, J.; Kulig, P.; Becher, B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Zalacain, M.; Bunuales, M.; Marrodan, L.; Labiano, S.; Gonzalez-Huarriz, M.; Martinez-Velez, N.; Laspidea, V.; Puigdelloses, M.; Garcia-Moure, M.; Gonzalez-Aparicio, M.; et al. Local administration of IL-12 with an HC vector results in local and metastatic tumor control in pediatric osteosarcoma. Mol. Ther. Oncolytics 2021, 20, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Starr, R.; Aguilar, B.; Shami, A.F.; Martinez, C.; D’Apuzzo, M.; Barish, M.E.; Forman, S.J.; Jensen, M.C. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin. Cancer Res. 2012, 18, 2199–2209. [Google Scholar] [CrossRef]
- Rechberger, J.S.; Porath, K.A.; Zhang, L.; Nesvick, C.L.; Schrecengost, R.S.; Sarkaria, J.N.; Daniels, D.J. IL-13Ralpha2 Status Predicts GB-13 (IL13.E13K-PE4E) Efficacy in High-Grade Glioma. Pharmaceutics 2022, 14, 922. [Google Scholar] [CrossRef]
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Potter, D.M.; Connelly, A.K.; Dibridge, S.A.; Whiteside, T.L.; Okada, H. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J. Clin. Oncol. 2014, 32, 2050–2058. [Google Scholar] [CrossRef]
- Burga, R.A.; Yvon, E.; Chorvinsky, E.; Fernandes, R.; Cruz, C.R.Y.; Bollard, C.M. Engineering the TGFbeta Receptor to Enhance the Therapeutic Potential of Natural Killer Cells as an Immunotherapy for Neuroblastoma. Clin. Cancer Res. 2019, 25, 4400–4412. [Google Scholar] [CrossRef]
- Fuchs, B.; Inwards, C.Y.; Janknecht, R. Vascular endothelial growth factor expression is up-regulated by EWS-ETS oncoproteins and Sp1 and may represent an independent predictor of survival in Ewing’s sarcoma. Clin. Cancer Res. 2004, 10, 1344–1353. [Google Scholar] [CrossRef]
- DuBois, S.G.; Marina, N.; Glade-Bender, J. Angiogenesis and vascular targeting in Ewing sarcoma: A review of preclinical and clinical data. Cancer 2010, 116, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.S.; Kleinerman, E.S. Tumor Vessel Development and Expansion in Ewing’s Sarcoma: A Review of the Vasculogenesis Process and Clinical Trials with Vascular-Targeting Agents. Sarcoma 2011, 2011, 165837. [Google Scholar] [CrossRef] [PubMed]
- Capasso, L.; Florio, M.; Lillo, M.; Basilico, M.; De Santis, V.; Ziranu, A.; Grasso, A.; Minutillo, F.; Maccauro, G. Vascular endothelial growth factor expression as a biomarker of prognosis in patients with chondrosarcoma, Ewing’s sarcoma and osteosarcoma. Current concepts. J. Biol. Regul. Homeost. Agents 2019, 33, 39–43. [Google Scholar] [PubMed]
- Ghanem, M.A.; van Steenbrugge, G.J.; Sudaryo, M.K.; Mathoera, R.B.; Nijman, J.M.; van der Kwast, T.H. Expression and prognostic relevance of vascular endothelial growth factor (VEGF) and its receptor (FLT-1) in nephroblastoma. J. Clin. Pathol. 2003, 56, 107–113. [Google Scholar] [CrossRef]
- Rowe, D.H.; Huang, J.; Kayton, M.L.; Thompson, R.; Troxel, A.; O’Toole, K.M.; Yamashiro, D.; Stolar, C.J.; Kandel, J.J. Anti-VEGF antibody suppresses primary tumor growth and metastasis in an experimental model of Wilms’ tumor. J. Pediatr. Surg. 2000, 35, 30–32; discussion 32–33. [Google Scholar] [CrossRef]
- Hong, B.; Dong, R. Research advances in the targeted therapy and immunotherapy of Wilms tumor: A narrative review. Transl. Cancer Res. 2021, 10, 1559–1567. [Google Scholar] [CrossRef]
- Tang, M.J.; Ma, X.L.; He, X.L.; Pan, W.H.; Zhang, X.H.; Jiang, S.Y.; Gao, J.; Li, F.; Yao, W.; Gu, S.; et al. A multicenter prospective study on the management of hepatoblastoma in children: A report from the Chinese Children’s Cancer Group. World J. Pediatr. 2023. [Google Scholar] [CrossRef]
- Li, M.; Li, H.; Li, C.; Wang, S.; Jiang, W.; Liu, Z.; Zhou, S.; Liu, X.; McNutt, M.A.; Li, G. Alpha-fetoprotein: A new member of intracellular signal molecules in regulation of the PI3K/AKT signaling in human hepatoma cell lines. Int. J. Cancer 2011, 128, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Dai, X.; Dai, J.; Ding, C.; Zhang, Z.; Lin, Z.; Hu, J.; Lu, M.; Wang, Z.; Qi, Y.; et al. AFP promotes HCC progression by suppressing the HuR-mediated Fas/FADD apoptotic pathway. Cell Death Dis. 2020, 11, 822. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Malogolowkin, M.H.; Furman, W.L.; Piao, J.; Krailo, M.D.; Chung, N.; Brock, L.; Towbin, A.J.; McCarville, E.B.; Finegold, M.J.; et al. Vincristine/irinotecan/temsirolimus upfront window treatment of high-risk hepatoblastoma: A report from the Children’s Oncology Group AHEP0731 Study Committee. Pediatr. Blood Cancer 2023, 70, e30365. [Google Scholar] [CrossRef] [PubMed]
- Hutzen, B.; Paudel, S.N.; Naeimi Kararoudi, M.; Cassady, K.A.; Lee, D.A.; Cripe, T.P. Immunotherapies for pediatric cancer: Current landscape and future perspectives. Cancer Metastasis Rev. 2019, 38, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Butler, E.; Ludwig, K.; Pacenta, H.L.; Klesse, L.J.; Watt, T.C.; Laetsch, T.W. Recent progress in the treatment of cancer in children. CA Cancer J. Clin. 2021, 71, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Reaman, G.H.; Hank, J.A.; Cairo, M.S.; Anderson, P.; Blazar, B.R.; Frierdich, S.; Sondel, P.M. A phase II trial of human recombinant interleukin-2 administered as a 4-day continuous infusion for children with refractory neuroblastoma, non-Hodgkin’s lymphoma, sarcoma, renal cell carcinoma, and malignant melanoma. A Childrens Cancer Group study. Cancer 1995, 75, 2959–2965. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Inaba, H.; Kang, G.; Gan, K.; Hartford, C.; Triplett, B.M.; Dallas, M.; Shook, D.; Gruber, T.; Pui, C.H.; et al. Natural killer cell therapy in children with relapsed leukemia. Pediatr. Blood Cancer 2015, 62, 1468–1472. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A.; et al. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children With High-risk First-Relapse B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 843–854. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef]
- Cripe, T.P.; Ngo, M.C.; Geller, J.I.; Louis, C.U.; Currier, M.A.; Racadio, J.M.; Towbin, A.J.; Rooney, C.M.; Pelusio, A.; Moon, A.; et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.E.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Bernhardt, M.B.; et al. Effect of Postreinduction Therapy Consolidation With Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults With First Relapse of B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Dinutuximab approved for high-risk neuroblastoma. Cancer Discov. 2015, 5, OF5. [CrossRef] [PubMed]
- Navid, F.; Santana, V.M.; Barfield, R.C. Anti-GD2 antibody therapy for GD2-expressing tumors. Curr. Cancer Drug Targets 2010, 10, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Yu, R.K.; Cheung, N.K. Ganglioside GD2 specificity of monoclonal antibodies to human neuroblastoma cell. Biochem. Biophys. Res. Commun. 1985, 127, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Uttenreuther-Fischer, M.M.; Huang, C.S.; Yu, A.L. Pharmacokinetics of human-mouse chimeric anti-GD2 mAb ch14.18 in a phase I trial in neuroblastoma patients. Cancer Immunol. Immunother. 1995, 41, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Barker, E.; Mueller, B.M.; Handgretinger, R.; Herter, M.; Yu, A.L.; Reisfeld, R.A. Effect of a chimeric anti-ganglioside GD2 antibody on cell-mediated lysis of human neuroblastoma cells. Cancer Res. 1991, 51, 144–149. [Google Scholar] [PubMed]
- Ladenstein, R.; Weixler, S.; Baykan, B.; Bleeke, M.; Kunert, R.; Katinger, D.; Pribill, I.; Glander, P.; Bauer, S.; Pistoia, V.; et al. Ch14.18 antibody produced in CHO cells in relapsed or refractory Stage 4 neuroblastoma patients: A SIOPEN Phase 1 study. MAbs 2013, 5, 801–809. [Google Scholar] [CrossRef]
- Navid, F.; Sondel, P.M.; Barfield, R.; Shulkin, B.L.; Kaufman, R.A.; Allay, J.A.; Gan, J.; Hutson, P.; Seo, S.; Kim, K.; et al. Phase I trial of a novel anti-GD2 monoclonal antibody, Hu14.18K322A, designed to decrease toxicity in children with refractory or recurrent neuroblastoma. J. Clin. Oncol. 2014, 32, 1445–1452. [Google Scholar] [CrossRef]
- Albertini, M.R.; Hank, J.A.; Gadbaw, B.; Kostlevy, J.; Haldeman, J.; Schalch, H.; Gan, J.; Kim, K.; Eickhoff, J.; Gillies, S.D.; et al. Phase II trial of hu14.18-IL2 for patients with metastatic melanoma. Cancer Immunol. Immunother. 2012, 61, 2261–2271. [Google Scholar] [CrossRef]
- Cheung, I.Y.; Kushner, B.H.; Modak, S.; Basu, E.M.; Roberts, S.S.; Cheung, N.V. Phase I trial of anti-GD2 monoclonal antibody hu3F8 plus GM-CSF: Impact of body weight, immunogenicity and anti-GD2 response on pharmacokinetics and survival. Oncoimmunology 2017, 6, e1358331. [Google Scholar] [CrossRef] [PubMed]
- Tassev, D.V.; Cheung, N.K. Monoclonal antibody therapies for solid tumors. Expert Opin. Biol. Ther. 2009, 9, 341–353. [Google Scholar] [CrossRef]
- Park, J.A.; Cheung, N.V. Targets and Antibody Formats for Immunotherapy of Neuroblastoma. J. Clin. Oncol. 2020, 38, 1836–1848. [Google Scholar] [CrossRef]
- Matthay, K.K.; George, R.E.; Yu, A.L. Promising therapeutic targets in neuroblastoma. Clin. Cancer Res. 2012, 18, 2740–2753. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.K.; Cheung, I.Y.; Kushner, B.H.; Ostrovnaya, I.; Chamberlain, E.; Kramer, K.; Modak, S. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J. Clin. Oncol. 2012, 30, 3264–3270. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef]
- Mody, R.; Naranjo, A.; Van Ryn, C.; Yu, A.L.; London, W.B.; Shulkin, B.L.; Parisi, M.T.; Servaes, S.E.; Diccianni, M.B.; Sondel, P.M.; et al. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): An open-label, randomised, phase 2 trial. Lancet Oncol. 2017, 18, 946–957. [Google Scholar] [CrossRef]
- Furman, W.L.; McCarville, B.; Shulkin, B.L.; Davidoff, A.; Krasin, M.; Hsu, C.W.; Pan, H.; Wu, J.; Brennan, R.; Bishop, M.W.; et al. Improved Outcome in Children With Newly Diagnosed High-Risk Neuroblastoma Treated With Chemoimmunotherapy: Updated Results of a Phase II Study Using hu14.18K322A. J. Clin. Oncol. 2022, 40, 335–344. [Google Scholar] [CrossRef]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef]
- Gupta, A.K.; Chopra, A.; Meena, J.P.; Singh, J.; Pandey, R.M.; Bakhshi, S.; Seth, R. Rituximab added to standard chemotherapy and its effect on minimal residual disease during induction in CD20 positive pediatric acute lymphoblastic leukemia: A pilot RCT. Am. J. Blood Res. 2021, 11, 571–579. [Google Scholar]
- FDA Expands Approval of Ipilimumab to Pediatric Patients 12 Years and Older. 2017. Available online: https://ascopost.com/issues/october-10-2017/fda-expands-approval-of-ipilimumab-to-pediatric-patients-12-years-and-older/ (accessed on 10 March 2024).
- Subbiah, V.; Solit, D.B.; Chan, T.A.; Kurzrock, R. The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB) >/=10: A decision centered on empowering patients and their physicians. Ann. Oncol. 2020, 31, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Hoshitsuki, K.; Zhou, Y.; Miller, A.M.; Choi, J.K.; Swanson, H.D.; Bhakta, N.H.; Jeha, S.; Karol, S.E.; Ribeiro, R.C.; Rubnitz, J.E.; et al. Rituximab administration in pediatric patients with newly diagnosed acute lymphoblastic leukemia. Leukemia 2023, 37, 1782–1791. [Google Scholar] [CrossRef]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Bergeron, C.; Gore, L.; Sender, L.; Dunkel, I.J.; Herzog, C.; Brochez, L.; Cruz, O.; Nysom, K.; Berghorn, E.; et al. Phase II study of ipilimumab in adolescents with unresectable stage III or IV malignant melanoma. Eur. J. Cancer 2017, 86, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.; Joechner, A.; Li, Z.; Yang, S.F.; Schlegel, P. Development of CAR T Cell Therapy in Children-A Comprehensive Overview. J. Clin. Med. 2022, 11, 2158. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Morgenstern, D.A.; Leruste, A.; Bourdeaut, F.; Davis, K.L. Checkpoint Immunotherapy in Pediatrics: Here, Gone, and Back Again. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sweet-Cordero, E.A.; Biegel, J.A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 2019, 363, 1170–1175. [Google Scholar] [CrossRef]
- Diefenbach, C.S.; Hong, F.; Ambinder, R.F.; Cohen, J.B.; Robertson, M.J.; David, K.A.; Advani, R.H.; Fenske, T.S.; Barta, S.K.; Palmisiano, N.D.; et al. Ipilimumab, nivolumab, and brentuximab vedotin combination therapies in patients with relapsed or refractory Hodgkin lymphoma: Phase 1 results of an open-label, multicentre, phase 1/2 trial. Lancet Haematol. 2020, 7, e660–e670. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Frey, N.V.; Engels, B.; Barrett, D.M.; Shestova, O.; Ravikumar, P.; Cummins, K.D.; Lee, Y.G.; Pajarillo, R.; Chun, I.; et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat. Med. 2021, 27, 842–850. [Google Scholar] [CrossRef]
- Jonus, H.C.; Lee, J.Y.; Silva, J.A.; Spencer, H.T.; Goldsmith, K.C. Abstract 4093: Dual targeted CAR immunotherapy for neuroblastoma using γδ T cells. Cancer Res. 2023, 83, 4093. [Google Scholar] [CrossRef]
- Dunkel, I.J.; Doz, F.; Foreman, N.K.; Hargrave, D.; Lassaletta, A.; Andre, N.; Hansford, J.R.; Hassall, T.; Eyrich, M.; Gururangan, S.; et al. Nivolumab with or without ipilimumab in pediatric patients with high-grade CNS malignancies: Safety, efficacy, biomarker, and pharmacokinetics-CheckMate 908. Neuro Oncol. 2023, 25, 1530–1545. [Google Scholar] [CrossRef]
- Turpin, B.; Vatner, R. A pilot study of pembrolizumab in combination with decitabine and hypofractionated index lesion radiation in pediatric and young adult patients with relapsed and refractory solid tumors or lymphoma. J. Clin. Oncol. 2019, 37, TPS25. [Google Scholar] [CrossRef]
- Short, S.S.; Kastenberg, Z.J.; Wei, G.; Bondoc, A.; Dasgupta, R.; Tiao, G.M.; Watters, E.; Heaton, T.E.; Lotakis, D.; La Quaglia, M.P.; et al. Histologic type predicts disparate outcomes in pediatric hepatocellular neoplasms: A Pediatric Surgical Oncology Research Collaborative study. Cancer 2022, 128, 2786–2795. [Google Scholar] [CrossRef] [PubMed]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and challenges of adoptive T-cell therapies for solid tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef] [PubMed]
- Theruvath, J.; Menard, M.; Smith, B.A.H.; Linde, M.H.; Coles, G.L.; Dalton, G.N.; Wu, W.; Kiru, L.; Delaidelli, A.; Sotillo, E.; et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat. Med. 2022, 28, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef]
- Upadhye, A.; Meza Landeros, K.E.; Ramirez-Suastegui, C.; Schmiedel, B.J.; Woo, E.; Chee, S.J.; Malicki, D.; Coufal, N.G.; Gonda, D.; Levy, M.L.; et al. Intra-tumoral T cells in pediatric brain tumors display clonal expansion and effector properties. Nat. Cancer 2024, 5, 791–807. [Google Scholar] [CrossRef]

{kind=link}
| Cytokines and Their Receptors | Tumor Type | Cell Source | Prognosis and Correlation with Treatment | Role in Tumor Progression | Mechanisms |
|---|---|---|---|---|---|
| Hematological malignancies: | |||||
| TNFα [90,98] | AML | Blast cells, LSC, TAMs | Worse OS | Promoting | Promotes tumor inflammation by activating NF-kB, PI3K/AKT, and MAPK signaling |
| IL3Rα (CD123) [99,100] | B-ALL | Blast cells | Good OS | Promoting | Alters CXCR4/SDF-1 interaction in the BM, promotes AML cell growth |
| IL-7Rα [101,102,103] | T-ALL | T cells | Worse OS | Promoting | Promotes JAK, STAT-5, PI3K signaling |
| IL-2 [104,105,106,107] | AML | T cells | Good OS | Inhibitory | Stimulates CTLs production |
| IL-6 [108,109] | AML | Blast cells, BMSCs | Worse OS | Promoting | IL-6-induced STAT3 pathway promotes AML progression |
| IL-10 [110,111,112,113] | ALL | Myeloid, lymphoid cells | Good OS | Inhibitory | IL-10 family cytokines maintain tissue balance by regulating inflammation, enhancing immunity, and aiding tissue repair. Children with ALL have low IL-10 levels. IL-10 inhibits AML blast proliferation, while its deficiency impairs B-cell development, increases DNA damage, and raises proinflammatory cytokines IL-1α, IL-6, IL-12p40, IL-13, MIP-1β/CCL4, and G-CSF. |
| IL-33, IL1RL1 [114,115,116] | AML | AML, BM, mast, ILC2, Tregs and TAM cells | Worse OS | Promoting | Activates p38 MAPK, Wnt, and Notch pathways, promoting cell survival and stemness |
| IL1β [92,117,118,119] | AML/ALL | Leukemia cells, monocytes | Worse OS | Promoting | Activates IL-1/p38MAPK pathway and promotes leukemia progression |
| Solid tumors: | |||||
| IL-1β and TNF-α [120] | NB | TAMs | Worse OS | Promoting | Promotes ARG2 expression via p38/ERK signaling |
| IL-2 [121,122,123,124] | NB | T and NK cells | Good OS | Inhibitory | promote CD8+ T, B, and NK cell cytotoxicity activity |
| IL-6 [125,126] | NB | BMSCs, MSC, CAFs, NB cells | Worse OS | Promoting | Promotes STAT-3, ERK signaling, inhibits NK functions |
| IL-6 [87,127,128] | Glioma | Glial, stromal, or TME cells | Worse OS | Promoting | Promotes oncogenic JAK/STAT3 signaling |
| IL-8 [129,130,131,132] | Osteosarcoma | Osteosarcoma cells, MSC, TAMs | Worse OS | Promoting | Promotes FAK signaling, ABCB1/MDR1 pathway and immune suppression |
| IL-10 [87,133,134] | MB, GCT, Glioma | Bregs | Worse OS | Promoting | IL-10 suppresses T and NK cell anti-tumor immunity and promotes cell proliferation. |
| IL-6, IL-10 [87,135,136] | MB | MB cells | Worse OS | Promoting | IL-6 induces STAT3 pathway and promotes MB growth |
| IL-12 [137,138,139] | Osteosarcoma | APCs | Good OS | Inhibiting | Activates NK, CTLs, and memory T cells, inhibiting tumor growth and metastasis |
| IL-13Rα2 [140,141,142] | Glioma | Glioma cells | Worse OS | Promoting | IL-13 and its receptor IL-13Rα2 signals through JAK/STAT and AP-1 pathways |
| TGF-β [126,143] | NB | CAFs, TAMs | Worse OS | Promoting | Activates TGF-β/IL-6 pathway in NB and MSC, promotes immune suppression |
| VEGF [144,145,146,147] | ES | ES cells | Worse OS | Promoting | Attracts BM-derived EPCs to drive tumor blood vessel growth |
| VEGF [148,149,150] | WT | WT and stromal cells | Worse OS | Promoting | Regulates angiogenesis by activating VEGFR-1 and VEGFR-2 |
| AFP [151,152,153,154] | Hepato blastoma | Liver cancer cells | Worse OS | Promoting | Promotes PI3K/AKT, suppresses Fas/FADD apoptotic pathway |
| Cancer Type | NCT; Phase | Monoclonal Antibodies or Immune Cells | Target | Combination Drugs | Status and Outcome |
|---|---|---|---|---|---|
| Hematological malignancies: | |||||
| Relapsed/Refractory Mature B-NHL | NCT05533775; I/II | Glofitamab | CD20 | Rituximab, Ifosfamide, Carboplatin, Etoposide | Recruiting |
| Ph-like ALL | NCT03571321; I | Ruxolitinib | JAK1 JAK2 | Rituximab, Cyclophosphamide, Cytarabine, Mercaptopurine, Vincristine, Pegaspargase, Methotrexate, dexamethasone, Doxorubicin, Thioguanine | Recruiting |
| Relapsed B-ALL | NCT05645718; II | Inotuzumab Ozogamicin, Blinatumomab and Rituximab | Tumor cells | Cyclophosphamide, Vincristine, and Dexamethasone | Recruiting |
| Aggressive B-cell Lymphoma | NCT03864419; I | Rituximab Hyaluronidase | CD20 | Cyclophosphamide, Vincristine, Methotrexate, Etoposide, Doxorubicin, Prednisone | Completed |
| B-ALL | NCT03150693; III | Inotuzumab Ozogamicin | CD22 | Rituximab, Allopurinol, Cytarabine, Daunorubicin, Vincristine, Dexamethasone, PLA, Methotrexate, Cyclophosphamide, Mercaptopurine, Doxorubicin, Thioguanine | Suspended due to high toxicity |
| Relapsed/Refractory HL [190] | NCT01896999; I/II | Brentuximab vedotin | CD30 | Nivolumab and Ipilimumab | CRR improved, Suspended |
| PD-L1+ve solid tumors, lymphoma [59] | NCT02332668; I/II | Pembrolizumab | PD-1 | Single agent | Recruiting |
| Relapsed/Refractory B-ALL or B-NHL | NCT04544592; I/II NCT04173988; I | CAR-T cells | CD19 | Single agent | Recruiting Not Recruiting |
| Relapsed/Refractory B-ALL [191] | NCT02650414; I/II | CAR-T cells | CD22 | Single agent | Recruiting |
| Relapsed/Refractory HL | NCT04268706; II | CAR-T cells | CD30 | Single agent | Recruiting |
| Relapsed/Refractory B-ALL, B-NHL | NCT03743246; I/II | JCAR017 (CAR-T cells) | CD19 | Fludarabine, Cyclophosphamide | Completed |
| Relapsed/ Refractory pre-B ALL | NCT03605589; I | Pembrolizumab | PD-1 | blinatumomab | Withdrawn due to low enrollment |
| Solid tumors: | |||||
| Relapsed/ Refractory NB | NCT04238819; 1b/2 | Abemaciclib | CDK4, CDK6 | Dinutuximab, GM-CSF, Irinotecan, Temozolomide | Active, Not recruiting |
| NCT03794349; II | Eflornithine | polyamines | Irinotecan, Temozolomide, Dinutuximab | Recruiting | |
| NCT02914405; I | Dinutuximab | GD2 | mlBG, Nivolumab (anti-PD1) antibody | Recruiting | |
| NCT05400603; I [192] | Allogeneic γδ T Cells | Tumor cells | Temozolomide, Irinotecan, Dinutuximab, Zoledronate | Recruiting | |
| INI1(-) or SMARCA4-def. tumors | NCT05407441; I/II | tazemetostat | EZH2 | Nivolumab and Ipilimumab | Recruiting |
| Recurrent or Progressive HGG | NCT04323046; I | nivolumab | PD-1 | Single agent. Given before and after surgery | Recruiting |
| High-grade primary CNS malignancies [193] | NCT03130959; 1b/II | Nivolumab | PD-1 | Ipilimumab | No clinical benefit in combination |
| Relapsed/Refractory solid tumors | NCT05302921; II | Nivolumab, Ipilimumab | PD-1, CTLA-4 | cryoablation therapy | Active, not recruiting |
| Relapsed, Refractory, or Progressive CNS solid tumors and lymphomas [194] | NCT03445858; I | Pembrolizumab | PD-1 | Decitabine and radiation therapy | Active, not recruiting |
| Refractory gliomas, MB | NCT02359565; I | Pembrolizumab | PD-1 | Single agent | Recruiting |
| Liver cancer [195] | NCT04134559; II | Pembrolizumab | PD-1 | Single agent | Recruiting |
| GD2+ve Brain tumors | NCT04099797; I | C7R-GD2.CAR T Cells | GD2 | Single agent | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathania, A.S. Immune Microenvironment in Childhood Cancers: Characteristics and Therapeutic Challenges. Cancers 2024, 16, 2201. https://doi.org/10.3390/cancers16122201
Pathania AS. Immune Microenvironment in Childhood Cancers: Characteristics and Therapeutic Challenges. Cancers. 2024; 16(12):2201. https://doi.org/10.3390/cancers16122201
Chicago/Turabian StylePathania, Anup Singh. 2024. "Immune Microenvironment in Childhood Cancers: Characteristics and Therapeutic Challenges" Cancers 16, no. 12: 2201. https://doi.org/10.3390/cancers16122201
APA StylePathania, A. S. (2024). Immune Microenvironment in Childhood Cancers: Characteristics and Therapeutic Challenges. Cancers, 16(12), 2201. https://doi.org/10.3390/cancers16122201