1. Introduction
Bladder cancer is one of the most common malignant tumors in the urinary system [
1]. According to cancer statistics, in 2023, the incidence rate of bladder cancer in the United States is estimated to be 82,290 cases, with an expected mortality rate of 16,170 cases [
2]. In China, cancer statistics show that 91,893 new cases and 42,973 deaths due to bladder cancer were recorded in 2020 [
3]. Bladder cancer is a very broad concept that ranges from low-risk non-muscle invasive bladder cancer (NMIBC) to high-risk muscle-invasive bladder cancer (MIBC). The most important problem for patients with low-risk and moderate-risk NMIBC is the high recurrence rate and five-year relapse-free survival rates of only 43% and 33%, respectively [
4]. By contrast, the main problem faced by patients with MIBC is the 50–70% tumor metastasis rate. The total five-year survival rate of patients with advanced MIBC is only 4.8% because of the extremely high metastasis rate [
5]. NMIBC is generally believed to be caused by hyperplasia of the urinary tract epithelium, with the loss of chromosome 9 at an early stage. Meanwhile, MIBC is known to be due to abnormal cell development and closely related to genetic instability [
6]. Although the two types of bladder cancer have noted differences in genetics and molecular biology, 10–15% of NMIBC still progress to MIBC [
7]. For NMIBC patients, the prognosis of MIBC is poor, and the five-year survival rate is extremely low. Therefore, the residual tumor cells treated after tumor resection have a significant impact on the quality of life, health, and economy of patients.
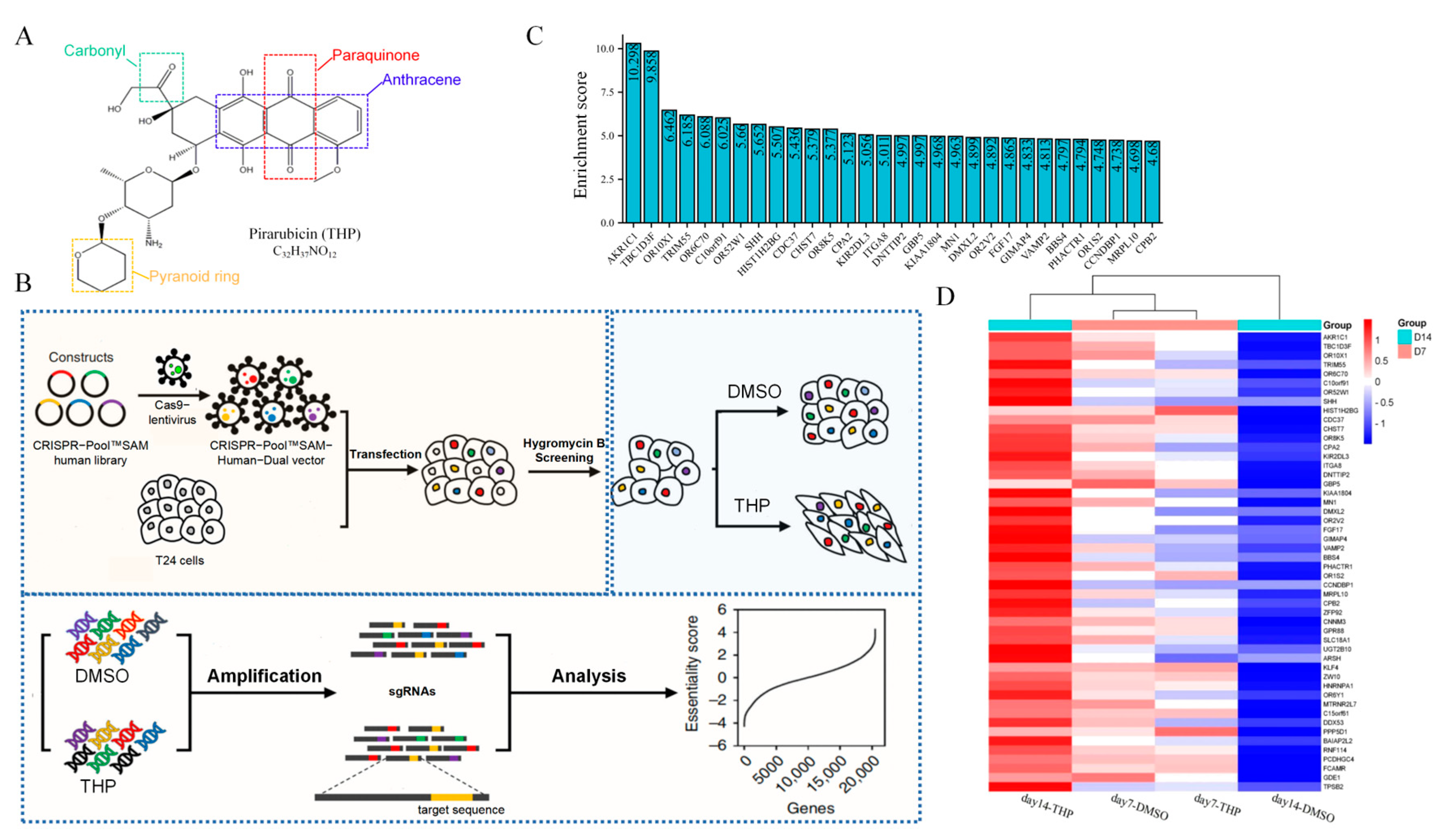
Pirarubicin (THP;
Figure 1A) is a bladder perfusion drug recommended by the guidelines of NMIBC and commonly used in clinical practice, with a usual dose of 30–50 mg. THP is a new generation of synthetic anthracycline anti-tumor drugs. It plays an antitumor role by inhibiting DNA polymerase to interfere with the synthesis of DNA in cells, causing cell cycle arrest, and promoting tumor cell apoptosis [
8]. The chemical structure of THP has another pyran ring on the basis of doxorubicin. This structural change not only enhances the anticancer activity of THP but also increases its distribution concentration in tumor cells and shortens its half-life to reduce its lethality to normal cells [
9]. Although the widespread use of THP significantly reduces the recurrence rate of NMIBC, 10–50% of patients still have tumor recurrence within 5 years after receiving THP infusion chemotherapy [
10].
The CRISPR/dCas9-SAM system is an improved technology based on CRISPR/Cas9 [
11]. The system fuses the nuclease-head mutants of Cas9 (dCas9) without endonuclease activity with the transcription activator VP64. Under the guidance of sgRNA, dCas9-VP64 fusion protein combines with the specific promoter of the genome and collects the transcription activation complex MS2-p63-HSF1. Thus, the transcription of endogenous genes is activated, realizing the expression and activation of specific genes. The system is named the CRISPR/dCas9-SAM system [
12], because it can recruit additional transcription activating factors as a synergistic activation mediator (SAM). Compared with the previous CRISPR/Cas9 system, which knocks out target genes as the mechanism, CRISPR/dCas9-SAM involves the overexpression of endogenous target genes. Therefore, the latter technology is widely used in the exploration of drug resistance genes in pathogenic microorganisms [
13].
The aldo-keto reductase family 1 member C1 (AKR1C1) is a member of the aldo-keto reductase (AKR) superfamily [
14]. This molecule can use nicotinamide adenine dinucleotide phosphate (NADPH) as a coenzyme to reduce carbonyl groups in aldehydes and ketones to hydroxyl groups, thus generating corresponding alcohols [
15]. AKRs are widely distributed in prokaryotes and eukaryotes, with 16 subfamilies and more than 190 family members [
16]. The AKR1C1 protein coding gene is located on chromosome 10.p15–p14 and consists of 12 exons [
16]. The AKR1C1 protein is composed of 323 amino acids with a relative molecular weight of 36 kDa [
16]. AKR1C1 participates in various normal physiological and biochemical reactions, and its highest mRNA expression level is observed in the lungs, followed by the liver, testes, breasts, endometrium, and brain [
17]. This protein is involved in the reduction of progesterone to 20-hydroxyprogesterone through NADPH catalysis and the reduction of progesterone concentration, which is closely related to premature delivery, endometriosis, and endometrial cancer. In addition, AKR1C1 is involved in the metabolism of multiple drugs. Different drugs, especially antitumor ones, can increase the reactive oxygen species (ROS) in cells, thus causing cell dysfunction or apoptosis through different signaling mechanisms. AKR1C1 can regulate drug metabolism by reducing the production of ROS, clearing oxygen free radicals, and inactivating the active components of various anticancer drugs [
17]. AKR1C1 also participates in the metabolism of chemical carcinogens, such as polycyclic aromatic hydrocarbons. AKR1C1 can participate in the metabolism of polycyclic aromatic hydrocarbons and reduce them to corresponding o-quinones with carcinogenic activity [
18]. AKR1C1 has been reported to be significantly overexpressed in the metastatic tissues of bladder cancer. Knocking down AKR1C1 in metastatic tissues can inhibit its invasion ability [
19]. However, in primary bladder cancer, the drug resistance mechanism of AKR1C1 and THP has not yet been elucidated.
In this study, the key genes of THP resistance in the T24 human bladder cancer cell line were screened through the CRISPR/dCas9-SAM genome-wide library. The results showed that AKR1C1 was the critical gene that caused THP resistance both in vivo and in vitro. The effect and mechanism of AKR1C1 on THP resistance in bladder cancer were explored using cell biology, molecular biology, and pharmacology and combined with AKR1C1 inhibitor (aspirin) and ROS inhibitor (scavenger) to rescue drug-resistant bladder cancer cells to recover their sensitivity to THP. The results provide a new theoretical basis for the exploration of the mechanism of THP resistance in bladder cancer. In addition, a novel theoretical foundation and therapeutic target for the prognostic classification and individualized treatment of bladder cancer have been established.
2. Materials and Methods
2.1. Crispr Library, Lentivirus, Plasmid, and Cell Lines
Library: CRISPR-Pool™ SAM human library was purchased from Shanghai Genechem Gene Chemical Co., Ltd. (Shanghai, China). The library contained 70,290 SAM-sgRNA targets and 23,430 transcripts of coding genes.
Lentivirus: CRISPR-Pool™ SAM-Human-Dual vector lentivirus was purchased from Shanghai Genechem. The lentivirus system was a two-vector system, which contained two different plasmids.
Tool plasmid: The lentivirus packaging process involved two auxiliary plasmids, namely, lentivirus packaging auxiliary plasmid 1.0 (pHelper 1.0) and lentivirus packaging auxiliary plasmid 2.0 (pHelper 2.0). The plasmids were purchased from Shanghai Genechem Gene Chemical Co., Ltd.
Cell lines: The cell line packed with lentivirus was HEK293T cell, which was an adherent-dependent epithelioid cell, and was cultured in DMEM containing 10% fetal bovine serum (FBS). The selected target cell line was T24 bladder cancer cell line, which was cultured in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% FBS. The RT4 cell line was cultured in McCoy’s 5A medium with 10% FBS. All cells were maintained in a 37 ℃ cell incubator with 5% CO2. The cells were resuscitated and subcultured twice before subsequent experiments. All cell lines were purchased from the cell bank of the Typical Culture Preservation Committee of the Chinese Academy of Sciences.
2.2. Clinical Tissues, Tissue Microarray, and Animals
The clinical tissues were from patients with bladder cancer who underwent surgery in the Affiliated Haikou Hospital of Xiangya Medical College, Central South University (
Table 1 lists the information of patients). After informed consent was signed by the patient and the patient’s family, the removed primary bladder cancer tissues and tumor tissue from recurrent bladder cancer were stored at the bladder cancer clinical sample bank. None of the patients with bladder cancer had other malignant tumors and underwent a biopsy. The pathological diagnosis was bladder cancer. None of the patients had received neoadjuvant radiotherapy, chemotherapy, or other special treatments before the first surgery. The tissue microarray of bladder cancer and normal tissues surrounding the tumor were purchased from Bioaitech Biotechnology Co., Ltd. (Xi’an, China, U812101). The microarray contained 23 cases of noninvasive urothelial carcinoma, 43 cases of invasive urothelial carcinoma, 4 cases of urothelial carcinoma with squamous tissue, and 11 cases of paraneoplastic tissue, of which 7 cases were paraneoplastic and tumor-matched (
Table 2). In this study, BALB/c nude mice (4–6 weeks old) were purchased from the Hunan SJA Laboratory Animal Co., Ltd. (Changsha, China). All animal care and related experiments outlined in this study were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals drafted by the US National Institutes of Health (NIH Publication, 8th Edition. 2011). The Institutional Animal Care and Use Committees of Xiangya Medical College and Central Southern University approved the animal protocols used in this study. Adult 4- to 6-week-old BALB/c mice weighing 20–22 g were housed under pathogen-free conditions in a temperature-controlled environment at 22–24 °C, humidity-controlled environment at 40–70% under a 12 h light/dark cycle with ad libitum access to water and food. Male and female mice were raised separately, with 3–5 mice per cage. If there was any mouse fighting behavior, these mice were divided into cages again. Mice were generally in good health before transplanting tumor cells.
Recipe for THP in vivo injection: THP storage solution was made from 1.2 mg of THP dissolved in 150 μL DMSO. Then, every 150 μL storage solution was mixed with 450 μL PEG3000, and then added to 300 μL Tween 80. Finally, the mixed compound contained 2100 μL ddH2O. Each mouse was injected with 0.1 mL of THP solution each time.
The clinical tumor tissues, animal experiments, and supporting fund projects have been discussed and approved by the Ethics Committee of the Affiliated Haikou Hospital of Xiangya Medical College, Central South University. All enrolled patients and their immediate family members fully understood and agreed that the surgically removed tissues would be used for this medical research and signed the informed consent. Written informed consent has been obtained from the patients to publish this paper.
2.3. CRISPR/dCas9-SAM Screening Process
The T24 cells were transduced with lentivirus carrying the CRISPR-Pool™ SAM library, with a multiplicity of infection of 1. Hygromycin B selection was carried out for 2 days. Then, the transduced T24 cells were treated with THP for 14 days, and the surviving cells at the end of days 7 and 14 were harvested. Genomic DNA was extracted and amplified. Then, the sgRNA sequence in the genome of the living cells was deeply sequenced. For data analysis, the enrichment score was calculated as Formula (1) [
20]. The sgRNAs used for validation were synthesized and constructed as described [
21]. The screening process is shown in
Figure 1B.
2.4. Off-Target Effect Examination
Off-target sites were predicted using an online search tool (
http://crispr.mit.edu, 20 May 2021). In addition, 3 bp mismatches compared with the target consensus sequence were allowed.
2.5. Quantitative Real-Time Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from tissues using the RNA isolater Total RNA Extraction Reagent (Vazyme, Nanjing, China, R401-01). Reverse transcription to cDNA was performed using HiScript®III All-in-one RT SuperMix Perfect for qPCR (Vazyme, R333). The cDNA was amplified with an Applied Biosystems QuantStudio 5 Real-Time PCR instrument (Thermo Fisher Scientific, Waltham, MA, USA) with ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711). The primer sequences used were as follows:
AKR1C1 forward prime: 5′-CTTCAAAGCTTTGGTGCAATTC-3′
AKR1C1 reverse prime: 5′-ATCTTTTGGGATCACTTCCTCA-3′
ACTB forward prime: 5′-CATGTACGTTGCTATCCAGGC-3′
ACTB reverse prime: 5′-CTCCTTAATGTCACGCACGAT-3′
The 2−∆∆Ct comparative method was applied to calculate the relative AKR1C1 mRNA expression.
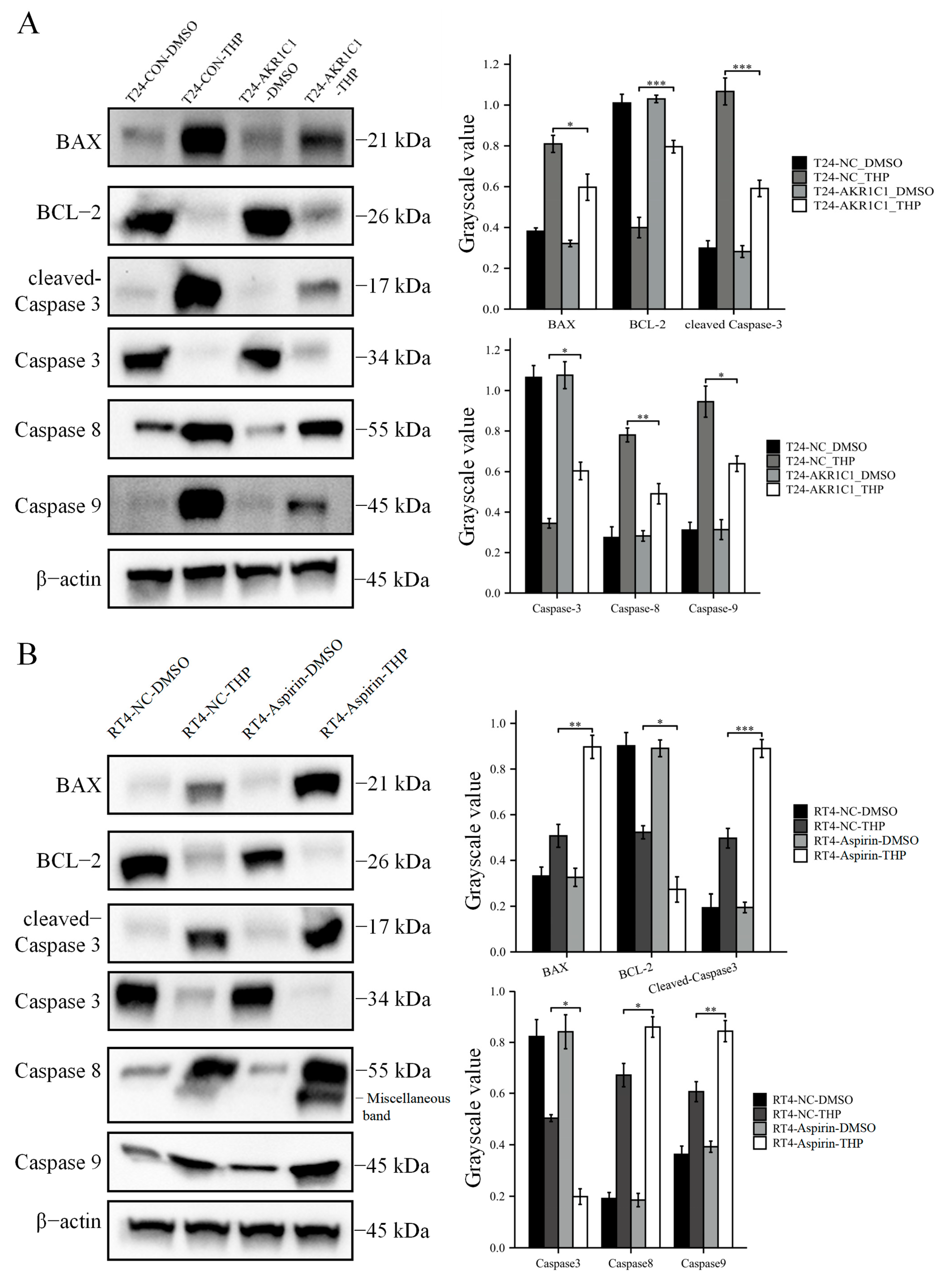
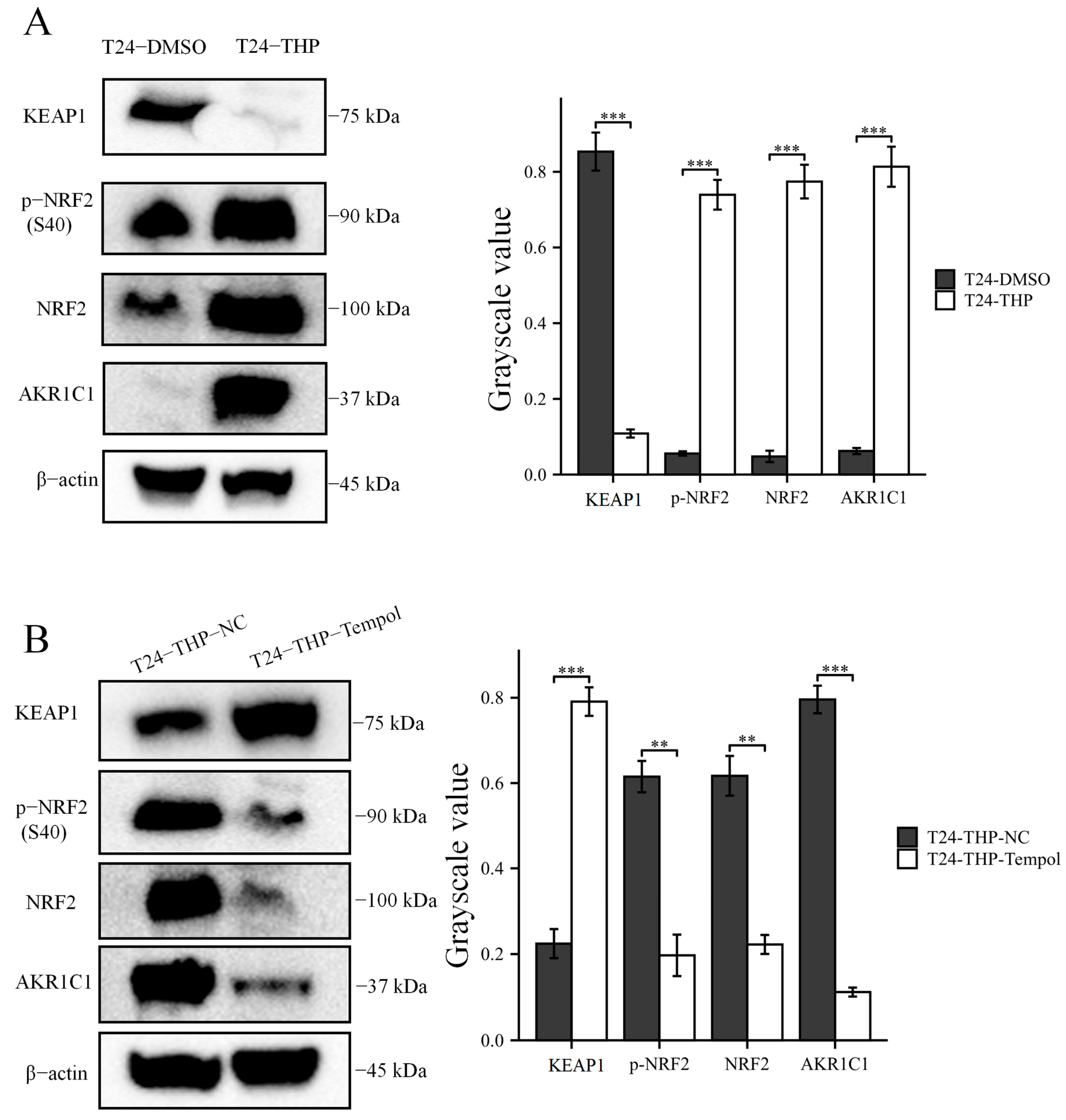
2.6. Immunoblot Analysis
The TPEB mixed reagents were made by phosphatase inhibitor cocktail (DI201, TransGen Biotech, Beijing, China), protease inhibitor cocktail (DI101, TransGen Biotech, Beijing, China), and mammalian total protein extraction kit (TPEB) (DE101, TransGen Biotech, Beijing, China) according to the protocols. Then, the cells were mixed with the TPEB mixed reagents on ice. The lysates were collected and centrifuged at 14,000× g for 10 min at 4 °C. Protein concentrations were analyzed using a bicinchoninic acid (BCA) kit (P0010, Beyotime Biotechnology, Shanghai, China). Equal amounts of protein samples were separated through electrophoresis on a 10% precast gel (M00664, GenScript Biotech, Nanjing, China). The proteins were then transferred to PVDF membranes. The membranes were blocked with 5% skimmed milk overnight at 4 °C. Afterward, the membranes were incubated for 2 h at room temperature with different primary antibodies, including AKR1C1 (ab192785, 1/10,000), β-actin (ab8227, 1/10,000), Bax (ab32503, 1/10,000), Bcl-2 (ab32124, 1/10,000), caspase-9 (ab32539, 1/1000), caspase-3 (ab32351, 1/1000), caspase-8 (ab32397, 1/1000), cleaved Caspase-3 (ab32042, 1/1000), Nrf2 (ab62352, 1/5000), p-Nrf2 (ab76026, 1/1000), and KEAP1 (ab227828, 1/10,000). The second antibody was HRP goat anti-rabbit (ab6721, 1/2000–1/20,000). The above antibodies were purchased from Abcam (Cambridge, MA, USA). The membranes were washed three times with TBST and incubated with the secondary antibodies (1/2000–1/20,000) for 1 h at room temperature. The target protein lane was imaged using an iBright 1500 (Invitrogen, Carlsbad, CA, USA) enhanced by chemiluminescent substrates (Merck Millipore, Darmstadt, Germany, WBKLS0500).
2.7. IC50 Assay
The target cells to be tested were digested, resuspended, and evenly planted in 96-well plates. PBS was added in the outer side of the circle to prevent edge effects. Cell adherent growth after 24 h. The THP (MCE, Belleville, NJ, USA, HY-13725) solution was diluted by dissolution in DMSO into gradient concentration with FBS-free medium. At this time, the DMSO content in the highest concentration group was 0.159%. The medium in the 96-well plate was discarded, and the drug-containing medium of each concentration group was added into the 96-well plate for incubation at 37 °C and 5% CO2. After 24 h, the drug-containing medium was discarded, and FBS-free medium containing 10% CCK-8 (Dojindo Laboratories, CK04) was added for incubation. After 2 h, the OD450 nm absorption was detected using the enzyme marker (Bio-Rad, Hercules, CA, USA). The experimental data (mean ± standard deviation) were plotted as histograms. Dose–response curves were generated using GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA) software, and the absolute 50% inhibitory concentration (IC50) was determined.
2.8. Hematoxylin/Eosin (HE) and Immunohistochemistry (IHC) Staining
HE staining: The tissue was embedded in paraffin and cut into a 5–7 μm sheet, spread in warm water, and then transferred to the glass to dry. First, the section was placed in xylene to remove the wax, then ethanol was used to remove the xylene, then the section was dyed with hematoxylin and eosin in turn, and then anhydrous alcohol was used to make the section transparent. Finally, 1–2 drops of neutral gum were dropped on the section and covered with cover glass.
IHC staining: IHC staining of the tissue microarray and the tissue samples for the in vivo experiments was performed in the following steps: dewaxing, blocking endogenous peroxidase, serum blocking, primary antibody binding, secondary antibody binding, DAB coloration, restaining the nucleus, and dehydration seal. The results were obtained using the H-score system, which is a scoring method widely used in immunohistochemical pathology research and can accurately evaluate the positive rate of each region [
22,
23].
2.9. Construction of AKR1C1 Overexpression Stable Transgenic Strain
The AKR1C1 overexpression stable transgenic strain was constructed in T24 cell line by embedding the overexpression plasmid with lentivirus. The vector plasmid was GV492 vector, and the sequence of elements was Ubi-MCS-3FLAG-CBh-gcGFP-IRES-puromycin. The primer sequence of the target gene fragment acquisition contained the exchange pair base, the restriction enzyme site, and the 5′-end of the target gene for PCR fishing of the target gene. The lentivirus vector plasmid was purchased from the Shanghai Genechem Gene Chemical Technology Co., Ltd. (Shanghai, China). The transfection step followed the protocol related to the reagent.
The gene acquisition sequences are given in
Table 3.
2.10. SiRNA Interference of AKR1C1 Expression in RT4 Cells
The expression level of AKR1C1 gene in the RT4 cell line was downregulated by small interfering RNA (siRNA). The siRNA was purchased from Guangzhou RiBo™ Biotechnology Co., Ltd. (Guangzhou, China) The transfection reagent was Lipofectamine 3000 (ThermoFisher, Waltham, MA, USA, L3000015). The interference experiment steps followed the reagent-related protocol.
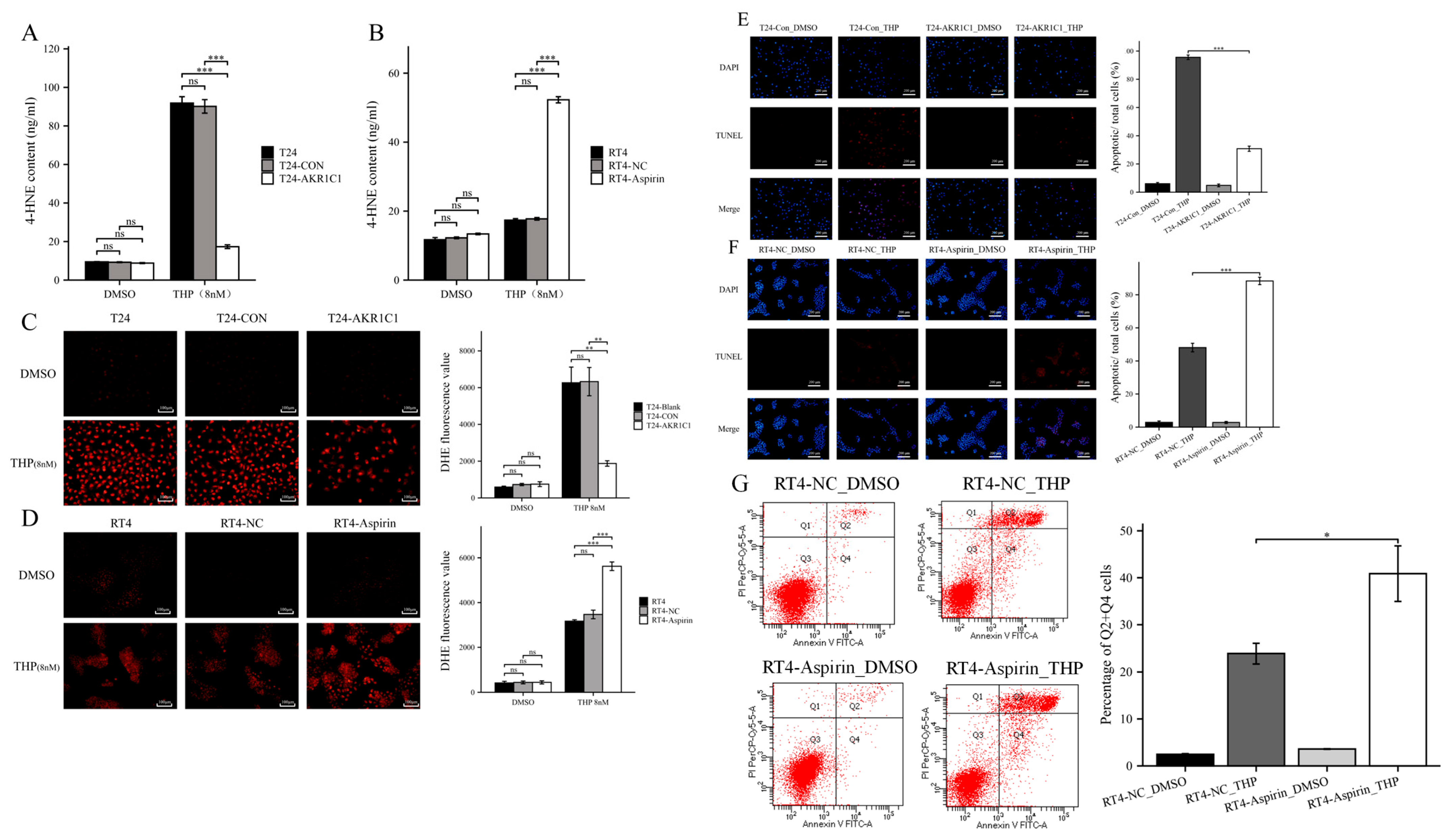
2.11. 4-Hydroxynonenal (4-HNE) Assay
4-HNE was detected by ELISA. After the culture medium was discarded and cells were washed with PBS, the cell samples tested were resuspended with lysate. The cells were broken by ultrasonic crusher on the ice 5 times, at 10 s each instance. Next, the sample was placed on ice for 30 min and then centrifuged at 12,000 rpm and 4 °C for 10 min. The supernatant was taken for testing. The experimental steps followed the 4-HNE ELISA reagent (Cusabio, Wuhan, China, E16214h) protocol. Finally, the OD450 nm absorption was detected by the enzyme marker, and the standard curve was drawn using Curve Expert v1.4 software to calculate the concentration of each sample to be measured.
2.12. ROS Assay
Intracellular ROS levels were measured using an intracellular ROS assay kit (Solarbio Science & Technology, Beijing, China, CA1420), and 0.5 × 104 cells/mL of T24 cells or 2.5 × 105 cells/mL of RT4 cells were inoculated in 6 wells plates, 2 mL/well. The cells were treated with 8 nM THP (DMSO was used in the control group) for 72 h, then washed with PBS 3 times to remove the original culture medium and treated with a 10 μM dichlorofluorescein diacetate probe for 20–30 min. The cells were washed three times with a serum-free medium, and their fluorescence intensity was measured using a fluorescence microplate reader at excitation wavelength 488 nm, emission wavelength 610 nm.
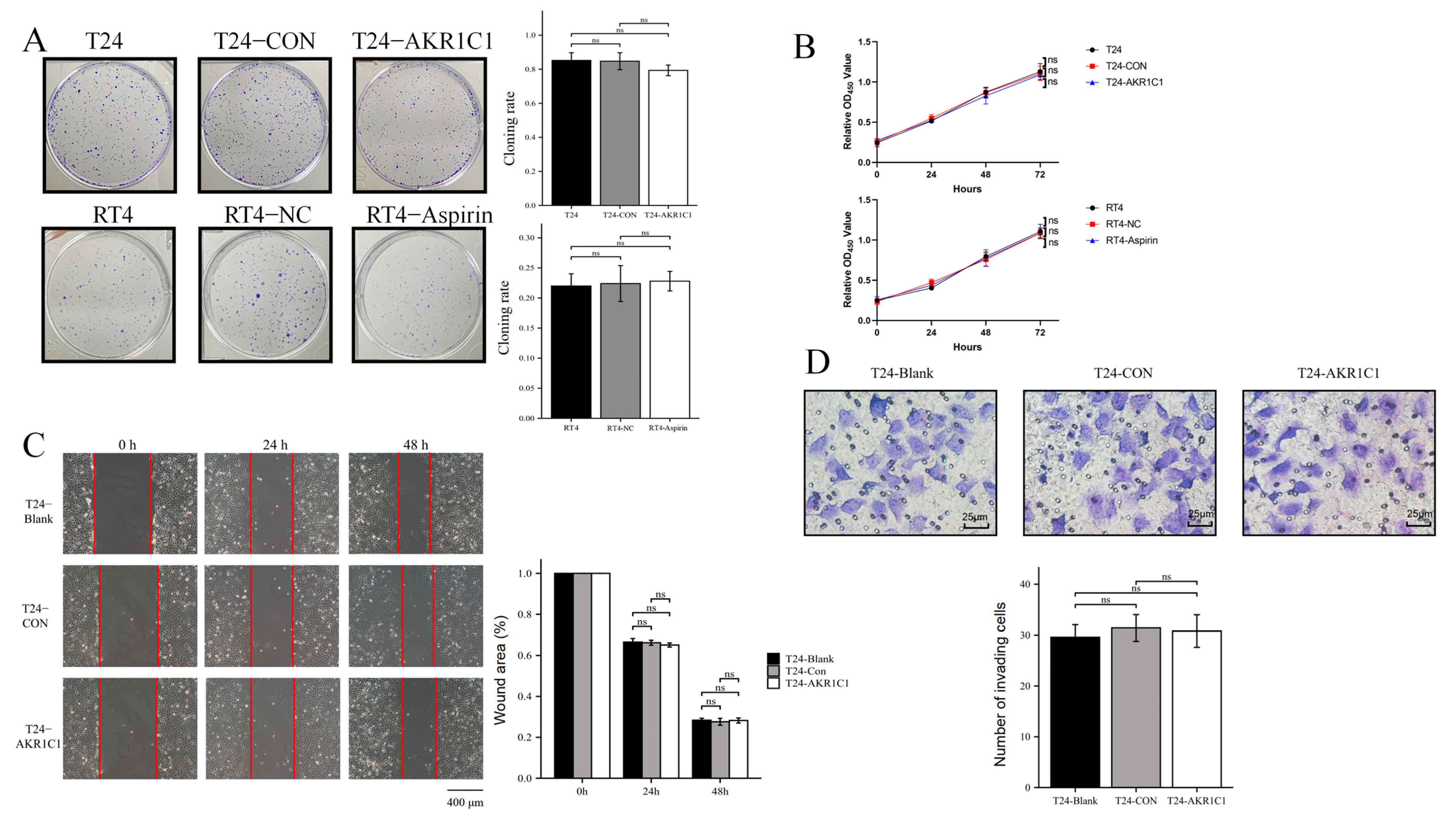
2.13. Colony Formation and Proliferation Assay
Colony formation assay: A total of 500 T24 cells or 1000 RT4 cells were resuspended in 0.2 mL of medium, evenly added to a 6-well plate containing 1.8 mL of 10% FBS medium, and cultured in a 5% CO
2 incubator at 37 °C for 2 weeks. During this period, the frequency of fluid change was reduced, changing of the fluid was avoided, or the cell culture medium was changed only once. After 2 weeks, the 6-well plate was taken out, and the cells were observed using a microscope to grow in a single colony. The culture medium was discarded, the PBS was gently washed three times, and the cells were dyed with 0.5% crystal violet for 15 min. After the colonies turned to purple, the cloning rate was calculated using Formula (2).
Cell proliferation assay: The cells in each group were resuspended with a culture medium containing 10% FBS, implanted into 96-well plate, and divided into groups of 0, 24, 48, and 72 h. After 6 h, cells showed adherent growth. At this time, the 96-well plate were taken out, and the culture medium of the 0 h group was discarded. The culture medium containing 10% CCK-8 was added and incubated for 2 h. Then, the OD450 nm absorption was detected using the enzyme marker. The above operations were repeated at 24, 48, and 72 h, and the growth curves were plotted according to the obtained values.
2.14. Wound Healing Assay
A total of 70 μL of T24 cells were inoculated at a density of 8 × 10
5 cells/mL into each insert of a culture-insert 2 well (Ibidi, Grafelfing, Germany) at the middle of a dish. After the cells were attached, the insert was removed. Incubation was continued, and the cells were removed at 0, 24, and 48 h for observation under a microscope (IX71, Olympus, Tokyo, Japan) to determine whether the peripheral cells had migrated to the central scratch area. The cells were photographed and recorded. The percentage of wound healing was analyzed using the ImageJ software and calculated as the ratio of the initial scratch area minus the partially healed area that had healed at a certain time to the initial area, using Formula (3).
2.15. Transwell Assay
The T24 cells were collected and adjusted to 5 × 104 cells/well. The cells were inoculated into the upper chamber of a Transwell plate, and the lower chamber was supplemented with 10% FBS medium. The plate was placed in the incubator for 24 h, washed twice with PBS, and fixed with methanol. Then, the cells were treated with 0.1% Giemsa staining solution, washed three times with PBS, and allowed to air dry. The number of migrated cells was recorded by photography under multiple high-magnification fields using a microscope (Etaluma, Inc., San Diego, CA, USA, LS720) and by counting the number of migrated cells.
2.16. Apoptosis Assay
Apoptosis was detected by TUNEL (Beyotime, Shanghai, China, C1090). After the original culture medium was discarded, the cells from each group were washed once with PBS. Then, the cells were fixed with an immune fixative (Beyotime, P0098) for 30 min, washed again after 30 min, added to an immune permeable solution (Beyotime, P0097), and incubated at room temperature for 5 min. The TUNEL solution was prepared according to the protocol and used to incubate cells at 37 ℃, away from light. After 1 h, the TUNEL solution was drawn, washed with PBS three times, and then restained with DAPI (Beyotime, C1002). TUNEL (red fluorescence) was detected at 550 nm, and DAPI (blue fluorescence) was detected at 360–440 nm.
Flow CytoMetry (FCM) is also used to detect apoptosis in RT4 cells. The RT4 cells were treated with different aspirin or normal saline (NC) for 24 h, then treated with THP or DMSO for 24 h, digested by using EDTA-free trypsin, collected, washed, and resuspended with PBS, and centrifuged to collect cell precipitates. The precipitates were resuspended again with a small amount of a binding buffer, mixed with an Annexin-V-FITC working solution, incubated for 5 min at room temperature under protection from light, and mixed with a PI reagent and PBS. The samples were analyzed using flow cytometry and the results were analyzed using Modfit software. Cells in the Q2 + Q4 region are generally considered apoptotic.
2.17. Statistical Analysis
SPSS 21.0 and GraphPad Prism 7.0 software were used for the statistical analyses. For the comparison between the two sets of data, the Student t-test was used for normal distribution data, and the Mann–Whitney U nonparametric test was used for non-normal distribution data. The comparison between multiple groups was conducted using one-way ANOVA. When the data met the normal distribution, the Bonferroni post hoc test was used for homogeneous variance data, and the Tamhane T2 post hoc test was used for uneven variance. The Kruskal–Wallis nonparametric test is used to compare data with a non-normal distribution among multiple groups. p < 0.05 was considered statistically significant.
4. Discussion
Chemotherapy resistance of bladder cancer is one of the most serious problems in clinical practice. The recurrence or progression of bladder cancer caused by drug resistance accounts for more than half of the total recurrence and progression. This phenomenon has become one of the important factors that restrict the prognosis and quality of life of patients with bladder cancer. THP is one of the most commonly used intravesical chemotherapy drugs for patients with NMIBC. However, THP resistance or tumor recurrence after the clinical THP treatment has been increasingly reported. CRISPR/dCas9 SAM library screening technology is an efficient, rapid, and accurate method for screening genes. At present, this technology has been widely used to screen the drug- or toxicant-resistant genes of pathogenic microorganisms and plants. However, this research method has not been widely applied in basic medicine, especially in tumor drug resistance. Its application on bladder cancer-related fields has not been reported yet. The full-text database screening method based on the CRISPR/Cas9 system has caused a technological revolution in biomedical research since it was discovered and used in basic research [
30]. This strategy has helped researchers worldwide to systematically verify the key genes and mechanisms behind a series of biochemical and molecular biological processes, including finding drug-sensitive genes [
31,
32], virus infection mechanism [
33,
34], and tumor proliferation and metastasis [
35]. Goodspeed et al. [
36] reported that MSH2 gene played a crucial role in cisplatin-mediated cell death in myometrial invasive bladder cancer. A CRISPR library screening strategy was also used in this study. These successful applications showed that CRISPR library screening will greatly promote the understanding of basic biological processes. However, given that the core principle of CRISPR/Cas9 was to knock out the target sequence, the MSH2 gene screened by Andrew et al. was actually a gene that mediated bladder cancer to be more sensitive to cisplatin, rather than a drug-resistant gene. Nevertheless, this study was the first to use the whole genome CRISPR/dCas9 SAM library in bladder cancer to find the mechanism behind pirarubicin resistance. Compared with previous studies, completely different findings were obtained by using the SAM library combined with THP to further screen the gene expression profile data of T24 bladder cancer cells. Through the screening of the CRISPR/dCas9 SAM library, 1493 genes that were positively screened were identified after THP treatment. Analysis of the difference in gene expression showed that AKR1C1 was the most obvious gene enriched on the 14th day of THP treatment. AKR1C1 gene is an aldehyde and ketone reductase that is widely distributed from lower prokaryotes to the human body and deeply involved in the metabolic regulation of various substances. In addition, AKR1C1 has been reported in various tumors that may lead to the tolerance of tumor cells to chemotherapy drugs, such as anthracycline compounds. For example, Han et al. [
37] identified that AKR1C1 was one of the genes that led to the resistance of glioblastoma to adriamycin through transcriptome sequencing. In addition, the results showed that AKR1B1, AKR1C2, and AKR1C3, which were also members of the aldosterone reductase family, were also involved in the mechanism of the resistance of glioblastoma to adriamycin. The overexpression of AKR1C1 was detected in both doxorubicin-resistant lung cancer cells [
38] and epirubicin-resistant breast cancer [
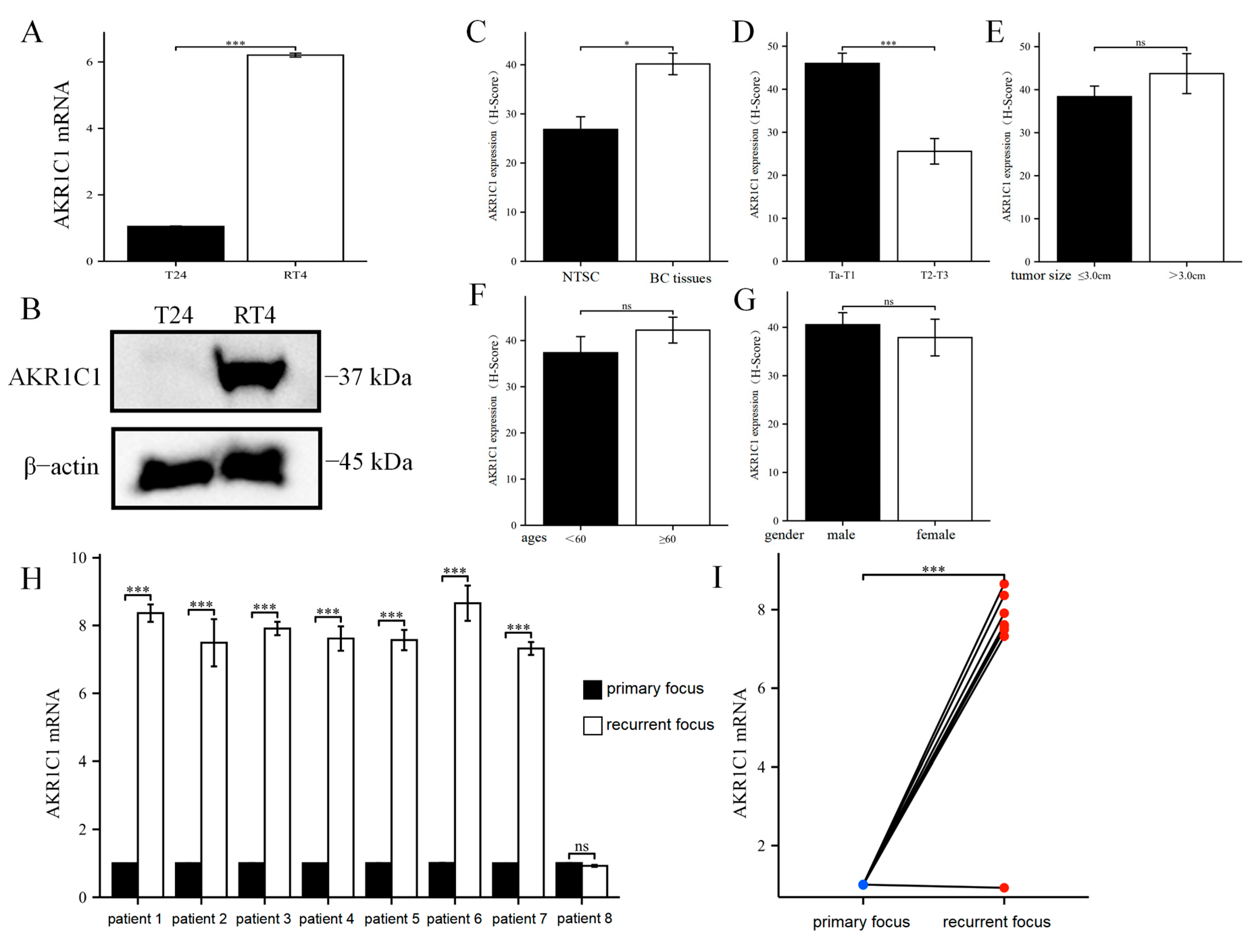
39]. These reports indicated that the proposed screening strategy for key genes of pirarubicin resistance in bladder cancer may be correct and effective. Through the study of tissue microarray and IHC methods, AKR1C1 was found to be significantly higher in the bladder cancer tissues than in adjacent tissues, which indicated that AKR1C1 was originally high in the bladder cancer tissues at the protein level. Therefore, compared with normal adjacent tissues, the high level of AKR1C1 in bladder cancer tissues was more capable of eliminating the anticancer activity of multiple drugs and gained certain advantages for the chemotherapy resistance of bladder cancer. In addition, the AKR1C1 content in the Ta/T1 bladder cancer tissue was higher than those in the T2/T3 patients. According to current clinical guidelines, only patients in Ta and T1 may receive pirarubicin single-drug infusion therapy, while patients in T2/T3 need to face a combination chemotherapy regimen based on cisplatin. Thus, patients in Ta/T1 may face more severe challenges in pirarubicin resistance. Among the eight patients with recurrent bladder cancer, in one patient, the expression of AKR1C1 in the tumor tissue at the time of recurrence was not significantly different from that at the time of initial onset. By contrast, in the remaining seven patients, the expression of AKR1C1 in the recurrent tumor tissue was significantly higher than those in the initial tumor tissue.
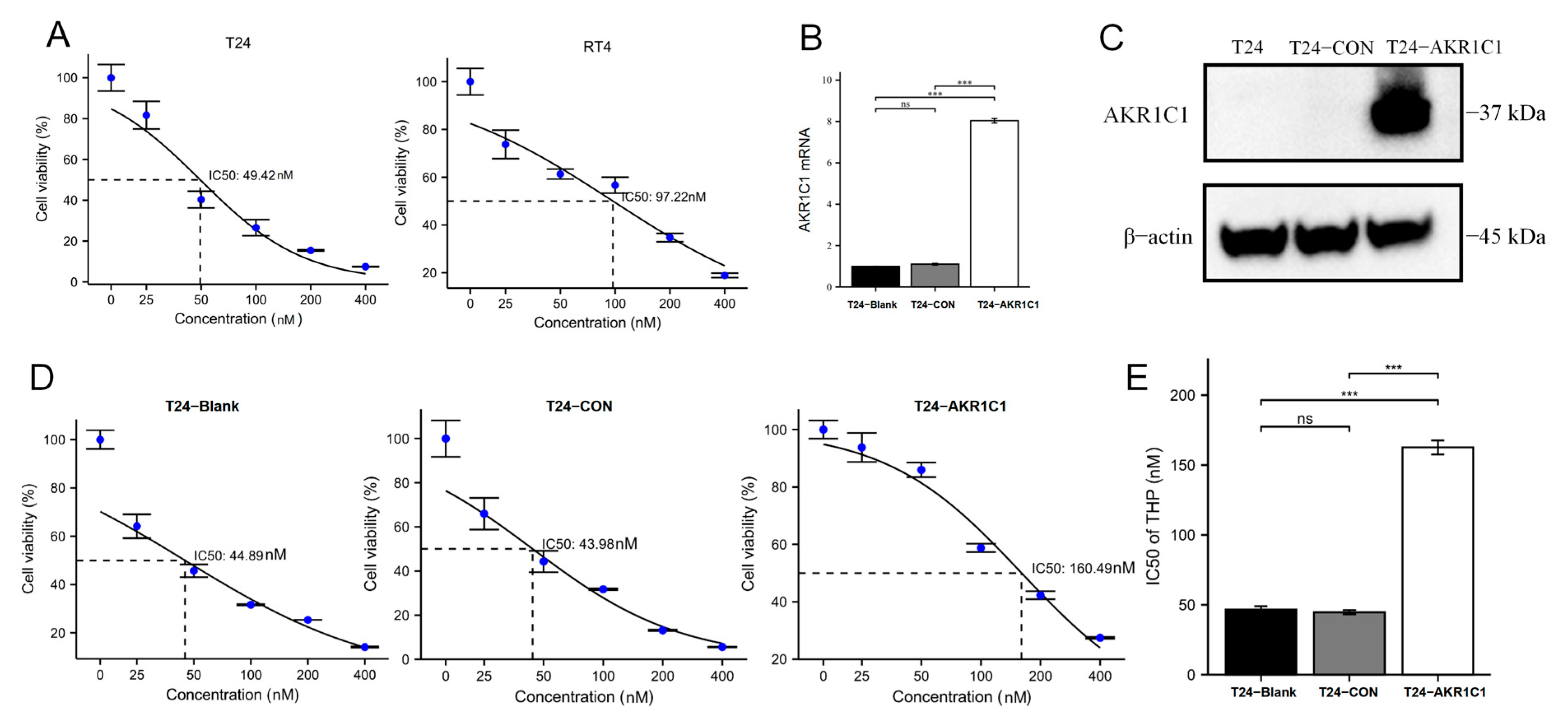
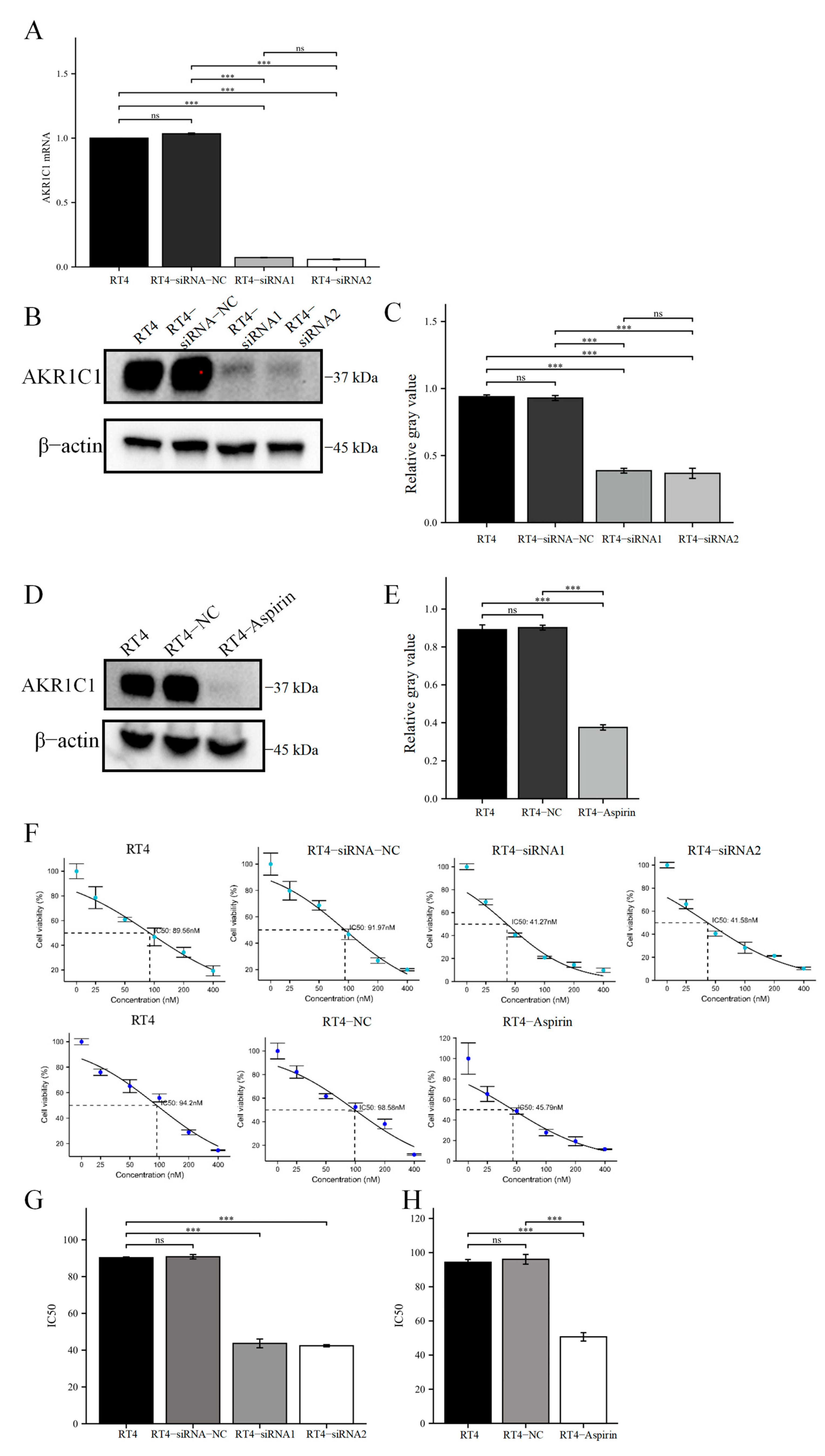
Then, the original expression level of AKR1C1 was changed in the different bladder cancer cell lines by overexpression of the lentivirus plasmid, interfering with siRNA or using inhibitors. Overexpression of AKR1C1 could increase the resistance of T24 bladder cancer cell lines to THP, while interference or inhibition of AKR1C1 could increase the sensitivity of RT4 bladder cancer cell lines to THP. However, the expression level of AKR1C1 would not affect the proliferation, invasion, or migration of bladder cancer cells. Overexpression of AKR1C1 could promote bladder cancer cell lines to resist THP-induced apoptosis, while inhibition of AKR1C1 could make bladder cancer cells more susceptible to THP treatment and apoptosis. This phenomenon may be related to the AKR1C1 reduction in 4-HNE and ROS levels. The long-term treatment of THP could upregulate the expression of AKR1C1 in T24 cells, which was related to the uncoupling of KEAP1/Nrf2 and the phosphorylation of Nrf2 caused by the increase of ROS after T24 treatment. The use of the ROS scavenger tempol, a SOD analog, can significantly save the THP-induced upregulation of AKR1C1 expression.
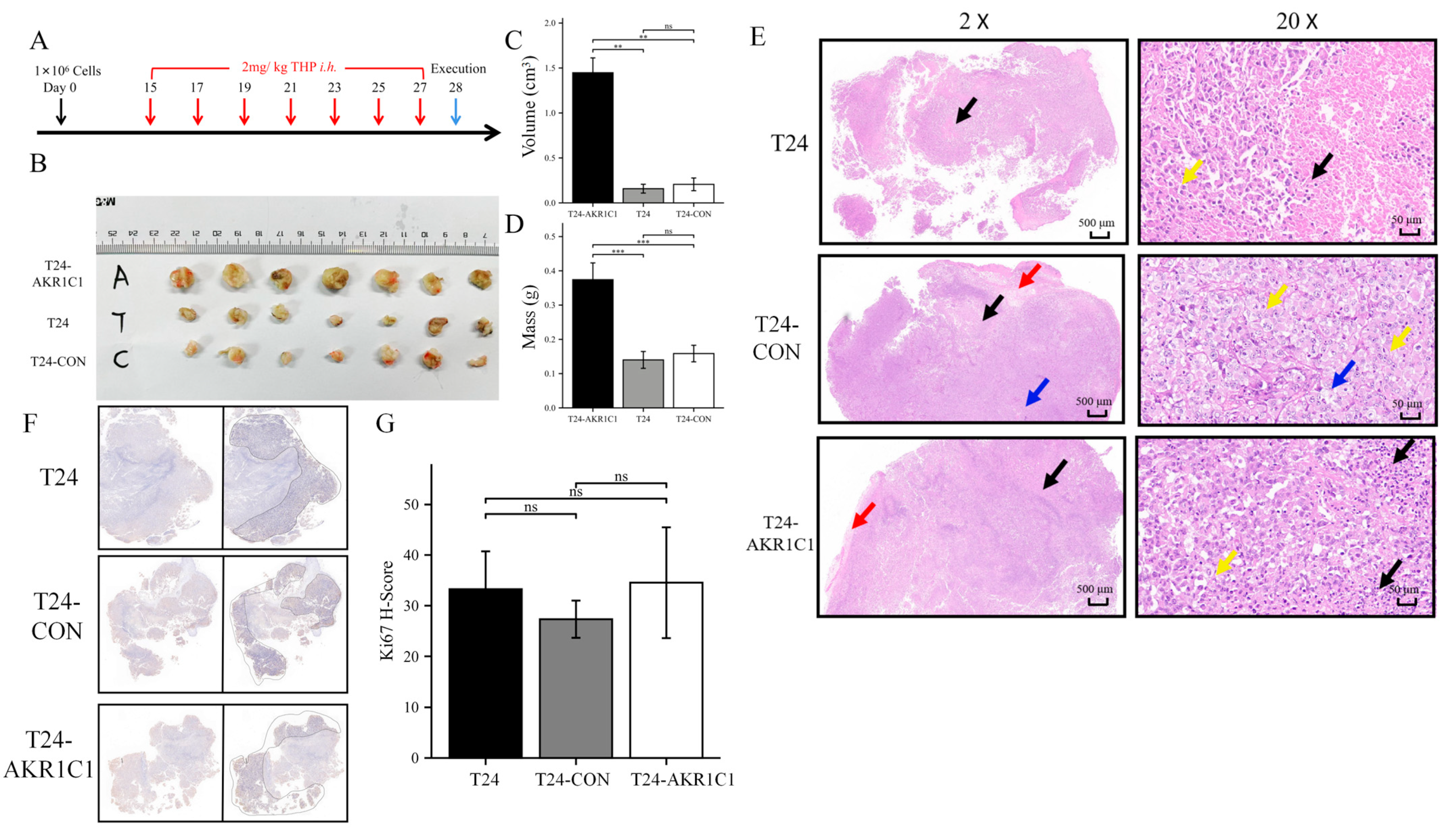
The BALB/c nude mice were randomly divided into three groups and subcutaneously transplanted with T24 bladder cancer cell line, T24-CON negative control cell line, and T24-AKR1C1 overexpression stable strain. Tumors were successfully formed and treated with THP. After 14 days of treatment, by comparing the mass and volume of tumor tissue in the three groups, the mass and volume of the tumor tissue in the AKR1C1 stable transfection group were significantly higher than those in the blank and the negative control groups. The AKR1C1 gene was shown to promote the drug resistance of tumor tissue to THP in vivo, which was similar to the results in vitro. After HE staining of the sections of the three groups of tumor tissues, the area of cell death in the AKR1C1 overexpression group was lesser than those in the blank and negative control groups after THP treatment. This difference reflected that the experimental group had better resistance to THP compared with the tumor tissue of the blank and the control groups and was more resistant to the cell death induced by THP. These results further showed that the overexpression of AKR1C1 could enhance the drug resistance of tumor tissue to THP in xenotransplantation mice in vivo. Ki67 is an important molecular marker for detecting the proliferative capacity of solid tumors [
40,
41]. The expression of Ki67 in NMIBC was significantly related to the grade, stage of tumor tissue, and the size and number of tumors [
42]. The results from the sections of tumor tissue in vitro and Ki67-IHC staining showed no statistical difference in the expression of Ki67 in the transplanted tumors of three groups of nude mice. The expression of AKR1C1 in the animal tissues did not cause a change in the Ki67 expression. Therefore, AKR1C1 would not promote the proliferation of tumor in vivo. These experimental results were similar to the conclusions in vivo.
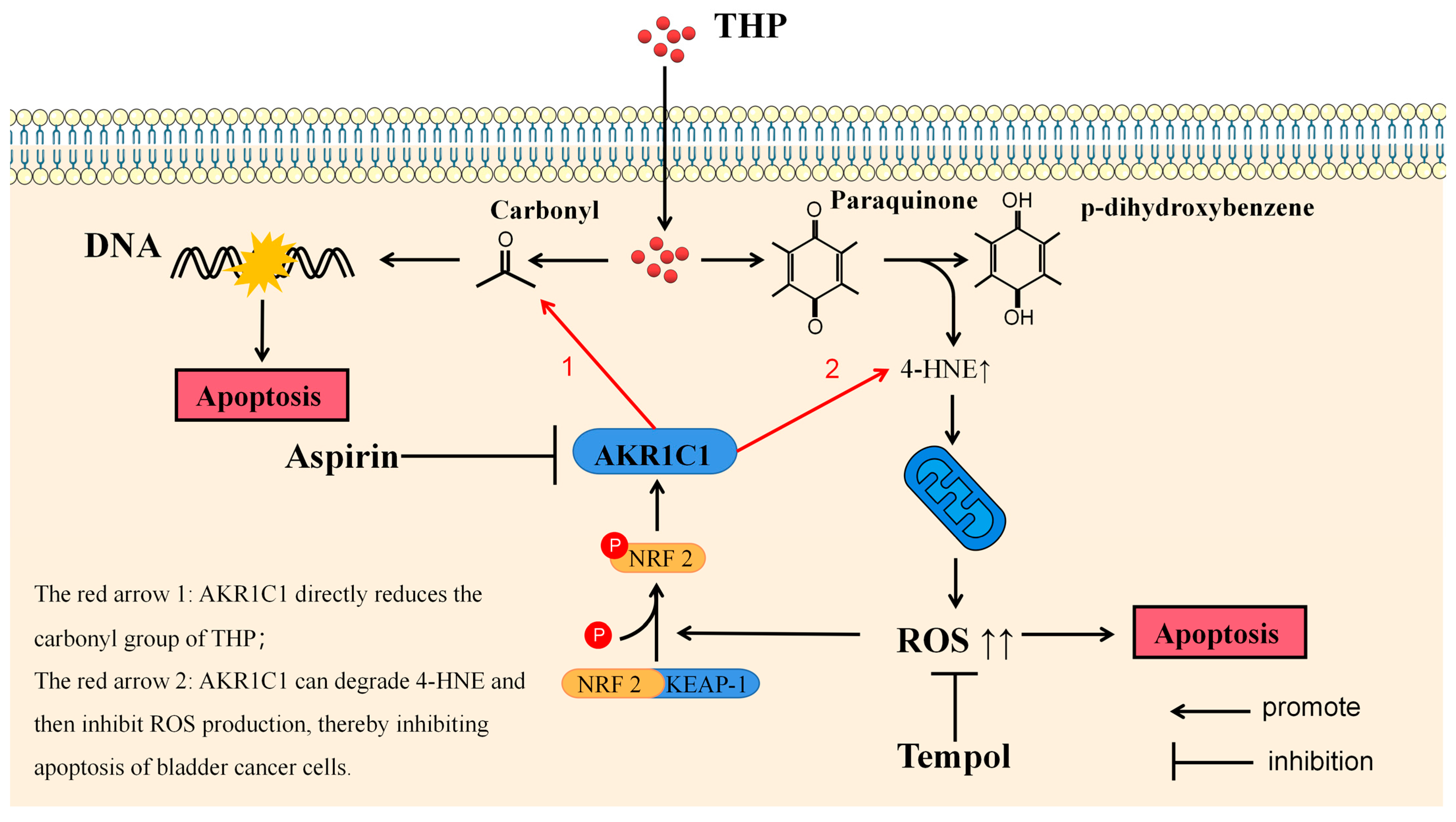
THP, as the representative drug of anthracyclines, has been proved to participate in the antitumor effect through various mechanisms. The most well-known anticancer pharmacological action of anthracycline drugs is to exert an anticancer effect by inhibiting the interference of DNA polymerase with the synthesis of DNA in cells, causing cell cycle arrest, and promoting tumor cell apoptosis. This effect depends on the carbonyl group in pirarubicin. In the final analysis, the anticancer effect of anthracycline drugs or the side effect represented by cardiotoxicity are all due to the carbonyl group in the structure [
43]. The metabolic inactivation of anthracycline drugs usually refers to their carbonyl reduction pathway. Another anticancer activity of anthracycline drugs is derived from the paraquinone (p-quinone) in the structure. P-quinone can be reduced to hydroquinone under the action of some reductases in the cell, and the surplus electrons are combined and transferred by various electrophilic groups in the cell. Finally, 4-HNE can be produced [
44]. 4-HNE is a type of aldehyde substance that can destroy the potential balance of mitochondrial inner and outer membrane and promote the increase of ROS after accumulation in tissues and cells, leading to tissue cell damage. ELISA results showed that THP could significantly increase 4-HNE and ROS after acting on T24 or RT4 cells of bladder cancer. However, if AKR1C1 is overexpressed in T24 cells, the elevated levels of 4-HNE and ROS could be significantly reduced. Thus, if the inhibitor was used in RT4 to inhibit the expression of AKR1C1, 4-HNE and ROS levels were promoted. This phenomenon was also related to the enzyme function of AKR1C1 itself, because AKR1C1 was an aldehyde and ketone reductase that could reduce aldehydes or ketones (both of which have a carbonyl). AKR1C1 could open the carbon–oxygen double bond of carbonyl, thus reducing a variety of endogenous and exogenous substances, such as 4-HNE, and protecting cells from the stress caused by these substances [
39]. Thus, AKR1C1 could destroy the structure of THP and its metabolites in two ways. The first one is the direct reduction of the carbonyl in the structure of THP, thereby destroying the function of THP insertion in DNA to inhibit DNA replication and promote cancer cell apoptosis. The second one is to reduce 4-HNE and inhibit its function of inducing ROS production to avoid cell apoptosis caused by ROS (
Figure 10). In the current study, the expression of apoptosis-related proteins among apoptosis-treated cells were detected by TUNEL and immunoblot assay. The results confirmed that high levels of AKR1C1 could inhibit the ability of THP to induce apoptosis in bladder cancer cells, thus causing bladder cancer cells to be resistant to THP. AKR1C1 inhibitor could enhance the ability of THP to induce apoptosis in bladder cancer cells, improve the anticancer ability of THP, promote tumor cell apoptosis, and reduce drug resistance of tumor cells.
This study also has some extended problems or limitations that require further study. This study was restricted by the complexity of the pathological type, grading, staging, and treatment methods of bladder cancer and the constraints of the research methods, tools, and sample size. The authors expect to increase the sample size and conduct prospective tracking analysis of the impact of AKR1C1 gene expression level on pirarubicin treatment in the future. In addition, whether AKR1C1 can be used to predict the prognosis of NMIBC or as a target for targeted treatment will be evaluated to explore the targeted treatment drugs and means for NMIBC. In this study, aspirin and tempol were used. Aspirin is a drug widely used clinically, and tempol has been used in many clinical trials to observe its antitumor effect. More in-depth research on these two drugs will be conducted in the future to find ways to prevent or reverse the drug resistance of NMIBC to pirarubicin. Due to the anatomical characteristics of the bladder and the tissue specificity of bladder cancer, the authors will build an animal model of bladder carcinoma in situ for a more accurate, objective, and comprehensive evaluation of the impact of AKR1C1 on the proliferation, invasion, and drug resistance of bladder cancer.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}