
NK3.3-Derived Extracellular Vesicles Penetrate and Selectively Kill Treatment-Resistant Tumor Cells

Abstract
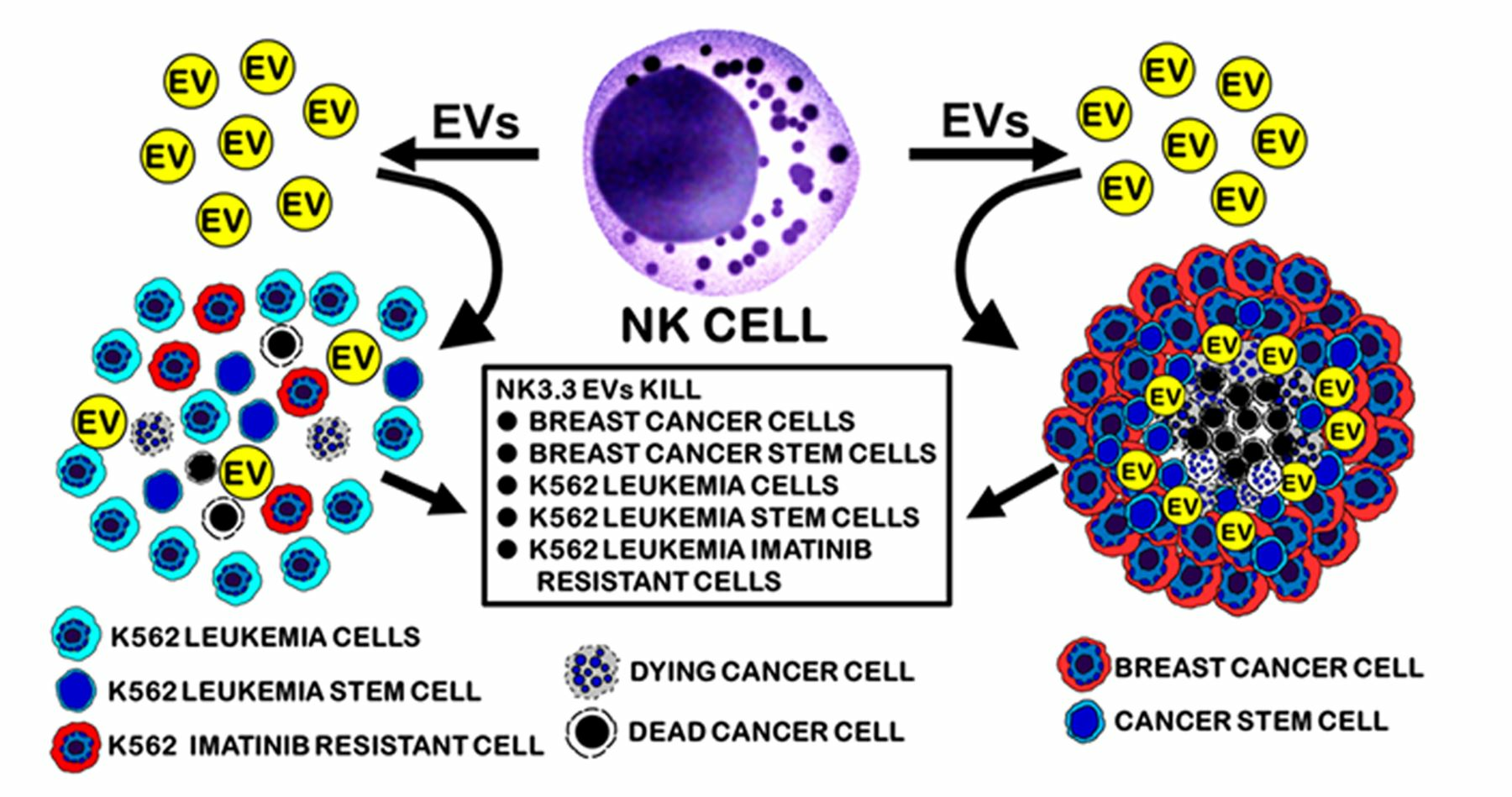
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Drug-Resistant K562 Cell Line Development
2.3. Sphere Formation
2.4. EV Generation and Isolation
2.5. Cell Growth/Viability Assays
2.6. Flow Cytometry
2.7. Caspase-3/7 Activity Assay
2.8. RNA Analysis
2.9. EV Uptake Assays
2.10. Statistical Analyses
3. Results
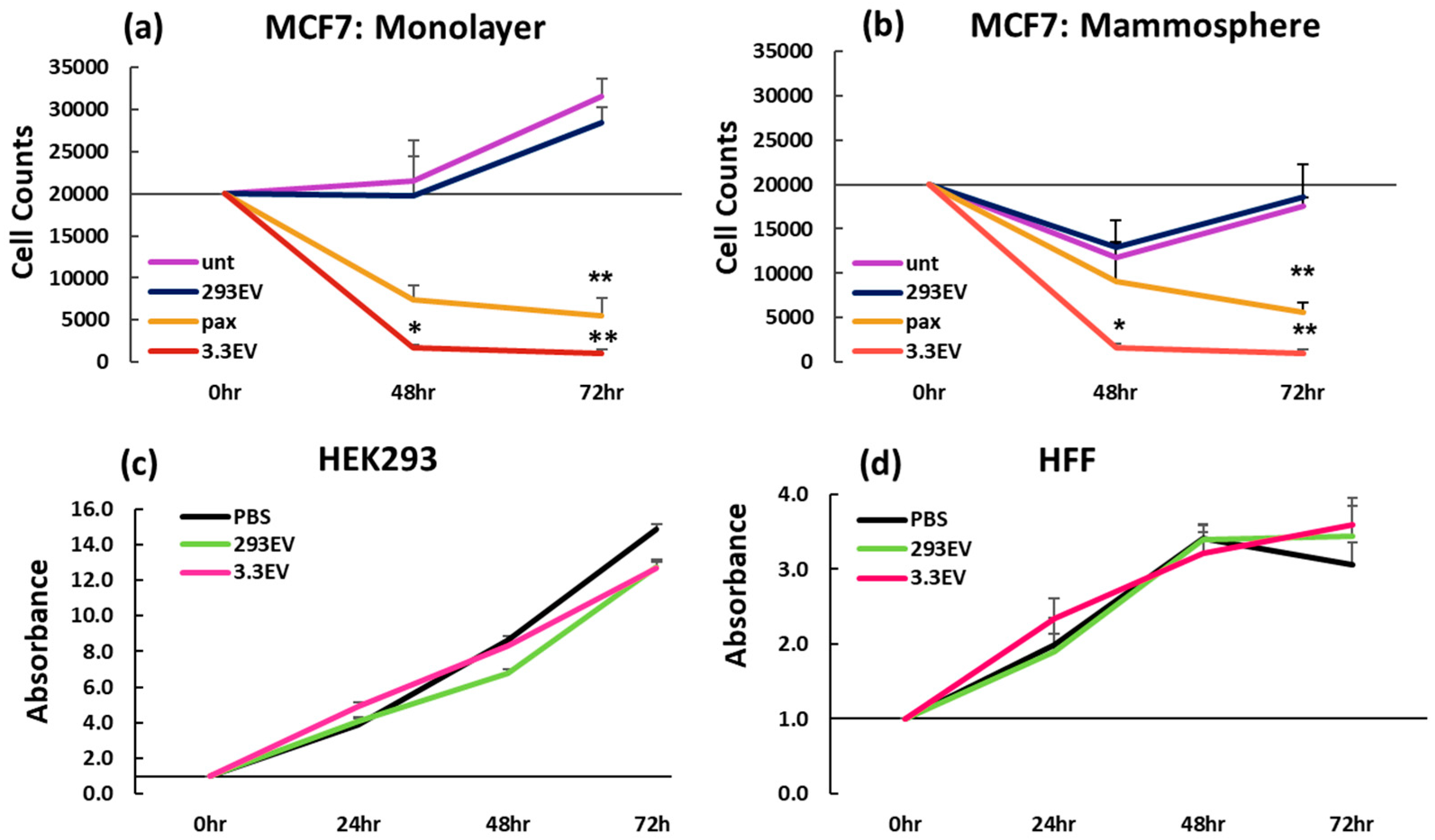
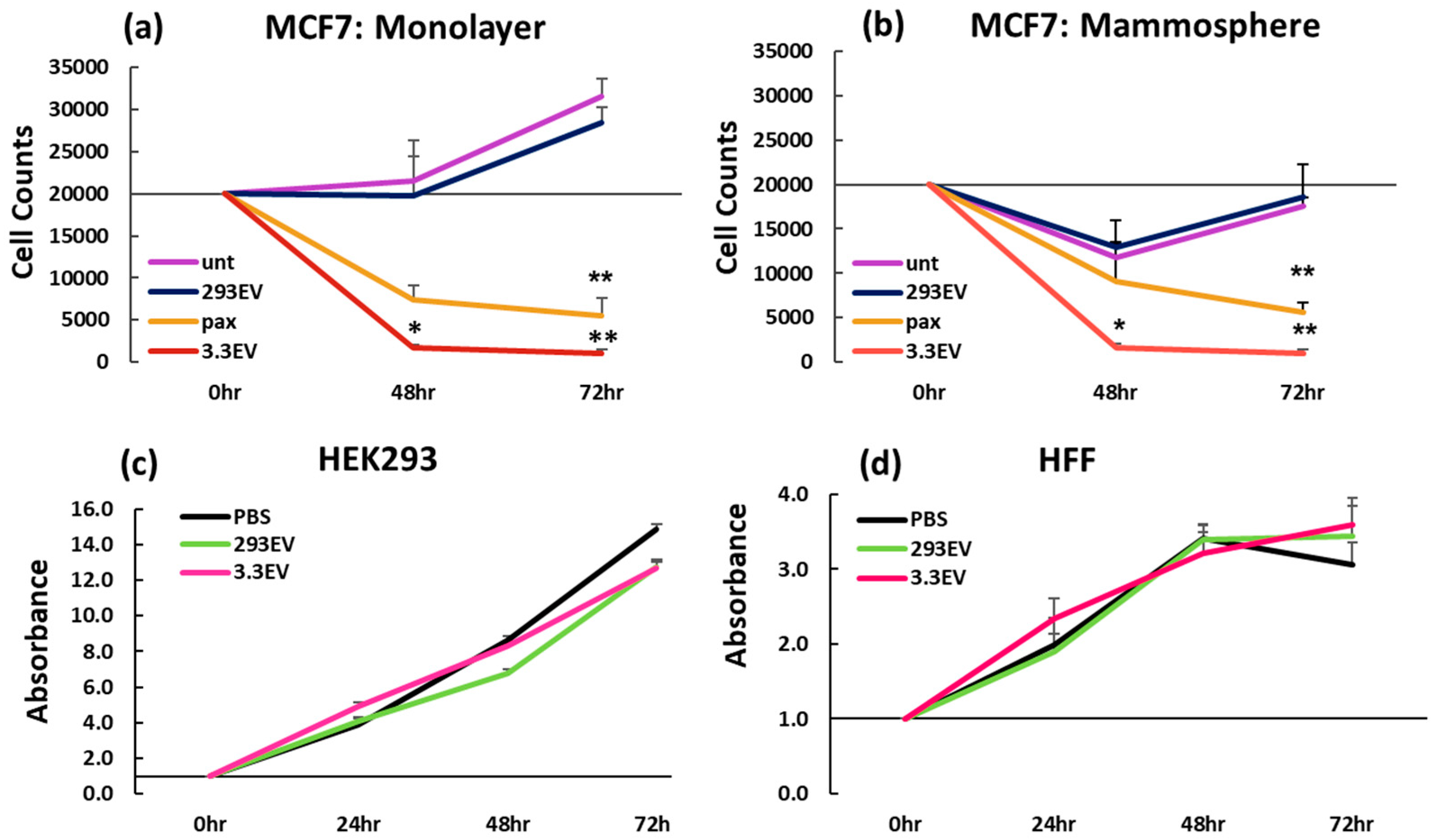
3.1. NK3.3-Derived EVs Kill Breast Cancer Cells in Monolayer and Mammosphere Cultures
3.2. NK3.3-Derived EVs Are Not Toxic to Non-Tumorigenic Cells
3.3. NK3.3-Derived EVs Induce Apoptosis of MCF7 Cells in Mammospheres
3.4. NK3.3-Derived EVs Are Taken up and Internalized by Cells Grown in Monolayers, Mammospheres, and 3D Cultures
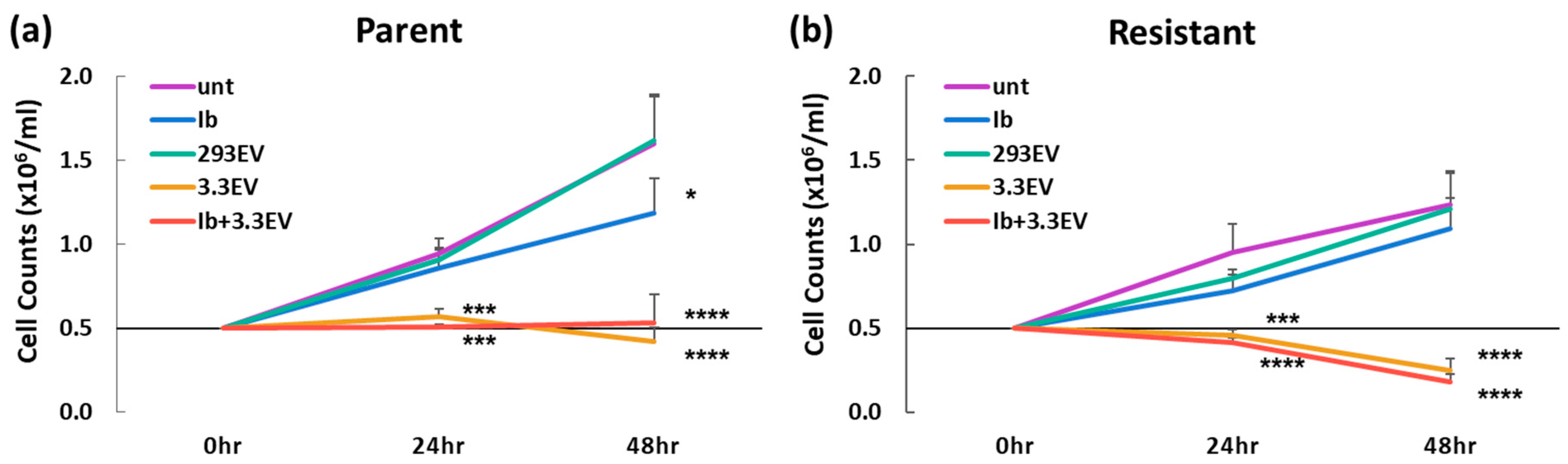
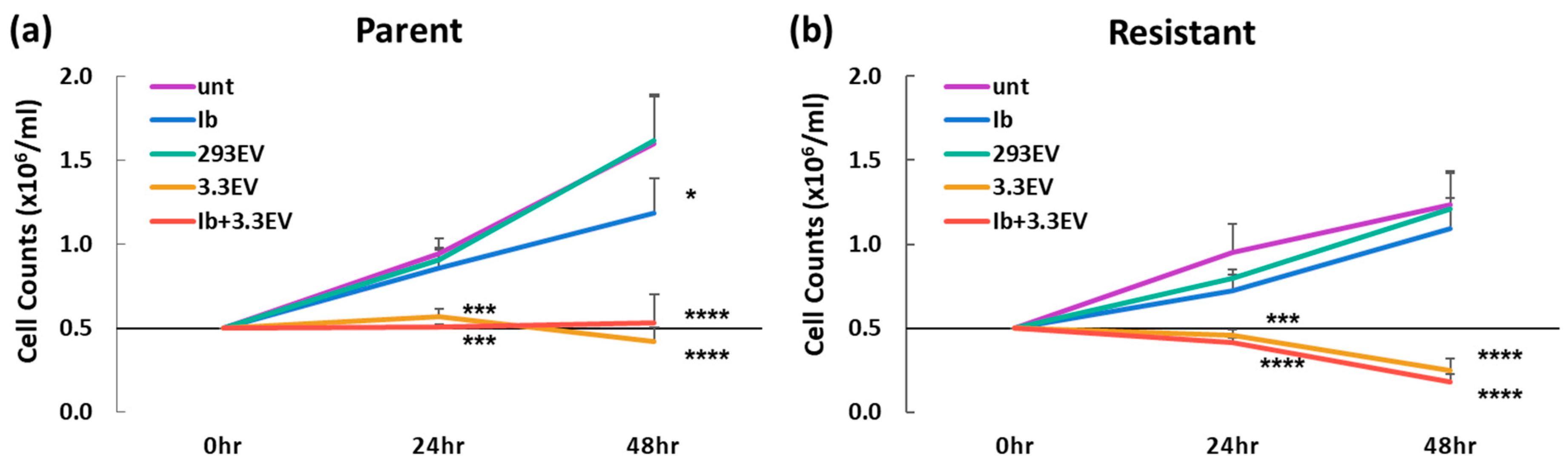
3.5. NK3.3-Derived EVs Kill Imatinib-Resistant K562 Cells
3.6. NK3.3-Derived EVs Induce Apoptotic Death in Imatinib-Resistant K562 Cells
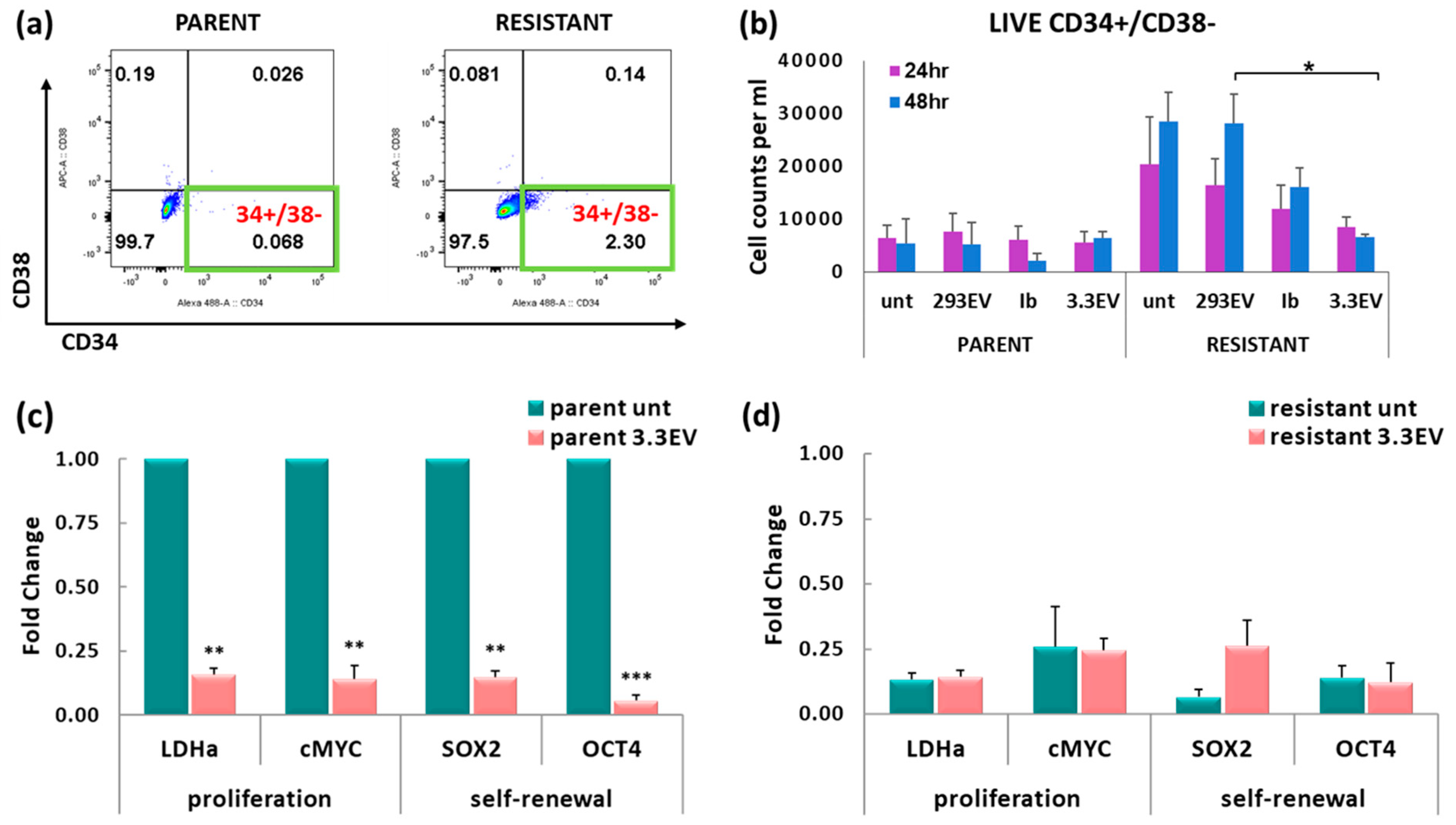
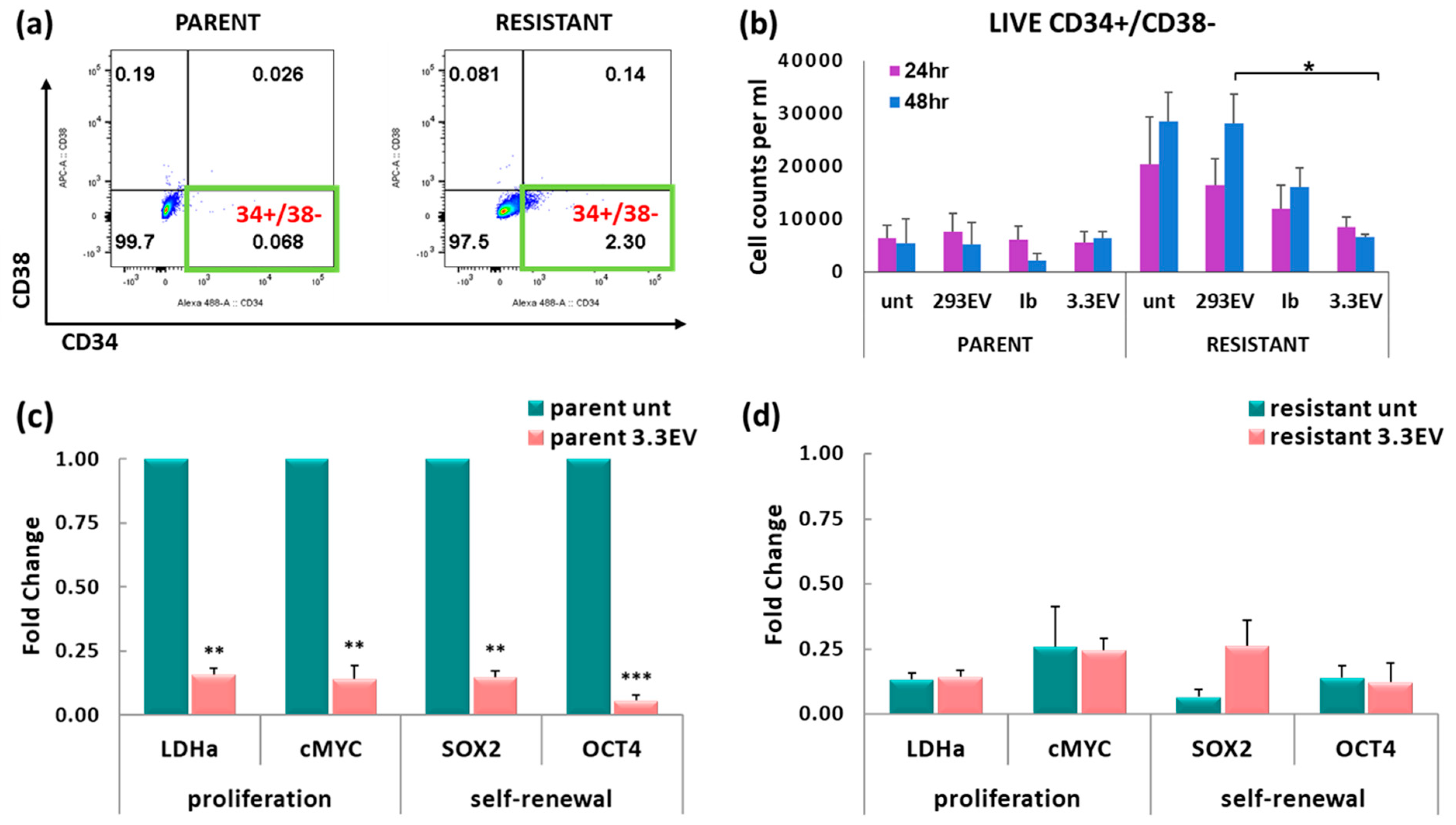
3.7. NK3.3-Derived EVs Reduce the Number of Imatinib-Resistant K562 Cells with a CSC-like Phenotype
3.8. Treatment of K562 with NK3.3-Derived EVs Reduces Tumor-Promoting Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Kocarnik, J.N.; Compton, K.; Dean, F.E.; Fu, W.; Gaw, B.L.; Harvey, J.D.; Henrikson, H.J.; Lu, D.; Pennini, A.; Xu, R.; et al. Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life Years for 29 Cancer Groups From 2010 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 2022, 8, 420–444. [Google Scholar] [PubMed]
- Costa, E.C.; Moreira, A.F.; de Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: An overview of the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning from 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Mukundan, S.; Jaramillo, M.; Oesterreich, S.; Sant, S. Three-Dimensional Breast Cancer Models Mimic Hallmarks of Size-Induced Tumor Progression. Cancer Res. 2016, 76, 3732–3743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, Z.; Dong, D.; Jang, T.; Knowles, J.C.; Kim, H.; Jin, G.; Xuan, Y. 3D culture technologies of cancer stem cells: Promising ex vivo tumor models. J. Tissue Eng. 2020, 11, 2041731420933407. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.; Lee, J.; Ahn, B. Enhancement of antitumor potency of extracellular vesicles derived from natural killer cells by IL-15 priming. Biomaterials 2019, 190–191, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Jong, A.Y.; Wu, C.; Li, J.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell. Vesicles 2017, 6, 1294368. [Google Scholar] [CrossRef] [PubMed]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Neviani, P.; Wise, P.M.; Murtadha, M.; Liu, C.W.; Wu, C.; Yong, A.Y.; Seeger, R.C.; Fabbri, M. Natural Killer-Derived Exosomal miR-186 Inhibits Neuroblastoma Growth and Immune Escape Mechanisms. Cancer Res. 2019, 79, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, J.; Li, L.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C.; Jong, A.Y. Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells. J. Extracell. Vesicles 2019, 8, 1588538. [Google Scholar] [CrossRef]
- Federici, C.; Shahaj, E.; Cecchetti, S.; Camerini, S.; Casella, M.; Iessi, E.; Camisaschi, C.; Paolino, G.; Calvieri, S.; Ferro, S.; et al. Natural-Killer-Derived Extracellular Vesicles: Immune Sensors and Interactors. Front. Immunol. 2020, 11, 262. [Google Scholar] [CrossRef]
- Cochran, A.M.; Kornbluth, J. Extracellular Vesicles from the Human Natural Killer Cell Line NK3.3 Have Broad and Potent Anti-Tumor Activity. Front. Cell Dev. Biol. 2021, 9, 698639. [Google Scholar] [CrossRef] [PubMed]
- Matchett, E.C.; Kornbluth, J. Extracellular vesicles derived from immortalized human natural killer cell lone NK3.3 as a novel therapeutic for multiple myeloma. Front. Immunol. 2023, 14, 1265101. [Google Scholar] [CrossRef] [PubMed]
- Di Pace, A.L.; Tumino, N.; Besi, F.; Alicata, C.; Conti, L.A.; Munari, E.; Maggi, E.; Vacca, P.; Moretta, L. Characterization of human NK cell-derived exosomes: Role of DNAM receptor in exosome-mediated cytotoxicity against tumor. Cancers 2020, 12, 661. [Google Scholar] [CrossRef] [PubMed]
- Aarsund, M.; Segers, F.M.; Wu, Y.; Inngjerdingen, M. Comparison of characteristics and tumor targeting properties of extracellular vesicles derived from primary NK cells or NK-cell lines stimulated with IL-15 or IL-12/15/18. Cancer Immunol. Immunother. 2022, 71, 2227–2238. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Min, H.; Song, H.; Yoo, A.; Lee, S.; Kim, K.; Park, J.; Choi, Y.H.; Cho, E. Delivery of human natural killer cell-derived exosomes for liver cancer therapy: An in vivo study in subcutaneous and orthotopic animal models. Drug Deliv. 2022, 29, 2897–2911. [Google Scholar] [CrossRef] [PubMed]
- Samara, A.; Anbar, M.; Shapira, S.; Zemlyansky, A.; Zozovsky, A.; Raanani, P.; Granot, G.; Rozovski, U. Using natural killer cell-derived exosomes as a cell-free therapy for leukemia. Hematol. Oncol. 2022, 41, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Xie, M.; Hun, M.; She, Z.; Li, C.; Luo, S.; Chen, X.Z.; Wan, W.; Wen, C.; Tian, J. Natural killer cell-derived extracellular vesicles: Novel players in cancer immunotherapy. Front. Immunol. 2021, 12, 658698. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, Y.; Li, P.; Jenkins, L.M.; Anastasakis, D.; Lyons, G.C.; Hafner, M.; Leonard, W.J. Cytokine-enhanced cytolytic activity of exosomes from NK cells. Cancer Gene Ther. 2022, 29, 734–749. [Google Scholar] [CrossRef] [PubMed]
- Farcas, M.; Inngjerdingen, M. Natural killer cell-derived extracellular vesicles in cancer therapy. Scand. J. Immunol. 2020, 92, e12938. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 2016, 95, S20–S25. [Google Scholar] [CrossRef] [PubMed]
- Cailleau, R.; Olivé, M.; Cruciger, Q.V. Long-term human breast carcinoma cell lines of metastatic origin: Preliminary characterization. In Vitro 1978, 14, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Abu Samaan, T.M.; Samec, M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar] [CrossRef] [PubMed]
- Baykal-Köse, S.; Acikgoz, E.; Yavuz, A.S.; Geyik, O.G.; Ates, H.; Sezerman, O.U.; Ozsan, G.H.; Yuce, Z. Adaptive phenotypic modulations lead to therapy resistance in chronic myeloid leukemia cells. PLoS ONE 2020, 15, e0229104. [Google Scholar] [CrossRef]
- Arrigoni, E.; Re, M.D.; Galimberti, S.; Restante, F.; Rofi, E.; Crucitta, S.; Barate, C.; Petrini, M.; Danesi, R.; Di Paolo, A. Concise Review: Chronic Myeloid Leukemia: Stem Cell Niche and Response to Pharmacologic Treatment. Stem Cells Transl. Med. 2018, 7, 305–314. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.; Suo, H.; Kitte, R.; Dluczek, S.; Straub, A.; Blaudszun, A.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Lozzio, C.B.; Lozzio, B.B. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 1975, 45, 321–334. [Google Scholar] [CrossRef] [PubMed]
- McGahon, A.J.; Brown, D.G.; Martin, S.J.; Amarante-Mendes, G.P.; Cotter, T.G.; Cohen, G.M.; Green, D.R. Downregulation of Bcr-Abl in K562 cells restores susceptibility to apoptosis: Characterization of the apoptotic death. Cell Death Differ. 1997, 4, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.F.; de Oliveira, G.L.V.; Tognon, R.; Collassanti, M.D.S.; Zanichelli, M.A.; Hamerschlak, N.; de Souza, A.M.; Covas, D.T.; Kashima, S.; de Castro, F.A. Apoptosis-related gene expression profile in chronic myeloid leukemia patients after imatinib mesylate and dasatinib therapy. Acta Haematol. 2015, 133, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Belloc, F.; Moureau-Gaudry, F.; Uhalde, M.; Cazalis, L.; Jeanneteau, L.; Lacombe, F.; Praloran, V.; Mahon, F. Imatinib and nilotinib induce apoptosis of chronic myeloid leukemia cells through a Bim-dependent pathway modulated by cytokines. Cancer Biol. Ther. 2007, 6, 912–919. [Google Scholar] [CrossRef]
- Deininger, M.W.; Druker, B.J. Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacol. Rev. 2003, 55, 401–423. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jiang, H.; Wang, K.; Liu, C.; Man, X.; Fu, Q. Hypoxia enhances the production and antitumor effect of exosomes derived from natural killer cells. Ann. Transl. Med. 2021, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Mojtahedi, H.; Yazdanpanah, N.; Rezaei, N. Chronic myeloid leukemia stem cells: Targeting therapeutic implications. Stem Cell Res. Ther. 2021, 12, 603. [Google Scholar] [CrossRef]
- Aktuna, A.Y. Tendency of K562 Chronic Myeloid Leukemia Cells Towards Cell Reprogramming. Turk. J. Haematol. 2018, 35, 260–264. [Google Scholar] [CrossRef]
- Lehne, G.; Grasmo-Wendler, U.; Berner, J.; Meza-Zepeda, L.A.; Adamsen, B.L.; Flack, A.; Reiner, A.; Clausen, O.P.F.; Hovig, E.; Myklebost, O. Upregulation of stem cell genes in multidrug resistant K562 leukemia cells. Leuk. Res. 2009, 33, 1379–1385. [Google Scholar] [CrossRef]
- Yong, A.S.; Keyvanfar, K.; Hensel, N.; Enaife, R.; Savani, B.N.; Berg, M.; Lundqvist, A.; Adams, S.; Sloand, E.M.; Goldman, J.M.; et al. Primitive quiescent CD34+ cells in chronic myeloid leukemia are targeted by in vitro expanded natural killer cells, which are functionally enhanced by bortezomib. Blood 2009, 113, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Ames, E.; Canter, R.J.; Grossenbacher, S.K.; Mac, S.; Chen, M.; Smith, R.C.; Hagino, T.; Perez-Cunningham, J.; Sckisel, G.D.; Urayama, S.; et al. NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. J. Immunol. 2015, 195, 4010–4019. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.; Arasteh, A.; Paranjpe, A.; Teruel, A.; Yang, W.; Behel, A.; Alva, J.A.; Walter, G.; Head, C.; Ishikawa, T.; et al. Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells. PLoS ONE 2010, 5, e11590. [Google Scholar] [CrossRef] [PubMed]
- Tremblay-McLean, A.; Coenraads, S.; Kiani, Z.; Dupuy, F.P.; Bernard, N.F. Expression of ligands for activating natural killer cell receptors on cell lines commonly used to assess natural killer cell function. BMC Immunol. 2019, 20, 8. [Google Scholar] [CrossRef]
- Luna, J.I.; Grossenbacher, S.K.; Murphy, W.J.; Canter, R.J. Targeting Cancer Stem Cells with Natural Killer Cell Immunotherapy. Expert Opin. Biol. Ther. 2017, 17, 313–324. [Google Scholar] [CrossRef]
- Kim, J.W.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef]
- van Schaijik, B.; Davis, P.F.; Wickremesekera, A.C.; Tan, S.T.; Itinteang, T. Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: A review. J. Clin. Pathol. 2018, 71, 88–91. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCune, A.; Kornbluth, J. NK3.3-Derived Extracellular Vesicles Penetrate and Selectively Kill Treatment-Resistant Tumor Cells. Cancers 2024, 16, 90. https://doi.org/10.3390/cancers16010090
McCune A, Kornbluth J. NK3.3-Derived Extracellular Vesicles Penetrate and Selectively Kill Treatment-Resistant Tumor Cells. Cancers. 2024; 16(1):90. https://doi.org/10.3390/cancers16010090
Chicago/Turabian StyleMcCune, Allyson, and Jacki Kornbluth. 2024. "NK3.3-Derived Extracellular Vesicles Penetrate and Selectively Kill Treatment-Resistant Tumor Cells" Cancers 16, no. 1: 90. https://doi.org/10.3390/cancers16010090
APA StyleMcCune, A., & Kornbluth, J. (2024). NK3.3-Derived Extracellular Vesicles Penetrate and Selectively Kill Treatment-Resistant Tumor Cells. Cancers, 16(1), 90. https://doi.org/10.3390/cancers16010090