HER2-Low Luminal Breast Carcinoma Is Not a Homogenous Clinicopathological and Molecular Entity

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design, Patients, and Samples
2.2. Histopathological Review
2.3. ER, PR, and Ki67 Protein Expression Analysis Using Immunohistochemistry (IHC)
2.4. HER2 Status Assessment
2.5. Molecular Analysis
2.5.1. DNA and RNA Extraction
2.5.2. DNA Sequencing
2.5.3. RNA Sequencing and Transcriptomic Analysis
2.6. Statistical Analysis
3. Results
3.1. Clinicopathological Characteristics of Patients and Carcinomas
3.2. Genomic Profiles
3.3. Transcriptomic Profiles
3.3.1. ERBB2 mRNA Expression
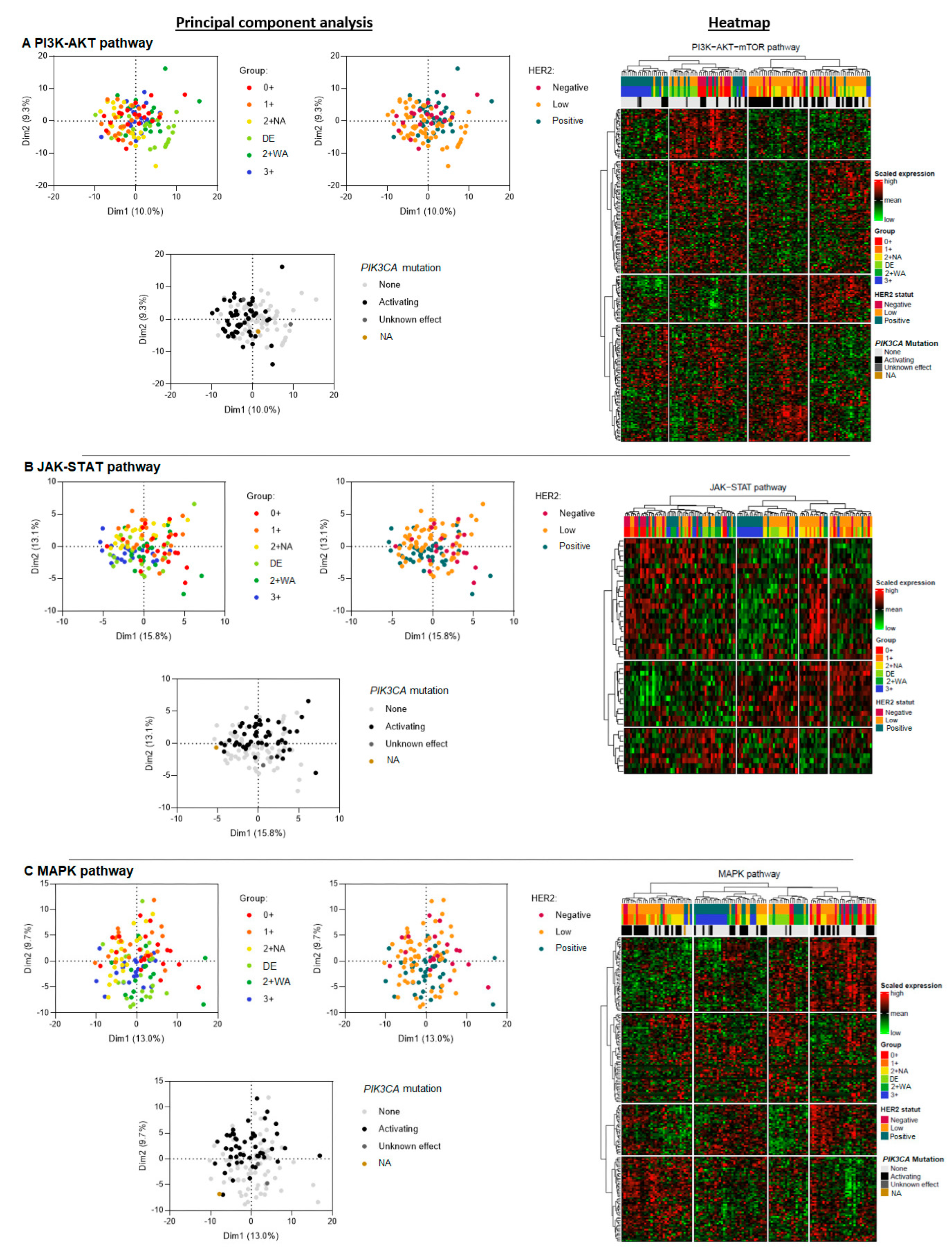
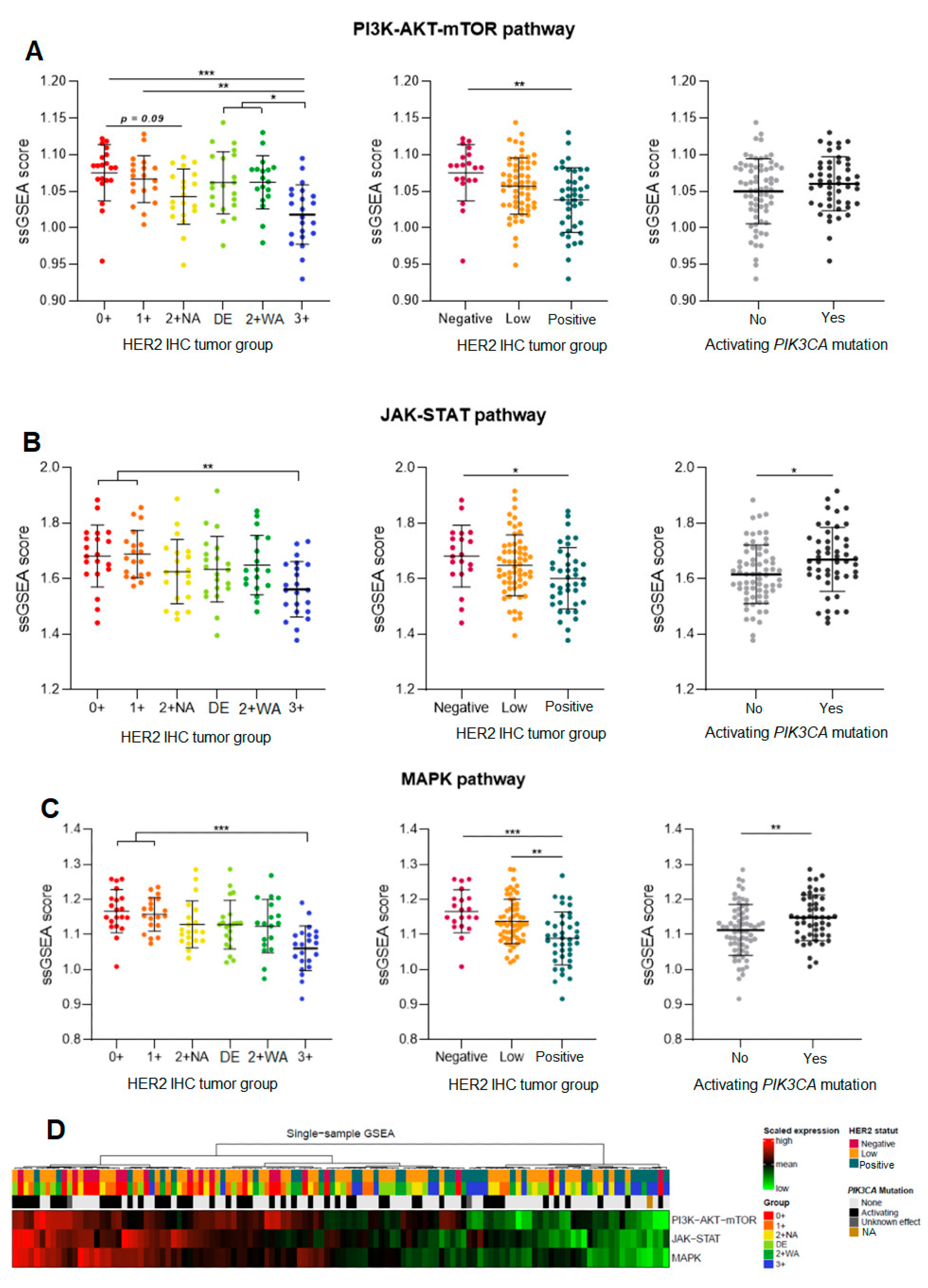
3.3.2. Gene Expression of the PI3K-AKT, JAK-STAT, and the MAPK Pathway
3.4. Global Gene Expression and Phenotypic Profiles within the Different Pathways, and the Impact of PIK3CA Activating Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.S.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.S.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W.; et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2018, 142, 1364–1382. [Google Scholar] [CrossRef]
- Franchet, C.; Djerroudi, L.; Maran-Gonzalez, A.; Abramovici, O.; Antoine, M.; Becette, V.; Berghian, A.; Blanc-Fournier, C.; Brabencova, E.; Charafe-Jauffret, E.; et al. Mise à jour 2021 des recommandations du GEFPICS pour l’évaluation du statut HER2 dans les cancers infiltrants du sein en France. Ann. Pathol. 2021, 41, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Schettini, F.; Chic, N.; Brasó-Maristany, F.; Paré, L.; Pascual, T.; Conte, B.; Martínez-Sáez, O.; Adamo, B.; Vidal, M.; Barnadas, E.; et al. Clinical, Pathological, and PAM50 Gene Expression Features of HER2-Low Breast Cancer. NPJ Breast Cancer 2021, 7, 1. [Google Scholar] [CrossRef]
- Schalper, K.A.; Kumar, S.; Hui, P.; Rimm, D.L.; Gershkovich, P. A Retrospective Population-Based Comparison of HER2 Immunohistochemistry and Fluorescence In Situ Hybridization in Breast Carcinomas: Impact of 2007 American Society of Clinical Oncology/College of American Pathologists Criteria. Arch. Pathol. Lab. Med. 2014, 138, 213–219. [Google Scholar] [CrossRef]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant Trastuzumab in HER2-Positive Breast Cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef]
- Cameron, D.; Piccart-Gebhart, M.J.; Gelber, R.D.; Procter, M.; Goldhirsch, A.; de Azambuja, E.; Castro, G.; Untch, M.; Smith, I.; Gianni, L.; et al. 11 Years’ Follow-up of Trastuzumab after Adjuvant Chemotherapy in HER2-Positive Early Breast Cancer: Final Analysis of the HERceptin Adjuvant (HERA) Trial. Lancet 2017, 389, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Romond, E.H.; Suman, V.J.; Jeong, J.-H.; Sledge, G.; Geyer, C.E.; Martino, S.; Rastogi, P.; Gralow, J.; Swain, S.M.; et al. Trastuzumab Plus Adjuvant Chemotherapy for Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer: Planned Joint Analysis of Overall Survival From NSABP B-31 and NCCTG N9831. JCO 2014, 32, 3744–3752. [Google Scholar] [CrossRef] [PubMed]
- Buzdar, A.U.; Ibrahim, N.K.; Francis, D.; Booser, D.J.; Thomas, E.S.; Theriault, R.L.; Pusztai, L.; Green, M.C.; Arun, B.K.; Giordano, S.H.; et al. Significantly Higher Pathologic Complete Remission Rate After Neoadjuvant Therapy with Trastuzumab, Paclitaxel, and Epirubicin Chemotherapy: Results of a Randomized Trial in Human Epidermal Growth Factor Receptor 2–Positive Operable Breast Cancer. JCO 2005, 23, 3676–3685. [Google Scholar] [CrossRef]
- Arnould, L.; Arveux, P.; Couturier, J.; Gelly-Marty, M.; Loustalot, C.; Ettore, F.; Sagan, C.; Antoine, M.; Penault-Llorca, F.; Vasseur, B.; et al. Pathologic Complete Response to Trastuzumab-Based Neoadjuvant Therapy Is Related to the Level of HER-2 Amplification. Clin. Cancer Res. 2007, 13, 6404–6409. [Google Scholar] [CrossRef]
- Choi, J.H.; Jeon, C.W.; Kim, Y.O.; Jung, S. Pathological Complete Response to Neoadjuvant Trastuzumab and Pertuzumab Therapy Is Related to Human Epidermal Growth Factor Receptor 2 (HER2) Amplification Level in HER2-Amplified Breast Cancer. Medicine 2020, 99, e23053. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Park, H.; Murthy, R.K.; Iwata, H.; Tamura, K.; Tsurutani, J.; Moreno-Aspitia, A.; Doi, T.; Sagara, Y.; Redfern, C.; et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients with HER2-Low–Expressing Advanced Breast Cancer: Results From a Phase Ib Study. JCO 2020, 38, 1887–1896. [Google Scholar] [CrossRef]
- Guarini, C.; Grassi, T.; Pezzicoli, G.; Porta, C. Beyond RAS and BRAF: HER2, a New Actionable Oncotarget in Advanced Colorectal Cancer. IJMS 2021, 22, 6813. [Google Scholar] [CrossRef]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Miller, L.; Lo, H.-W. EGFR and HER2 Signaling in Breast Cancer Brain Metastasis. Front. Biosci. 2016, 8, 245. [Google Scholar]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board. Breast Tumours: WHO Classification of Tumours (Medicine), 5th ed.; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Elston, C.W.; Ellis, I.O. Pathological Prognostic Factors in Breast Cancer. I. The Value of Histological Grade in Breast Cancer: Experience from a Large Study with Long-Term Follow-Up. Histopathology 1991, 19, 403–410. [Google Scholar] [CrossRef]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The Evaluation of Tumor-Infiltrating Lymphocytes (TILs) in Breast Cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: ASCO/CAP Guideline Update. JCO 2020, 38, 1346–1366. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Picard Tools—By Broad Institute. Available online: https://broadinstitute.github.io/picard/ (accessed on 23 April 2022).
- Muller, K.E.; Marotti, J.D.; de Abreu, F.B.; Peterson, J.D.; Miller, T.W.; Chamberlin, M.D.; Tsongalis, G.J.; Tafe, L.J. Targeted Next-Generation Sequencing Detects a High Frequency of Potentially Actionable Mutations in Metastatic Breast Cancers. Exp. Mol. Pathol. 2016, 100, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain w 1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics—FastQC a Quality Control tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 23 April 2022).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Eiger, D.; Agostinetto, E.; Saúde-Conde, R.; de Azambuja, E. The Exciting New Field of HER2-Low Breast Cancer Treatment. Cancers 2021, 13, 1015. [Google Scholar] [CrossRef] [PubMed]
- Rossi, V.; Sarotto, I.; Maggiorotto, F.; Berchialla, P.; Kubatzki, F.; Tomasi, N.; Redana, S.; Martinello, R.; Valabrega, G.; Aglietta, M.; et al. Moderate Immunohistochemical Expression of HER-2 (2+) without HER-2 Gene Amplification Is a Negative Prognostic Factor in Early Breast Cancer. Oncologist 2012, 17, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Katerji, H.; Turner, B.M.; Audeh, W.; Hicks, D.G. HER2-Low Breast Cancers: Incidence, HER2 Staining Patterns, Clinicopathologic Features, MammaPrint and BluePrint Genomic Profiles. Mod. Pathol. 2022, 35, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Marchiò, C.; Annaratone, L.; Marques, A.; Casorzo, L.; Berrino, E.; Sapino, A. Evolving Concepts in HER2 Evaluation in Breast Cancer: Heterogeneity, HER2-Low Carcinomas and Beyond. Semin. Cancer Biol. 2021, 72, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Shen, J.; Guo, W.; Zhao, W.; Zhuang, Y.; Wang, L. Impact of the 2018 ASCO/CAP HER2 Guidelines Update for HER2 Testing by FISH in Breast Cancer. Pathol.-Res. Pract. 2019, 215, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Gandini, S.; Nicolò, E.; Trillo, P.; Giugliano, F.; Zagami, P.; Vivanet, G.; Bellerba, F.; Trapani, D.; Marra, A.; et al. Evolution of Low HER2 Expression between Early and Advanced-Stage Breast Cancer. Eur. J. Cancer 2022, 163, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Seither, F.; Schneeweiss, A.; Link, T.; Blohmer, J.-U.; Just, M.; Wimberger, P.; Forberger, A.; Tesch, H.; Jackisch, C.; et al. Clinical and Molecular Characteristics of HER2-Low-Positive Breast Cancer: Pooled Analysis of Individual Patient Data from Four Prospective, Neoadjuvant Clinical Trials. Lancet Oncol. 2021, 22, 1151–1161. [Google Scholar] [CrossRef]
- Li, Y.; Abudureheiyimu, N.; Mo, H.; Guan, X.; Lin, S.; Wang, Z.; Chen, Y.; Chen, S.; Li, Q.; Cai, R.; et al. In Real Life, Low-Level HER2 Expression May Be Associated with Better Outcome in HER2-Negative Breast Cancer: A Study of the National Cancer Center, China. Front. Oncol. 2022, 11, 774577. [Google Scholar] [CrossRef]
- Hoda, R.S.; Brogi, E.; Xu, J.; Ventura, K.; Ross, D.S.; Dang, C.; Robson, M.; Norton, L.; Morrow, M.; Wen, H.Y. Impact of the 2018 American Society of Clinical Oncology/College of American Pathologists HER2 Guideline Updates on HER2 Assessment in Breast Cancer with Equivocal HER2 Immunohistochemistry Results with Focus on Cases with HER2/CEP17 Ratio <2.0 and Average HER2 Copy Number ≥4.0 and <6.0. Arch. Pathol. Lab. Med. 2020, 144, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.W.; Lee, K.; Chung, Y.R.; Jang, M.H.; Ahn, S.; Park, S.Y. The Updated 2018 American Society of Clinical Oncology/College of American Pathologists Guideline on Human Epidermal Growth Factor Receptor 2 Interpretation in Breast Cancer: Comparison with Previous Guidelines and Clinical Significance of the Proposed in Situ Hybridization Groups. Hum. Pathol. 2020, 98, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Rajendra, J.; Dikshit, R.; Dutt, S. HER2 Borderline Is a Negative Prognostic Factor for Primary Malignant Breast Cancer. Breast Cancer Res. Treat. 2020, 181, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Bethune, G.C.; Veldhuijzen van Zanten, D.; MacIntosh, R.F.; Rayson, D.; Younis, T.; Thompson, K.; Barnes, P.J. Impact of the 2013 American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Human Epidermal Growth Factor Receptor 2 (HER2) Testing of Invasive Breast Carcinoma: A Focus on Tumours Assessed as ‘Equivocal’ for HER2 Gene Amplification by Fluorescence in-Situ Hybridization. Histopathology 2015, 67, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Hamilton, E.; Tolaney, S.M.; Cortes, J.; Morganti, S.; Ferraro, E.; Marra, A.; Viale, G.; Trapani, D.; Cardoso, F.; et al. HER2-Low Breast Cancer: Pathological and Clinical Landscape. JCO 2020, 38, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Agostinetto, E.; Rediti, M.; Fimereli, D.; Debien, V.; Piccart, M.; Aftimos, P.; Sotiriou, C.; de Azambuja, E. HER2-Low Breast Cancer: Molecular Characteristics and Prognosis. Cancers 2021, 13, 2824. [Google Scholar] [CrossRef] [PubMed]
- Crimini, E.; Repetto, M.; Aftimos, P.; Botticelli, A.; Marchetti, P.; Curigliano, G. Precision Medicine in Breast Cancer: From Clinical Trials to Clinical Practice. Cancer Treat. Rev. 2021, 98, 102223. [Google Scholar] [CrossRef] [PubMed]
- Mukohara, T. PI3K Mutations in Breast Cancer: Prognostic and Therapeutic Implications. BCTT 2015, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.X.; Polley, E.; Lipkowitz, S. New Insights on PI3K/AKT Pathway Alterations and Clinical Outcomes in Breast Cancer. Cancer Treat. Rev. 2016, 45, 87–96. [Google Scholar] [CrossRef]
- Roy-Chowdhuri, S.; de Melo Gagliato, D.; Routbort, M.J.; Patel, K.P.; Singh, R.R.; Broaddus, R.; Lazar, A.J.; Sahin, A.; Alvarez, R.H.; Moulder, S.; et al. Multigene Clinical Mutational Profiling of Breast Carcinoma Using Next-Generation Sequencing. Am. J. Clin. Pathol. 2015, 144, 713–721. [Google Scholar] [CrossRef]
- Kalinsky, K.; Jacks, L.M.; Heguy, A.; Patil, S.; Drobnjak, M.; Bhanot, U.K.; Hedvat, C.V.; Traina, T.A.; Solit, D.; Gerald, W.; et al. PIK3CA Mutation Associates with Improved Outcome in Breast Cancer. Clin. Cancer Res. 2009, 15, 5049–5059. [Google Scholar] [CrossRef] [PubMed]
- Tserga, A.; Chatziandreou, I.; Michalopoulos, N.V.; Patsouris, E.; Saetta, A.A. Mutation of Genes of the PI3K/AKT Pathway in Breast Cancer Supports Their Potential Importance as Biomarker for Breast Cancer Aggressiveness. Virchows Arch. 2016, 469, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Tenorio, G.; Stål, O.; Southeast Sweden Breast Cancer Group. Activation of AKT/PKB in Breast Cancer Predicts a Worse Outcome among Endocrine Treated Patients. Br. J. Cancer 2002, 86, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Cuorvo, L.V.; Verderio, P.; Ciniselli, C.M.; Girlando, S.; Decarli, N.; Leonardi, E.; Ferro, A.; Caldara, A.; Triolo, R.; Eccher, C.; et al. PI3KCA Mutation Status Is of Limited Prognostic Relevance in ER-Positive Breast Cancer Patients Treated with Hormone Therapy. Virchows Arch. 2014, 464, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Miyoshi, Y. Mechanism of Resistance to Endocrine Therapy in Breast Cancer: The Important Role of PI3K/Akt/mTOR in Estrogen Receptor-Positive, HER2-Negative Breast Cancer. Breast Cancer 2018, 25, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, E.; Kimura, Y.; Mashino, K.; Oki, E.; Kataoka, A.; Ohno, S.; Morita, M.; Kakeji, Y.; Baba, H.; Maehara, Y. Activation of PI3K/Akt Signaling and Hormone Resistance in Breast Cancer. Breast Cancer 2006, 13, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Witton, C.J.; McGlynn, L.M.; Tovey, S.M.; Dunne, B.; Lyon, A.; Bartlett, J.M. AKT Activation Predicts Outcome in Breast Cancer Patients Treated with Tamoxifen. J. Pathol. 2005, 207, 139–146. [Google Scholar] [CrossRef] [PubMed]
- McAuliffe, P.F.; Meric-Bernstam, F.; Mills, G.B.; Gonzalez-Angulo, A.M. Deciphering the Role of PI3K/Akt/mTOR Pathway in Breast Cancer Biology and Pathogenesis. Clin. Breast Cancer 2010, 10, S59–S65. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A Functional Genetic Approach Identifies the PI3K Pathway as a Major Determinant of Trastuzumab Resistance in Breast Cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef]
- Esteva, F.J.; Guo, H.; Zhang, S.; Santa-Maria, C.; Stone, S.; Lanchbury, J.S.; Sahin, A.A.; Hortobagyi, G.N.; Yu, D. PTEN, PIK3CA, p-AKT, and p-p70S6K Status. Am. J. Pathol. 2010, 177, 1647–1656. [Google Scholar] [CrossRef]
- Baselga, J.; Im, S.-A.; Iwata, H.; Cortés, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus Fulvestrant versus Placebo plus Fulvestrant in Postmenopausal, Hormone Receptor-Positive, HER2-Negative, Advanced Breast Cancer (BELLE-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Saura, C.; Richards, D.A.; Krop, I.E.; Cervantes, A.; Bedard, P.L.; Patel, M.R.; Pusztai, L.; Oliveira, M.; Cardenas, A.K.; et al. Phase II Study of Taselisib (GDC-0032) in Combination with Fulvestrant in Patients with HER2-Negative, Hormone Receptor–Positive Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 4380–4387. [Google Scholar] [CrossRef] [PubMed]
- Mollon, L.E.; Anderson, E.J.; Dean, J.L.; Warholak, T.L.; Aizer, A.; Platt, E.A.; Tang, D.H.; Davis, L.E. A Systematic Literature Review of the Prognostic and Predictive Value of PIK3CA Mutations in HR+/HER2− Metastatic Breast Cancer. Clin. Breast Cancer 2020, 20, e232–e243. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the Phosphatidylinositol 3-Kinase Pathway: Role in Tumor Progression and Therapeutic Implications in Breast Cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| p-Values | |||||||
|---|---|---|---|---|---|---|---|
| H2L (n = 62) | HER2-Negative (n = 20) | HER2-Positive (n = 43) | All | 0+ vs. HER2-Positive | 0+ vs. H2L | HER2-Positive vs. H2L | |
| n (%) | n (%) | n (%) | |||||
| Patient age | 0.9054 | ||||||
| Mean ± SD | 65.0 ± 12.6 | 65.3 ± 12.8 | 64.0 ± 13.5 | ||||
| Median [min–max] | 67.0 [34.0–93.0] | 68.5 [36.0–91.0] | 64.0 [37.0–89.0] | ||||
| Patient menopausal status | 0.4422 | ||||||
| Peri/premenopausal | 10 (16.1) | 5 (25.0) | 11 (25.6) | ||||
| Postmenopausal | 52 (83.9) | 15 (75.0) | 32 (74.4) | ||||
| Tumor size (US, mm) | 0.126 | ||||||
| Mean ± SD | 18.9 ± 14.1 | 14.9 ± 12.3 | 18.7 ± 11.4 | ||||
| Median [min–max] | 15.0 [5.0–70.0] | 11.3 [3.5–50.0] | 15.0 [4.0–58.0] | ||||
| Node status (US) | 0.5397 | ||||||
| N0 | 59 (95.2) | 18 (90.0) | 38 (88.4) | ||||
| N1 | 3 (4.8) | 2 (10.0) | 4 (9.3) | ||||
| N3 | 0 (0) | 0 (0) | 1 (2.3) | ||||
| Multifocality | 0.5519 | ||||||
| Unifocal | 50 (80.6) | 18 (90.0) | 37 (86.0) | ||||
| Bifocal | 12 (19.4) | 2 (10.0) | 6 (14.0) | ||||
| Tumor size (clinical, mm) | 0.8995 | ||||||
| Mean ± SD | 18.1 ± 11.9 | 18.1 ± 11.7 | 18.9 ± 11.6 | ||||
| Median [min–max] | 15.0 [4.5–70.0] | 15.5 [6.0–55.0] | 15.0 [5.8–58.0] | ||||
| Node status | 0.0146 | 0.0497 | 0.0531 | 0.6476 | |||
| pN0 | 41 (66.1) | 9 (45.0) | 34 (79.1) | ||||
| pN1 | 19 (30.6) | 6 (30.0) | 7 (16.3) | ||||
| pN2 | 2 (3.2) | 4 (20.0) | 2 (4.7) | ||||
| pN3 | 0 (0) | 1 (5.0) | 0 (0) | ||||
| E&E grade | 0.0005 | 0.0005 | 0.9656 | 0.0017 | |||
| I | 25 (40.3) | 10 (50.0) | 3 (7.0) | ||||
| II | 31 (50.0) | 10 (50.0) | 31 (72.1) | ||||
| III | 6 (9.7) | 0 (0) | 9 (20.9) | ||||
| Glandular differentiation | 0.1734 | ||||||
| 1 | 3 (4.8) | 1 (5.0) | 0 (0) | ||||
| 2 | 30 (48.4) | 9 (45.0) | 14 (32.6) | ||||
| 3 | 29 (46.8) | 10 (50.0) | 29 (67.4) | ||||
| Nuclear grade | 0.0348 | 0.1398 | 1 | 0.0756 | |||
| 1 | 2 (3.2) | 0 (0) | 0 (0) | ||||
| 2 | 54 (87.1) | 19 (95.0) | 31 (72.1) | ||||
| 3 | 6 (9.7) | 1 (5.0) | 12 (27.9) | ||||
| Mitosis score | 0.0003 | 0.0001 | 0.0651 | 0.0537 | |||
| 1 | 39 (62.9) | 19 (95.0) | 15 (34.9) | ||||
| 2 | 14 (22.6) | 1 (5.0) | 18 (41.9) | ||||
| 3 | 9 (14.5) | 0 (0) | 10 (23.3) | ||||
| Mitotic index (/mm²) | 0.0002 | 0.0002 | 0.4007 | 0.0042 | |||
| Mean ± SD | 2.9 ± 2.6 | 1.8 ± 1.6 | 4.8 ± 3.2 | ||||
| Median [min–max] | 1.8 [0.4–10.5] | 0.9 [0.4–6.6] | 4.4 [0.4–15.1] | ||||
| Histologic subtype | 0.5616 | ||||||
| Micropapillary | 2 (3.2) | 0 (0) | 1 (2.3) | ||||
| Mucinous | 0 (0) | 1 (5.0) | 0 (0) | ||||
| NST | 57 (91.9) | 18 (90.0) | 41 (95.3) | ||||
| NST + micropapillary | 3 (4.8) | 1 (5.0) | 1 (2.3) | ||||
| Lymphovascular emboli | 0.9027 | ||||||
| No | 41 (66.1) | 13 (65.0) | 30 (69.8) | ||||
| Yes | 21 (33.9) | 7 (3.0) | 13 (30.2) | ||||
| sTIL (%) | 0.1407 | ||||||
| Mean ± SD | 7.0 ± 7.6 | 8.2 ± 9.3 | 10.6 ± 9.6 | ||||
| Median [min–max] | 5.0 [1.0–50.0] | 4.0 [1.0–30.0] | 5.0 [1.0–40.0] | ||||
| sTIL (≤10%) | 0.0507 | ||||||
| No | 8 (12.9) | 4 (20.0) | 14 (32.6) | ||||
| Yes | 54 (87.1) | 16 (80.0) | 29 (67.4) | ||||
| sTILs (>40%) | 1 | ||||||
| No | 61 (98.4) | 20 (100.0) | 42 (97.7) | ||||
| Yes | 1 (1.6) | 0 (0) | 1 (2.3) | ||||
| ER (I × %) | 0.5524 | ||||||
| Mean ± SD | 285.2 ± 34.0 | 280.0 ± 41.9 | 277.3 ± 43.2 | ||||
| Median [min–max] | 300.0 [180.0–300.0] | 300.0 [160.0–300.0] | 300.0 [140.0–300.0] | ||||
| PR (I × %) | 0.0281 | 0.0757 | 0.7856 | 0.0567 | |||
| Mean ± SD | 198.4 ± 99.4 | 212.3 ± 100.2 | 151.0 ± 110.4 | ||||
| Median [min–max] | 210.0 [0.0–300.0] | 247.5 [20.0–300.0] | 140.0 [0.0–300.0] | ||||
| Ki67 (%) | <0.0001 | 0.0002 | 0.3919 | 0.0003 | |||
| n | 62 | 20 | 42 * | ||||
| Mean ± SD | 15.7 ± 10.2 | 14.4 ± 13.5 | 23.8 ± 11.4 | ||||
| Median [min–max] | 14.5 [2.0–60.0] | 11.0 [2.0–60.0] | 21.5 [5.0–60.0] | ||||
| p-Values | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| HER2 DE (n = 22) | HER2 0+ (n = 20) | HER2 1+ (n = 20) | HER2 2+ NA (n = 20) | HER2 2+ WA (n = 20) | HER2 3+ (n = 23) | All | DE vs. 0+ | DE vs. 1+ | |
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | ||||
| Patient age | 0.1129 | ||||||||
| Mean ± SD | 69.5 ± 9.1 | 65.3 ± 12.8 | 66.7 ± 11.5 | 58.3 ± 14.6 | 63.3 ± 13.3 | 64.7 ± 13.9 | |||
| Median [min–max] | 70.5 [50.0–85.0] | 68.5 [36.0–91.0] | 71.0 [42.0–83.0] | 53.0 [34.0–93.0] | 62.0 [37.0–84.0] | 64.0 [38.0–89.0] | |||
| Patient menopausal status | 0.5713 | ||||||||
| Peri/premenopausal | 2 (9.1%) | 5 (25.0%) | 3 (15.0%) | 5 (25.0%) | 6 (30.0%) | 5 (21.7%) | |||
| Postmenopausal | 20 (90.9%) | 15 (75.0%) | 17 (85.0%) | 15 (75.0%) | 14 (70.0%) | 18 (78.3%) | |||
| Tumor size (US, mm) | 0.0533 | ||||||||
| Mean ± SD | 20.2 ± 14.1 | 14.9 ± 12.3 | 13.4 ± 8.4 | 22.9 ± 17.3 | 16.6 ± 8.3 | 20.5 ± 13.4 | |||
| Median [min–max] | 15.0 [6.0–70.0] | 11.3 [3.5–50.0] | 11.0 [5.0–38.0] | 16.5 [6.0–70.0] | 14.5 [4.0–30.0] | 15.0 [7.0–58.0] | |||
| Node status (US) | 0.3889 | ||||||||
| N0 | 19 (86.4%) | 18 (90.0%) | 20 (100.0%) | 20 (100.0%) | 18 (90.0%) | 20 (87.0%) | |||
| N1 | 3 (13.6%) | 2 (10.0%) | 0 (0.0%) | 0 (0.0%) | 2 (10.0%) | 2 (8.7%) | |||
| N3 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (4.3%) | |||
| Multifocality | 0.1893 | ||||||||
| Unifocal | 18 (81.8%) | 18 (90.0%) | 14 (70.0%) | 18 (90.0%) | 15 (75.0%) | 22 (95.7%) | |||
| Bifocal | 4 (18.2%) | 2 (10.0%) | 6 (30.0%) | 2 (10.0%) | 5 (25.0%) | 1 (4.3%) | |||
| Tumor size (clinical, mm) | 0.6973 | ||||||||
| Mean ± SD | 17.5 ± 6.7 | 18.1 ± 11.7 | 14.6 ± 7.3 | 22.1 ± 17.9 | 17.8 ± 8.7 | 19.9 ± 13.7 | |||
| Median [min–max] | 16.0 [8.0–30.0] | 15.5 [6.0–55.0] | 12.3 [4.5–32.0] | 17.2 [5.0–70.0] | 14.8 [7.0–35.0] | 15.0 [5.8–58.0] | |||
| Node status | 0.1845 | ||||||||
| pN0 | 12 (54.5%) | 9 (45.0%) | 14 (70.0%) | 15 (75.0%) | 15 (75.0%) | 19 (82.6%) | |||
| pN1 | 9 (40.9%) | 6 (30.0%) | 6 (30.0%) | 4 (20.0%) | 4 (20.0%) | 3 (13.0%) | |||
| pN2 | 1 (4.5%) | 4 (20.0%) | 0 (0.0%) | 1 (5.0%) | 1 (5.0%) | 1 (4.3%) | |||
| pN3 | 0 (0.0%) | 1 (5.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |||
| E&E grade | <0.0001 | 0.2706 | 0.0577 | ||||||
| I | 3 (13.6%) | 10 (50.0%) | 12 (60.0%) | 10 (50.0%) | 2 (10.0%) | 1 (4.3%) | |||
| II | 17 (77.3%) | 10 (50.0%) | 7 (35.0%) | 7 (35.0%) | 14 (70.0%) | 17 (73.9%) | |||
| III | 2 (9.1%) | 0 (0.0%) | 1 (5.0%) | 3 (15.0%) | 4 (20.0%) | 5 (21.7%) | |||
| Glandular differentiation | 0.0166 | 1 | 0.398 | ||||||
| 1 | 1 (4.5%) | 1 (5.0%) | 0 (0.0%) | 2 (10.0%) | 0 (0.0%) | 0 (0.0%) | |||
| 2 | 5 (22.7%) | 9 (45.0%) | 12 (60.0%) | 13 (65.0%) | 7 (35.0%) | 7 (30.4%) | |||
| 3 | 16 (72.7%) | 10 (50.0%) | 8 (40.0%) | 5 (25.0%) | 13 (65.0%) | 16 (69.6%) | |||
| Nuclear grade | 0.0349 | 1 | 1 | ||||||
| 1 | 0 (0.0%) | 0 (0.0%) | 1 (5.0%) | 1 (5.0%) | 0 (0.0%) | 0 (0.0%) | |||
| 2 | 20 (90.9%) | 19 (95.0%) | 18 (90.0%) | 16 (80.0%) | 17 (85.0%) | 14 (60.9%) | |||
| 3 | 2 (9.1%) | 1 (5.0%) | 1 (5.0%) | 3 (15.0%) | 3 (15.0%) | 9 (39.1%) | |||
| Mitosis score | <0.0001 | 0.0156 | 0.2428 | ||||||
| 1 | 10 (45.5%) | 19 (95.0%) | 17 (85.0%) | 12 (60.0%) | 6 (30.0%) | 9 (39.1%) | |||
| 2 | 9 (40.9%) | 1 (5.0%) | 3 (15.0%) | 2 (10.0%) | 9 (45.0%) | 9 (39.1%) | |||
| 3 | 3 (13.6%) | 0 (0.0%) | 0 (0.0%) | 6 (30.0%) | 5 (25.0%) | 5 (21.7%) | |||
| Mitotic index (/mm²) | <0.0001 | 0.0065 | 0.1072 | ||||||
| Mean ± SD | 4.2 ± 2.6 | 1.8 ± 1.6 | 2.4 ± 2.7 | 2.0 ± 2.1 | 4.9 ± 3.3 | 4.6 ± 3.3 | |||
| Median [min–max] | 4.1 [0.9–10.5] | 0.9 [0.4–6.6] | 1.3 [0.4–9.6] | 0.7 [0.4–7.1] | 5.0 [0.4–11.3] | 4.0 [0.5–15.1] | |||
| Histologic subtype | 0.0988 | ||||||||
| Micropapillary | 2 (9.1%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (5.0%) | 0 (0.0%) | |||
| Mucinous | 0 (0.0%) | 1 (5.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |||
| NST | 17 (77.3%) | 18 (90.0%) | 20 (100.0%) | 20 (100.0%) | 19 (95.0%) | 22 (95.7%) | |||
| NST + micropapillary | 3 (13.6%) | 1 (5.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (4.3%) | |||
| Lymphovascular emboli | 0.1758 | ||||||||
| No | 11 (50.0%) | 13 (65.0%) | 17 (85.0%) | 13 (65.0%) | 12 (60.0%) | 18 (78.3%) | |||
| Yes | 11 (50.0%) | 7 (35.0%) | 3 (15.0%) | 7 (35.0%) | 8 (40.0%) | 5 (21.7%) | |||
| sTIL (%) | 0.0429 | 1 | 0.6094 | ||||||
| Mean ± SD | 6.4 ± 5.8 | 8.2 ± 9.3 | 10.0 ± 10.6 | 4.7 ± 4.7 | 10.2 ± 11.2 | 11.0 ± 8.3 | |||
| Median [min–max] | 5.0 [1.0–20.0] | 4.0 [1.0–30.0] | 6.5 [1.0–50.0] | 2.0 [1.0–20.0] | 5.0 [1.0–40.0] | 10.0 [2.0–25.0] | |||
| sTIL (≤10%) | 0.1314 | ||||||||
| No | 3 (13.6%) | 4 (20.0%) | 4 (20.0%) | 1 (5.0%) | 5 (25.0%) | 9 (39.1%) | |||
| Yes | 19 (86.4%) | 16 (80.0%) | 16 (80.0%) | 19 (95.0%) | 15 (75.0%) | 14 (60.9%) | |||
| sTILs (>40%) | 0.4702 | ||||||||
| No | 22 (100.0%) | 20 (100.0%) | 19 (95.0%) | 20 (100.0%) | 19 (95.0%) | 23 (100.0%) | |||
| Yes | 0 (0.0%) | 0 (0.0%) | 1 (5.0%) | 0 (0.0%) | 1 (5.0%) | 0 (0.0%) | |||
| ER (I × %) | 0.0299 | 0.9142 | 1 | ||||||
| Mean ± SD | 292.7 ± 22.5 | 280.0 ± 41.9 | 292.0 ± 23.5 | 270.0 ± 47.4 | 290.0 ± 30.8 | 266.3 ± 49.8 | |||
| Median [min–max] | 300.0 [200.0–300.0] | 300.0 [160.0–300.0] | 300.0 [200.0–300.0] | 300.0 [180.0–300.0] | 300.0 [200.0–300.0] | 300.0 [140.0–300.0] | |||
| PR (I × %) | 0.0407 | 0.833 | 1 | ||||||
| Mean ± SD | 180.5 ± 102.5 | 212.3 ± 100.2 | 184.0 ± 95.5 | 232.5 ± 95.7 | 133.1 ± 119.6 | 166.5 ± 101.8 | |||
| Median [min–max] | 170.0 [0.0–300.0] | 247.5 [20.0–300.0] | 160.0 [30.0–300.0] | 270.0 [10.0–300.0] | 130.0 [0.0–300.0] | 160.0 [20.0–300.0] | |||
| Ki67 (%) | <0.0001 | 0.0175 | 0.0011 | ||||||
| n | 22 | 20 | 20 | 20 | 20 | 22 * | |||
| Mean ± SD | 20.8 ± 10.4 | 14.4 ± 13.5 | 10.6 ± 5.7 | 15.2 ± 11.1 | 20.0 ± 7.8 | 27.2 ± 13.1 | |||
| Median [min–max] | 20.0 [8.0–60.0] | 11.0 [2.0–60.0] | 9.0 [5.0–25.0] | 13.5 [2.0–40.0] | 20.0 [5.0–40.0] | 25.0 [8.0–60.0] | |||
| Controls | p-Value | |||||
|---|---|---|---|---|---|---|
| Gene | Mutation Impact * | H2L (n = 62) | HER2-Negative (n = 20) | HER2-Positive (n = 40) | All | H2L vs. HER2-Positive |
| n (%) | n (%) | n (%) | ||||
| PIK3CA | Gain of function | 28 (45.2) | 8 (40.0) | 6 (15.0) | 0.0063 | 0.0048 |
| AKT1 | Gain of function | 5 (8.1) | 0 | 0 | 0.1104 | - |
| PTEN | Loss of function | 1 (1.6) | 0 | 1 (2.5) | 1 | - |
| TP53 | Loss of function | 3 (4.8) | 0 | 12 (30.0) | 0.0003 | 0.0028 |
| BRCA1 | - | 0 | 0 | 0 | - | - |
| BRCA2 | Loss of function | 3 (4.8) | 0 | 2 (5.0) | 0.854 | - |
| PALB2 | - | 0 | 0 | 0 | - | - |
| ARID1A | Loss of function | 0 | 0 | 1 (2.5) | 0.4918 | - |
| KRAS | - | 0 | 0 | 0 | - | - |
| NRAS | - | 0 | 0 | 0 | - | - |
| BRAF | - | 0 | 0 | 0 | - | - |
| Controls | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Mutation Impact * | HER2 DE (n = 22) | HER2 0+ (n = 20) | HER2 1+ (n = 20) | HER2 2+ NA (n = 20) | HER2 2+ WA (n = 20) | HER2 3+ (n = 20) | p-Value |
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |||
| PIK3CA | Gain of function | 8 (36.4) | 8 (40.0) | 10 (50.0) | 10 (50.0) | 5 (25.0) | 1 (5.0) | 0.0227 |
| AKT1 | Gain of function | 0 | 0 | 2 (10.0) | 3 (15) | 0 | 0 | 0.0368 |
| PTEN | Loss of function | 0 | 0 | 0 | 1 (5.0) | 1 (5.0) | 0 | 0.7019 |
| TP53 | Loss of function | 1 (4.5) | 0 | 0 | 2 (10.0) | 4 (20.0) | 8 (40.0) | 0.0004 |
| BRCA1 | - | 0 | 0 | 0 | 0 | 0 | 0 | |
| BRCA2 | Loss of function | 2 (9.1) | 0 | 0 | 1 (5.0) | 1 (5.0) | 1 (5.0) | 0.8997 |
| PALB2 | - | 0 | 0 | 0 | 0 | 0 | 0 | |
| ARID1A | Loss of function | 0 | 0 | 0 | 0 | 0 | 1 (5.0) | 0.8197 |
| KRAS | - | 0 | 0 | 0 | 0 | 0 | 0 | |
| NRAS | - | 0 | 0 | 0 | 0 | 0 | 0 | |
| BRAF | - | 0 | 0 | 0 | 0 | 0 | 0 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
André, C.; Bertaut, A.; Ladoire, S.; Desmoulins, I.; Jankowski, C.; Beltjens, F.; Charon-Barra, C.; Bergeron, A.; Richard, C.; Boidot, R.; et al. HER2-Low Luminal Breast Carcinoma Is Not a Homogenous Clinicopathological and Molecular Entity. Cancers 2024, 16, 2009. https://doi.org/10.3390/cancers16112009
André C, Bertaut A, Ladoire S, Desmoulins I, Jankowski C, Beltjens F, Charon-Barra C, Bergeron A, Richard C, Boidot R, et al. HER2-Low Luminal Breast Carcinoma Is Not a Homogenous Clinicopathological and Molecular Entity. Cancers. 2024; 16(11):2009. https://doi.org/10.3390/cancers16112009
Chicago/Turabian StyleAndré, Céline, Aurélie Bertaut, Sylvain Ladoire, Isabelle Desmoulins, Clémentine Jankowski, Françoise Beltjens, Céline Charon-Barra, Anthony Bergeron, Corentin Richard, Romain Boidot, and et al. 2024. "HER2-Low Luminal Breast Carcinoma Is Not a Homogenous Clinicopathological and Molecular Entity" Cancers 16, no. 11: 2009. https://doi.org/10.3390/cancers16112009
APA StyleAndré, C., Bertaut, A., Ladoire, S., Desmoulins, I., Jankowski, C., Beltjens, F., Charon-Barra, C., Bergeron, A., Richard, C., Boidot, R., & Arnould, L. (2024). HER2-Low Luminal Breast Carcinoma Is Not a Homogenous Clinicopathological and Molecular Entity. Cancers, 16(11), 2009. https://doi.org/10.3390/cancers16112009