
Role of Hypoxia and Rac1 Inhibition in the Metastatic Cascade

Abstract
Simple Summary
Abstract
1. Introduction
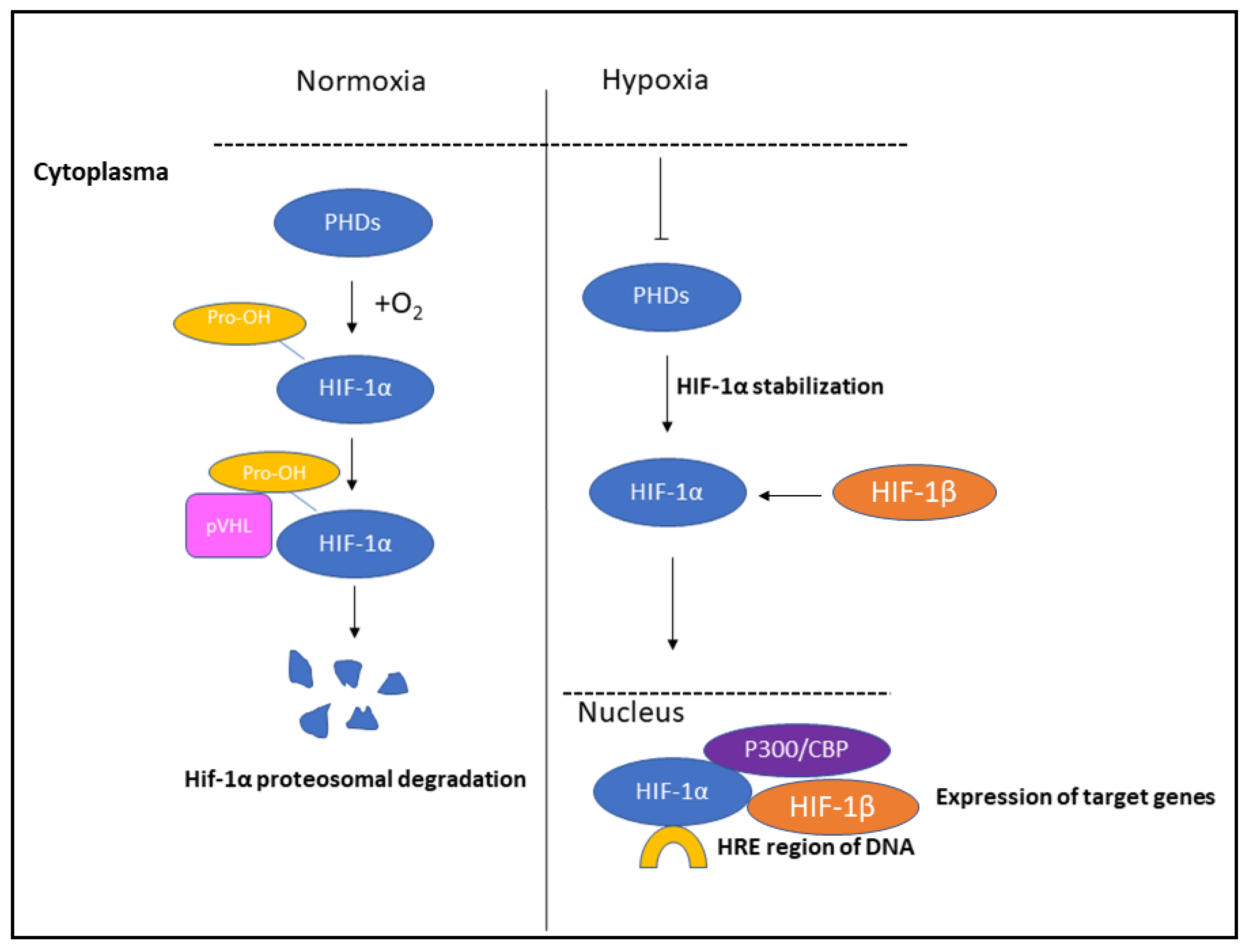
2. Role of HIFs in Hypoxia
3. Hypoxia-Mediated Tumor Progression and Metastatic Process
3.1. Angiogenesis
3.2. Immune System Evasion
3.3. Invasion and Intravasation
3.4. Extravasation
3.5. Colonization of Distant Tissue
3.6. Growth of Metastasis
4. Therapy Resistance
5. Anti-HIF Therapies
6. Role of Small G Proteins and Cytoskeleton in Tumor Cell Metastasis under Hypoxia
Efficacy and Limitations of Rac1 Inhibition
- (1)
- (2)
- (3)
- Regarding the importance of post-translational modifications mediating Rac1 subcellular localization and activation, several compounds which can block these lipid modifications have been developed. The geranylgeranyl transferases type I (GGTI) inhibitor demonstrated promising in vitro and preclinical results [126], exerting anti-tumorigenic effects in human pancreatic and non-small-cell lung cancer xenograft mouse models [127,128].
- (4)
- The interaction between Rac1–effector was the most effective for blocking Rac1 without affecting other downstream signaling pathways [106]. The best described Rac1 effectors are the PAKs as they showed the sensibilization of the Rac1 P29S mutant melanoma cell lines and xenografts [129]. However, the clinical application is controversial using PAKs inhibitors. Targeting ARP2/3 or formins could be used in the treatment of Rac1 mutant tumors [112]. Selective PI3K inhibitors were able to prevent melanoma cell proliferation and migration driven by mutant Rac1 [130]. Overall, the therapeutical advantage of targeting Rac1–effector inhibition is proven, while more potent Rac1 inhibitors are still necessary.
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| AMPK | Adenosine monophosphate-activated protein kinase |
| Ang1 | Angiopoietin-1 |
| ANGPT2 | Angiopoietin-2 |
| ANGPTL4 | Angiopoietin-like 4 |
| α-SMA | alpha smooth muscle actin |
| ARNT | Aryl hydrocarbon receptor nuclear translocator (HIF-1β) |
| Arp2/3 | Actin-related protein 2/3 complex |
| BNIP3L | BCL2 interacting protein 3 like |
| BC | Breast cancer |
| CCL-5 | C-C motif ligand-5 |
| Cdc42 | Cell division control protein homolog |
| CH1 | Cysteine/histidine-rich 1 domain |
| CML | Chronic myeloid leukemia |
| COP9 | Constitutive photomorphogenesis mutant 9 |
| COX2 | Cyclooxygenase-2 |
| CRC | Colorectal carcinoma |
| CSN5 | COP9 signalosome subunit 5 |
| C-TAD | C-terminal transactivation domain |
| CTC | Circulating tumor cell |
| CXCR4 | CXC-motif chemokine receptor 4 |
| Dia | Formin Diaphanous |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| FSP1 | Ferroptosis suppressor protein 1 |
| GAP | GTP-activated protein |
| GDI | Guanine nucleotide dissociation inhibitor |
| GDP | Guanosine–diphosphate |
| GEF | Guanine nucleotide exchange factor |
| GTP | Guanosine–triphosphate |
| GYS1 | Glycogen synthase 1 |
| HAF | Hypoxia-associated factor |
| HCC | Hepatocellular carcinoma |
| HER2 | Human epidermal growth factor receptor 2 |
| HIF | Hypoxia-inducible factor |
| HLH | Helix Loop Helix |
| HRE | Hypoxia-Response Element |
| LDHA | Lactate dehydrogenase A |
| LIMK | LIM domain kinase |
| LOX | Lysyl-oxidase |
| MAPK | Mitogen-activated protein kinase |
| MXI1 | MAX-interacting Protein 1 |
| MET | Mesenchymal–epithelial transition |
| MMP | Matrix metalloproteinase |
| MSCs | Mesenchymal stem cells |
| mTOR | Mammalian target of rapamycin |
| NDRG1 | N-Myc downstream regulated 1 |
| NF-κB | Nuclear factor k-light-chain-enhancer of activated B cells |
| LINC | Linker of nucleoskeleton and cytoskeleton NRF2—Nuclear factor erythroid 2-related factor 2 |
| OCC | Oral squamous cancer cell |
| ODDD | Oxygen-dependent degradation domain |
| PAK | Serine/threonine protein kinase |
| PAS | Per ARNT Sim |
| PDGF-B | Platelet-derived growth factor subunit B |
| PGF | Placental growth factor |
| PGK1 | Phosphoglycerate kinase 1 |
| PI3K | Phosphoinositide 3 kinase |
| PFKP | Phosphofructokinase, platelet |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RhoA | Ras homolog family member A |
| RNASE4 | Ribonuclease 4 |
| ROCK | Rho-associated protein kinase |
| ROS | Reactive oxygen species |
| SCF | Stem cell factor |
| SCL2A1 | Solute carrier family 2 member 1 |
| SDF-1 | Stromal cell-derived factor 1 |
| SMAD | Small worm phenotype (Caenorhabditis elegans) and MAD family (Mothers Against Decapentaplegic genes in Drosophila) |
| TADs | Transactivation domains |
| TIMP-1 | Tissue inhibitor matrix metalloproteinase-1 |
| TKI | Tyrosine kinase inhibitor |
| Treg | Regulatory T cell |
| VEGF | Vascular endothelial growth factor |
| VHL | von Hippel–Lindau |
| ZO-1 | Zonula Occludens 1 |
References
- Vajda, J.; Milojević, M.; Maver, U.; Vihar, B. Microvascular Tissue Engineering-A Review. Biomedicines 2021, 9, 589. [Google Scholar] [CrossRef]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why Is the Partial Oxygen Pressure of Human Tissues a Crucial Parameter? Small Molecules and Hypoxia. J. Cell Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Tu, K.; Xu, H.; Wang, L.; Yuan, X.; Qin, X.; Kong, L.; Chu, Q.; Zhang, Z. Improving Tumor Hypoxia and Radiotherapy Resistance via in Situ Nitric Oxide Release Strategy. Eur. J. Pharm. Biopharm. 2020, 150, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Tátrai, E.; Bartal, A.; Gacs, A.; Paku, S.; Kenessey, I.; Garay, T.; Hegedűs, B.; Molnár, E.; Cserepes, M.T.; Hegedűs, Z.; et al. Cell Type-Dependent HIF1 α-Mediated Effects of Hypoxia on Proliferation, Migration and Metastatic Potential of Human Tumor Cells. Oncotarget 2017, 8, 44498–44510. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic Control of Metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological Mechanisms of the Antitumor Effects of Supplemental Oxygenation. Sci. Transl. Med. 2015, 7, 277ra30. [Google Scholar] [CrossRef]
- Patel, U.; Pandey, M.; Kannan, S.; Samant, T.A.; Gera, P.; Mittal, N.; Rane, S.; Patil, A.; Noronha, V.; Joshi, A.; et al. Prognostic and Predictive Significance of Nuclear HIF1α Expression in Locally Advanced HNSCC Patients Treated with Chemoradiation with or without Nimotuzumab. Br. J. Cancer 2020, 123, 1757–1766. [Google Scholar] [CrossRef]
- Irshad, K.; Mohapatra, S.K.; Srivastava, C.; Garg, H.; Mishra, S.; Dikshit, B.; Sarkar, C.; Gupta, D.; Chandra, P.S.; Chattopadhyay, P.; et al. A Combined Gene Signature of Hypoxia and Notch Pathway in Human Glioblastoma and Its Prognostic Relevance. PLoS ONE 2015, 10, e0118201. [Google Scholar] [CrossRef]
- Cowman, S.J.; Fuja, D.G.; Liu, X.-D.; Tidwell, R.S.S.; Kandula, N.; Sirohi, D.; Agarwal, A.M.; Emerson, L.L.; Tripp, S.R.; Mohlman, J.S.; et al. Macrophage HIF-1α Is an Independent Prognostic Indicator in Kidney Cancer. Clin. Cancer Res. 2020, 26, 4970–4982. [Google Scholar] [CrossRef]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The Role of Hypoxia in the Tumor Microenvironment and Development of Cancer Stem Cell: A Novel Approach to Developing Treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Almendros, I.; Gozal, D. Intermittent Hypoxia and Cancer: Undesirable Bed Partners? Respir. Physiol. Neurobiol. 2018, 256, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Galappathi, K.; Mathiesen, B.; Ruud, E.-B.M. Fluctuating and Diffusion-Limited Hypoxia in Hypoxia-Induced Metastasis. Clin. Cancer Res. 2007, 13, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Gaustad, J.-V.; Simonsen, T.G.; Roa, A.M.A.; Rofstad, E.K. Tumors Exposed to Acute Cyclic Hypoxia Show Increased Vessel Density and Delayed Blood Supply. Microvasc. Res. 2013, 85, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, V.K.; Mohanam, I.; Rao, J.S.; Mohanam, S. Intermittent Hypoxia Regulates Stem-like Characteristics and Differentiation of Neuroblastoma Cells. PLoS ONE 2012, 7, e30905. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.-F.; Zhao, T.-T.; Wang, Z.-N.; Xu, Y.-Y.; Mao, X.-Y.; Wu, J.-H.; Liu, X.-Y.; Xu, H.; You, Y.; Xu, H.-M. Influence of Different Hypoxia Models on Metastatic Potential of SGC-7901 Gastric Cancer Cells. Tumour Biol. 2014, 35, 6801–6808. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.W.; So, D.; Min, S.; Kim, J.; Lee, M.; Khalmuratova, R.; Cho, C.-H.; Park, J.-W.; Shin, H.-W. Accelerated Tumor Growth under Intermittent Hypoxia Is Associated with Hypoxia-Inducible Factor-1-Dependent Adaptive Responses to Hypoxia. Oncotarget 2017, 8, 61592–61603. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors: Mediators of Cancer Progression and Targets for Cancer Therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Tsai, Y.-P.; Wu, K.-J. Hypoxia-Regulated Target Genes Implicated in Tumor Metastasis. J. Biomed. Sci. 2012, 19, 102. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Batty-Stuart, S.; Lee, J.E.; Ohh, M. HIF-1α Hydroxyprolines Modulate Oxygen-Dependent Protein Stability Via Single VHL Interface With Comparable Effect on Ubiquitination Rate. J. Mol. Biol. 2021, 433, 167244. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 Is an Asparaginyl Hydroxylase Enzyme That Regulates the Transcriptional Activity of Hypoxia-Inducible Factor. Genes. Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.A.; Zhou, C.; Wee, S. The COP9 Signalosome: An Assembly and Maintenance Platform for Cullin Ubiquitin Ligases? Nat. Cell Biol. 2003, 5, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Powis, G. HAF: The New Player in Oxygen-Independent HIF-1alpha Degradation. Cell Cycle 2009, 8, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, F.; Yang, H.; Wang, Z. Action Sites and Clinical Application of HIF-1α Inhibitors. Molecules 2022, 27, 3426. [Google Scholar] [CrossRef] [PubMed]
- Albadari, N.; Deng, S.; Li, W. The Transcriptional Factors HIF-1 and HIF-2 and Their Novel Inhibitors in Cancer Therapy. Expert. Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Zhao, J.; Du, F.; Shen, G.; Zheng, F.; Xu, B. The Role of Hypoxia-Inducible Factor-2 in Digestive System Cancers. Cell Death Dis. 2015, 6, e1600. [Google Scholar] [CrossRef]
- Fitzpatrick, S.F. Immunometabolism and Sepsis: A Role for HIF? Front. Mol. Biosci. 2019, 6, 85. [Google Scholar] [CrossRef]
- Duan, C. Hypoxia-Inducible Factor 3 Biology: Complexities and Emerging Themes. Am. J. Physiol. Cell Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular Characterization and Chromosomal Localization of a Third Alpha-Class Hypoxia Inducible Factor Subunit, HIF3alpha. Gene Expr. 1998, 7, 205–213. [Google Scholar] [PubMed]
- Yamashita, T.; Ohneda, O.; Nagano, M.; Iemitsu, M.; Makino, Y.; Tanaka, H.; Miyauchi, T.; Goto, K.; Ohneda, K.; Fujii-Kuriyama, Y.; et al. Abnormal Heart Development and Lung Remodeling in Mice Lacking the Hypoxia-Inducible Factor-Related Basic Helix-Loop-Helix PAS Protein NEPAS. Mol. Cell Biol. 2008, 28, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jeong, D.; Han, Y.-S.; Baek, M.J. Pivotal Role of Vascular Endothelial Growth Factor Pathway in Tumor Angiogenesis. Ann. Surg. Treat. Res. 2015, 89, 1–8. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia (Auckl) 2015, 3, 83–92. [Google Scholar] [CrossRef]
- de la Puente, P.; Muz, B.; Azab, F.; Azab, A.K. Cell Trafficking of Endothelial Progenitor Cells in Tumor Progression. Clin. Cancer Res. 2013, 19, 3360–3368. [Google Scholar] [CrossRef]
- Döme, B.; Hendrix, M.J.C.; Paku, S.; Tóvári, J.; Tímár, J. Alternative Vascularization Mechanisms in Cancer: Pathology and Therapeutic Implications. Am. J. Pathol. 2007, 170, 1–15. [Google Scholar] [CrossRef]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing Cancer Immunotherapy Using Antiangiogenics: Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Donato, C.; Kunz, L.; Castro-Giner, F.; Paasinen-Sohns, A.; Strittmatter, K.; Szczerba, B.M.; Scherrer, R.; Di Maggio, N.; Heusermann, W.; Biehlmaier, O.; et al. Hypoxia Triggers the Intravasation of Clustered Circulating Tumor Cells. Cell Rep. 2020, 32, 108105. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New Insights into Cancer Immunoediting and Its Three Component Phases--Elimination, Equilibrium and Escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Bae, H.C.; Noh, K.H.; Song, K.-H.; Ye, S.; Mao, C.-P.; Lee, K.-M.; Wu, T.-C.; Kim, T.W. Gain of HIF-1α under Normoxia in Cancer Mediates Immune Adaptation through the AKT/ERK and VEGFA Axes. Clin. Cancer Res. 2015, 21, 1438–1446. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 Regulates CD47 Expression in Breast Cancer Cells to Promote Evasion of Phagocytosis and Maintenance of Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef]
- Kumar, V.; Gabrilovich, D.I. Hypoxia-Inducible Factors in Regulation of Immune Responses in Tumour Microenvironment. Immunology 2014, 143, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Vignjevic, D.M. Modes of Cancer Cell Invasion and the Role of the Microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Tumour-Cell Invasion and Migration: Diversity and Escape Mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Chen, B.-J.; Wu, J.-S.; Tang, Y.-J.; Tang, Y.-L.; Liang, X.-H. What Makes Leader Cells Arise: Intrinsic Properties and Support from Neighboring Cells. J. Cell Physiol. 2020, 235, 8983–8995. [Google Scholar] [CrossRef] [PubMed]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer Cell Invasion and EMT Marker Expression: A Three-Dimensional Study of the Human Cancer-Host Interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, Y.; Ji, W.; Li, X.; Sun, B.; Gao, Q.; Su, C. Anti-Tumor Activities of Matrine and Oxymatrine: Literature Review. Tumour Biol. 2014, 35, 5111–5119. [Google Scholar] [CrossRef] [PubMed]
- Serganova, I.; Mayer-Kukuck, P.; Huang, R.; Blasberg, R. Molecular Imaging: Reporter Gene Imaging. Handb. Exp. Pharmacol. 2008, 185/2, 167–223. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Malec, V.; Gottschald, O.R.; Li, S.; Rose, F.; Seeger, W.; Hänze, J. HIF-1 Alpha Signaling Is Augmented during Intermittent Hypoxia by Induction of the Nrf2 Pathway in NOX1-Expressing Adenocarcinoma A549 Cells. Free Radic. Biol. Med. 2010, 48, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Bocci, F.; Tripathi, S.C.; Vilchez Mercedes, S.A.; George, J.T.; Casabar, J.P.; Wong, P.K.; Hanash, S.M.; Levine, H.; Onuchic, J.N.; Jolly, M.K. NRF2 Activates a Partial Epithelial-Mesenchymal Transition and Is Maximally Present in a Hybrid Epithelial/Mesenchymal Phenotype. Integr. Biol. (Camb) 2019, 11, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia Promotes Fibrogenesis in Vivo via HIF-1 Stimulation of Epithelial-to-Mesenchymal Transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Gilkes, D.M.; Takano, N.; Semenza, G.L. Hypoxia-Inducible Factor-Dependent Signaling between Triple-Negative Breast Cancer Cells and Mesenchymal Stem Cells Promotes Macrophage Recruitment. Proc. Natl. Acad. Sci. USA 2014, 111, E2120–E2129. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A Guide to the Composition and Functions of the Extracellular Matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhöfer, N.; Kong, C.; Le, Q.-T.; Chi, J.-T.A.; Jeffrey, S.S.; Giaccia, A.J. Lysyl Oxidase Is Essential for Hypoxia-Induced Metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef]
- Hielscher, A.; Qiu, C.; Porterfield, J.; Smith, Q.; Gerecht, S. Hypoxia Affects the Structure of Breast Cancer Cell-Derived Matrix to Support Angiogenic Responses of Endothelial Cells. J. Carcinog. Mutagen. 2013, Suppl. 13, 005. [Google Scholar] [CrossRef]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Guedj, N.; Clapéron, A.; Nguyen Ho-Bouldoires, T.H.; Paradis, V.; Fouassier, L. Epithelial-Mesenchymal Transition in Cholangiocarcinoma: From Clinical Evidence to Regulatory Networks. J. Hepatol. 2017, 66, 424–441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bai, X.; Chen, W.; Ma, T.; Hu, Q.; Liang, C.; Xie, S.; Chen, C.; Hu, L.; Xu, S.; et al. Wnt/β-Catenin Signaling Enhances Hypoxia-Induced Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma via Crosstalk with Hif-1α Signaling. Carcinogenesis 2013, 34, 962–973. [Google Scholar] [CrossRef]
- Lei, J.; Ma, J.; Ma, Q.; Li, X.; Liu, H.; Xu, Q.; Duan, W.; Sun, Q.; Xu, J.; Wu, Z.; et al. Hedgehog Signaling Regulates Hypoxia Induced Epithelial to Mesenchymal Transition and Invasion in Pancreatic Cancer Cells via a Ligand-Independent Manner. Mol. Cancer 2013, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Dekker, Y.; Le Dévédec, S.E.; Danen, E.H.J.; Liu, Q. Crosstalk between Hypoxia and Extracellular Matrix in the Tumor Microenvironment in Breast Cancer. Genes 2022, 13, 1585. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Brockmeier, U.; Otterbach, F.; Metzen, E. New Insight into the SDF-1/CXCR4 Axis in a Breast Carcinoma Model: Hypoxia-Induced Endothelial SDF-1 and Tumor Cell CXCR4 Are Required for Tumor Cell Intravasation. Mol. Cancer Res. 2012, 10, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Long, Y.; Li, M.; He, Q. Emerging Nanomedicine-Based Therapeutics for Hematogenous Metastatic Cascade Inhibition: Interfering with the Crosstalk between “Seed and Soil”. Acta Pharm. Sin. B 2021, 11, 2286–2305. [Google Scholar] [CrossRef]
- Pienta, K.J.; Robertson, B.A.; Coffey, D.S.; Taichman, R.S. The Cancer Diaspora: Metastasis beyond the Seed and Soil Hypothesis. Clin. Cancer Res. 2013, 19, 5849–5855. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating Tumor Cells: Biology and Clinical Significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Haber, D.A.; Maheswaran, S. Cancer Metastasis through the Prism of Epithelial-to-Mesenchymal Transition in Circulating Tumor Cells. Mol. Oncol. 2017, 11, 770–780. [Google Scholar] [CrossRef]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-Mapping Post-Hypoxic Tumor Cells Reveals a ROS-Resistant Phenotype That Promotes Metastasis. Nat. Commun. 2019, 10, 4862. [Google Scholar] [CrossRef]
- Reiterer, M.; Colaço, R.; Emrouznejad, P.; Jensen, A.; Rundqvist, H.; Johnson, R.S.; Branco, C. Acute and Chronic Hypoxia Differentially Predispose Lungs for Metastases. Sci. Rep. 2019, 9, 10246. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Rafii, S.; Lyden, D. Preparing the “Soil”: The Premetastatic Niche. Cancer Res. 2006, 66, 11089–11093. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.-T.; Giaccia, A.J. Hypoxia-Induced Lysyl Oxidase Is a Critical Mediator of Bone Marrow Cell Recruitment to Form the Premetastatic Niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Ghobrial, I.M.; Azab, A.K. Hypoxia Promotes Dissemination and Colonization in New Bone Marrow Niches in Waldenström Macroglobulinemia. Mol. Cancer Res. 2015, 13, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Luderer, M.; Azab, A.K. The Role of Hypoxia and Exploitation of the Hypoxic Environment in Hematologic Malignancies. Mol. Cancer Res. 2014, 12, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of Hypoxia in Cancer Therapy by Regulating the Tumor Microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef]
- Legendre, C.; Hori, T.; Loyer, P.; Aninat, C.; Ishida, S.; Glaise, D.; Lucas-Clerc, C.; Boudjema, K.; Guguen-Guillouzo, C.; Corlu, A.; et al. Drug-Metabolising Enzymes Are down-Regulated by Hypoxia in Differentiated Human Hepatoma HepaRG Cells: HIF-1alpha Involvement in CYP3A4 Repression. Eur. J. Cancer 2009, 45, 2882–2892. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug Penetration in Solid Tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Zieseniss, A. Hypoxia and the Modulation of the Actin Cytoskeleton—Emerging Interrelations. Hypoxia (Auckl) 2014, 2, 11–21. [Google Scholar] [CrossRef]
- Pollard, T.D.; Cooper, J.A. Actin, a Central Player in Cell Shape and Movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, R.; Pedersen, E.D.; Wang, Z.; Brakebusch, C. Rho GTPase Function in Tumorigenesis. Biochim. Biophys. Acta 2009, 1796, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.D.M.; Medina, J.I.; Velazquez, L.; Dharmawardhane, S. Targeting Rac and Cdc42 GEFs in Metastatic Cancer. Front. Cell Dev. Biol. 2020, 8, 201. [Google Scholar] [CrossRef] [PubMed]
- Stengel, K.R.; Zheng, Y. Essential Role of Cdc42 in Ras-Induced Transformation Revealed by Gene Targeting. PLoS ONE 2012, 7, e37317. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Iden, S.; Collard, J.G. Crosstalk between Small GTPases and Polarity Proteins in Cell Polarization. Nat. Rev. Mol. Cell Biol. 2008, 9, 846–859. [Google Scholar] [CrossRef]
- Turcotte, S.; Desrosiers, R.R.; Béliveau, R. HIF-1alpha mRNA and Protein Upregulation Involves Rho GTPase Expression during Hypoxia in Renal Cell Carcinoma. J. Cell Sci. 2003, 116, 2247–2260. [Google Scholar] [CrossRef] [PubMed]
- Kotelevets, L.; Chastre, E. Rac1 Signaling: From Intestinal Homeostasis to Colorectal Cancer Metastasis. Cancers 2020, 12, 665. [Google Scholar] [CrossRef]
- Hong, J.; Min, Y.; Wuest, T.; Lin, P.C. Vav1 Is Essential for HIF-1α Activation via a Lysosomal VEGFR1-Mediated Degradation Mechanism in Endothelial Cells. Cancers 2020, 12, 1374. [Google Scholar] [CrossRef]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. JNK Pathway Mediates Low Oxygen Level Induced Epithelial-Mesenchymal Transition and Stemness Maintenance in Colorectal Cancer Cells. Cancers 2020, 12, 224. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPases and Actin Dynamics in Membrane Protrusions and Vesicle Trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Maciver, S.K.; Hussey, P.J. The ADF/Cofilin Family: Actin-Remodeling Proteins. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-G.; Zhou, H.-M.; Zhang, X.; Mu, W.; Hu, J.-N.; Liu, G.-L.; Li, Q. Hypoxic Induction of Vasculogenic Mimicry in Hepatocellular Carcinoma: Role of HIF-1 α, RhoA/ROCK and Rac1/PAK Signaling. BMC Cancer 2020, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.; Liao, G.; Wang, H.; Kuang, J.; Tang, L. Rac1 Expression in Epithelial Ovarian Cancer: Effect on Cell EMT and Clinical Outcome. Med. Oncol. 2015, 32, 329. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, G.-Z.; Wang, J.-F.; Du, Y.-Y. SNHG3 Silencing Suppresses the Malignant Development of Triple-Negative Breast Cancer Cells by Regulating miRNA-326/Integrin A5 Axis and Inactivating Vav2/Rac1 Signaling Pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5481–5492. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Lin, J.; Su, J.; Oyang, L.; Wang, H.; Tan, S.; Tang, Y.; Chen, X.; Liu, W.; Luo, X.; et al. Diallyl Disulfide Inhibits Colon Cancer Metastasis by Suppressing Rac1-Mediated Epithelial-Mesenchymal Transition. Onco Targets Ther. 2019, 12, 5713–5728. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, Y.; Huan, L.; Zhao, J.; Zhou, Y.; Xu, L.; Hu, Z.; Liu, Y.; Chen, Z.; Wang, L.; et al. An LTR Retrotransposon-Derived Long Noncoding RNA lncMER52A Promotes Hepatocellular Carcinoma Progression by Binding P120-Catenin. Cancer Res. 2020, 80, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.V.; Caggia, S.; Gambrell-Sanders, D.; Khan, S.A. Differential Roles and Activation of Mammalian Target of Rapamycin Complexes 1 and 2 during Cell Migration in Prostate Cancer Cells. Prostate 2020, 80, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Hein, A.L.; Etekpo, A.; Burchett, K.M.; Lin, C.; Enke, C.A.; Batra, S.K.; Cowan, K.H.; Ouellette, M.M. Inhibition of RAC1 GTPase Sensitizes Pancreatic Cancer Cells to γ-Irradiation. Oncotarget 2014, 5, 10251–10270. [Google Scholar] [CrossRef]
- Levay, M.; Krobert, K.A.; Wittig, K.; Voigt, N.; Bermudez, M.; Wolber, G.; Dobrev, D.; Levy, F.O.; Wieland, T. NSC23766, a Widely Used Inhibitor of Rac1 Activation, Additionally Acts as a Competitive Antagonist at Muscarinic Acetylcholine Receptors. J. Pharmacol. Exp. Ther. 2013, 347, 69–79. [Google Scholar] [CrossRef]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernández, E.; Humphries-Bickley, T.; De la Mota-Peynado, A.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, Novel Small Molecule Inhibitor of Rac GTPase. J. Biol. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef]
- Yu, M.; Gong, D.; Lim, M.; Arutyunyan, A.; Groffen, J.; Heisterkamp, N. Lack of Bcr and Abr Promotes Hypoxia-Induced Pulmonary Hypertension in Mice. PLoS ONE 2012, 7, e49756. [Google Scholar] [CrossRef] [PubMed]
- Cardama, G.A.; Comin, M.J.; Hornos, L.; Gonzalez, N.; Defelipe, L.; Turjanski, A.G.; Alonso, D.F.; Gomez, D.E.; Menna, P.L. Preclinical Development of Novel Rac1-GEF Signaling Inhibitors Using a Rational Design Approach in Highly Aggressive Breast Cancer Cell Lines. Anticancer. Agents Med. Chem. 2014, 14, 840–851. [Google Scholar] [CrossRef]
- Trebucq, L.L.; Cardama, G.A.; Lorenzano Menna, P.; Golombek, D.A.; Chiesa, J.J.; Marpegan, L. Timing of Novel Drug 1A-116 to Circadian Rhythms Improves Therapeutic Effects against Glioblastoma. Pharmaceutics 2021, 13, 1091. [Google Scholar] [CrossRef]
- Hampsch, R.A.; Shee, K.; Bates, D.; Lewis, L.D.; Désiré, L.; Leblond, B.; Demidenko, E.; Stefan, K.; Huang, Y.H.; Miller, T.W. Therapeutic Sensitivity to Rac GTPase Inhibition Requires Consequential Suppression of mTORC1, AKT, and MEK Signaling in Breast Cancer. Oncotarget 2017, 8, 21806–21817. [Google Scholar] [CrossRef] [PubMed]
- Colón-Bolea, P.; García-Gómez, R.; Casar, B. RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma. Biomolecules 2021, 11, 1554. [Google Scholar] [CrossRef]
- Melendez, J.; Memtsa, M.; Stavroulis, A.; Fakokunde, A.; Yoong, W. The Best Way to Determine the Best Way to Undertake a Hysterectomy. BJOG 2009, 116, 1539–1540. [Google Scholar] [CrossRef]
- Irie, H.Y.; Pearline, R.V.; Grueneberg, D.; Hsia, M.; Ravichandran, P.; Kothari, N.; Natesan, S.; Brugge, J.S. Distinct Roles of Akt1 and Akt2 in Regulating Cell Migration and Epithelial-Mesenchymal Transition. J. Cell Biol. 2005, 171, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hirose, H.; Du, G.; Chong, K.; Kiyohara, E.; Witz, I.P.; Hoon, D.S.B. P-REX1 Amplification Promotes Progression of Cutaneous Melanoma via the PAK1/P38/MMP-2 Pathway. Cancer Lett. 2017, 407, 66–75. [Google Scholar] [CrossRef]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor Cell Invadopodia: Invasive Protrusions That Orchestrate Metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef]
- Revach, O.-Y.; Winograd-Katz, S.E.; Samuels, Y.; Geiger, B. The Involvement of Mutant Rac1 in the Formation of Invadopodia in Cultured Melanoma Cells. Exp. Cell Res. 2016, 343, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.S.; Dean, K.M.; Isogai, T.; Kasitinon, S.Y.; Murali, V.S.; Roudot, P.; Groisman, A.; Reed, D.K.; Welf, E.S.; Han, S.J.; et al. Enhanced Dendritic Actin Network Formation in Extended Lamellipodia Drives Proliferation in Growth-Challenged Rac1(P29S) Melanoma Cells. Dev. Cell 2019, 49, 444–460.e9. [Google Scholar] [CrossRef]
- Colón-Bolea, P.; García-Gómez, R.; Shackleton, S.; Crespo, P.; Bustelo, X.R.; Casar, B. RAC1 Induces Nuclear Alterations through the LINC Complex to Enhance Melanoma Invasiveness. Mol. Biol. Cell 2020, 31, 2768–2778. [Google Scholar] [CrossRef] [PubMed]
- Smalley, T.; Islam, S.M.A.; Apostolatos, C.; Apostolatos, A.; Acevedo-Duncan, M. Analysis of PKC-ζ Protein Levels in Normal and Malignant Breast Tissue Subtypes. Oncol. Lett. 2019, 17, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Abu-Kaoud, N.; Abdesselem, H.; Madani, A.; Hoarau-Véchot, J.; Thawadi, H.A.; Vidal, F.; Couderc, B.; Favre, G.; Rafii, A. SDF-1alpha Concentration Dependent Modulation of RhoA and Rac1 Modifies Breast Cancer and Stromal Cells Interaction. BMC Cancer 2015, 15, 569. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shukla, S.; Farhan, M.; Sinha, S.; Lakra, A.D.; Penta, D.; Kannan, A.; Meeran, S.M. Centchroman Prevents Metastatic Colonization of Breast Cancer Cells and Disrupts Angiogenesis via Inhibition of RAC1/PAK1/β-Catenin Signaling Axis. Life Sci. 2020, 256, 117976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, B.; You, W.; Li, P.; Kuang, Y. Rab23 Promotes Hepatocellular Carcinoma Cell Migration Via Rac1/TGF-β Signaling. Pathol. Oncol. Res. 2020, 26, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chen, J.; Peng, M.; Duan, S.; Hu, Y.; Guo, D.; Geng, J.; Zhou, J. POTEE Promotes Colorectal Carcinoma Progression via Activating the Rac1/Cdc42 Pathway. Exp. Cell Res. 2020, 390, 111933. [Google Scholar] [CrossRef] [PubMed]
- Al-Koshab, M.; Alabsi, A.M.; Bakri, M.M.; Naicker, M.S.; Seyedan, A. Chemopreventive Activity of Tualang Honey against Oral Squamous Cell Carcinoma-in Vivo. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2020, 129, 484–492. [Google Scholar] [CrossRef]
- Gastonguay, A.; Berg, T.; Hauser, A.D.; Schuld, N.; Lorimer, E.; Williams, C.L. The Role of Rac1 in the Regulation of NF-κB Activity, Cell Proliferation, and Cell Migration in Non-Small Cell Lung Carcinoma. Cancer Biol. Ther. 2012, 13, 647–656. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Westhoff, M.-A.; Zhou, S.; Nonnenmacher, L.; Dwucet, A.; Kast, R.E.; Bachem, M.G.; Wirtz, C.R.; Debatin, K.-M.; Halatsch, M.-E. Combined Inhibition of HER1/EGFR and RAC1 Results in a Synergistic Antiproliferative Effect on Established and Primary Cultured Human Glioblastoma Cells. Mol. Cancer Ther. 2013, 12, 1783–1795. [Google Scholar] [CrossRef]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational Design and Characterization of a Rac GTPase-Specific Small Molecule Inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef]
- Marei, H.; Malliri, A. Rac1 in Human Diseases: The Therapeutic Potential of Targeting Rac1 Signaling Regulatory Mechanisms. Small GTPases 2017, 8, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Dütting, S.; Heidenreich, J.; Cherpokova, D.; Amin, E.; Zhang, S.-C.; Ahmadian, M.R.; Brakebusch, C.; Nieswandt, B. Critical Off-Target Effects of the Widely Used Rac1 Inhibitors NSC23766 and EHT1864 in Mouse Platelets. J. Thromb. Haemost. 2015, 13, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and Mechanism of Action of EHT 1864, a Novel Small Molecule Inhibitor of Rac Family Small GTPases. J Biol Chem 2007, 282, 35666–35678. [Google Scholar] [CrossRef]
- Navarro-Lérida, I.; Sánchez-Perales, S.; Calvo, M.; Rentero, C.; Zheng, Y.; Enrich, C.; Del Pozo, M.A. A Palmitoylation Switch Mechanism Regulates Rac1 Function and Membrane Organization. EMBO J. 2012, 31, 534–551. [Google Scholar] [CrossRef]
- Zimonjic, D.B.; Chan, L.N.; Tripathi, V.; Lu, J.; Kwon, O.; Popescu, N.C.; Lowy, D.R.; Tamanoi, F. In Vitro and in Vivo Effects of Geranylgeranyltransferase I Inhibitor P61A6 on Non-Small Cell Lung Cancer Cells. BMC Cancer 2013, 13, 198. [Google Scholar] [CrossRef]
- Lu, J.; Chan, L.; Fiji, H.D.G.; Dahl, R.; Kwon, O.; Tamanoi, F. In Vivo Antitumor Effect of a Novel Inhibitor of Protein Geranylgeranyltransferase-I. Mol. Cancer Ther. 2009, 8, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Araiza-Olivera, D.; Feng, Y.; Semenova, G.; Prudnikova, T.Y.; Rhodes, J.; Chernoff, J. Suppression of RAC1-Driven Malignant Melanoma by Group A PAK Inhibitors. Oncogene 2018, 37, 944–952. [Google Scholar] [CrossRef]
- Uribe-Alvarez, C.; Guerrero-Rodríguez, S.L.; Rhodes, J.; Cannon, A.; Chernoff, J.; Araiza-Olivera, D. Targeting Effector Pathways in RAC1(P29S)-Driven Malignant Melanoma. Small GTPases 2021, 12, 273–281. [Google Scholar] [CrossRef]
- Pickering, K.A.; Gilroy, K.; Cassidy, J.W.; Fey, S.K.; Najumudeen, A.K.; Zeiger, L.B.; Vincent, D.F.; Gay, D.M.; Johansson, J.; Fordham, R.P.; et al. A RAC-GEF Network Critical for Early Intestinal Tumourigenesis. Nat. Commun. 2021, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ding, X.; Xie, M.; Huang, Z.; Han, P.; Tian, D.; Xia, L. CAMSAP2-Mediated Noncentrosomal Microtubule Acetylation Drives Hepatocellular Carcinoma Metastasis. Theranostics 2020, 10, 3749–3766. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Pichardo, L.; Humphries-Bickley, T.; De La Parra, C.; Forestier-Roman, I.; Martinez-Ferrer, M.; Hernandez, E.; Vlaar, C.; Ferrer-Acosta, Y.; Washington, A.V.; Cubano, L.A.; et al. The Rac Inhibitor EHop-016 Inhibits Mammary Tumor Growth and Metastasis in a Nude Mouse Model. Transl. Oncol. 2014, 7, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Onesto, C.; Shutes, A.; Picard, V.; Schweighoffer, F.; Der, C.J. Characterization of EHT 1864, a Novel Small Molecule Inhibitor of Rac Family Small GTPases. Methods Enzymol 2008, 439, 111–129. [Google Scholar] [CrossRef]
- Liang, J.; Oyang, L.; Rao, S.; Han, Y.; Luo, X.; Yi, P.; Lin, J.; Xia, L.; Hu, J.; Tan, S.; et al. Rac1, A Potential Target for Tumor Therapy. Front. Oncol. 2021, 11, 674426. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Rac1 Inhibitors | Description | References |
|---|---|---|
| NSC23766 | The compound blocks activation by the guanine nucleotide exchange factors Trio and Tiam1, but does not affect interactions with RhoA or Cdc42. It blocks ADP-mediated platelet aggregation. | [100] |
| EHop-016 | Derived from NSC23766. It has a lower IC50 than NSC23766. | [101] |
| Z62954982 | Potent, selective and cell-permeable Rac1 inhibitor that is 4 times more effective than NSC23766. It disrupts the Rac1/Tiam1 complex and decreases cytoplasmic levels of active Rac1 (GTP-bound Rac1) without affecting the activity of other Rho GTPases (such as Cdc42 or RhoA). | [102] |
| ZINC69391 | Specific Rac1 inhibitor. It acts by interfering with the interaction of Rac1 with Dock180, a relevant Rac1 activator in glioma invasion, and by reducing Rac1-GTP levels. | [103] |
| 1A-116 | Derived from ZINC69391. It is a Rac1 inhibitor, with antitumoral and antimetastatic effects in several types of cancer, such as breast cancer. It prevents Rac1-regulated processes involved in the primary tumorigenesis and metastatic processes. | [104] |
| EHT-1864 | Inhibitor of Rac family GTPases. Blocks activation by direct binding to Rac1, Rac1b, Rac2 and Rac3. Inhibits Rac-, Ras- and Tiam-induced growth transformation of NIH-3T3 fibroblasts. Reduces β-amyloid peptide production in vivo. | [105] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tátrai, E.; Ranđelović, I.; Surguta, S.E.; Tóvári, J. Role of Hypoxia and Rac1 Inhibition in the Metastatic Cascade. Cancers 2024, 16, 1872. https://doi.org/10.3390/cancers16101872
Tátrai E, Ranđelović I, Surguta SE, Tóvári J. Role of Hypoxia and Rac1 Inhibition in the Metastatic Cascade. Cancers. 2024; 16(10):1872. https://doi.org/10.3390/cancers16101872
Chicago/Turabian StyleTátrai, Enikő, Ivan Ranđelović, Sára Eszter Surguta, and József Tóvári. 2024. "Role of Hypoxia and Rac1 Inhibition in the Metastatic Cascade" Cancers 16, no. 10: 1872. https://doi.org/10.3390/cancers16101872
APA StyleTátrai, E., Ranđelović, I., Surguta, S. E., & Tóvári, J. (2024). Role of Hypoxia and Rac1 Inhibition in the Metastatic Cascade. Cancers, 16(10), 1872. https://doi.org/10.3390/cancers16101872