
Review of the Tumor Microenvironment in Basal and Squamous Cell Carcinoma

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Basal Cell Carcinoma
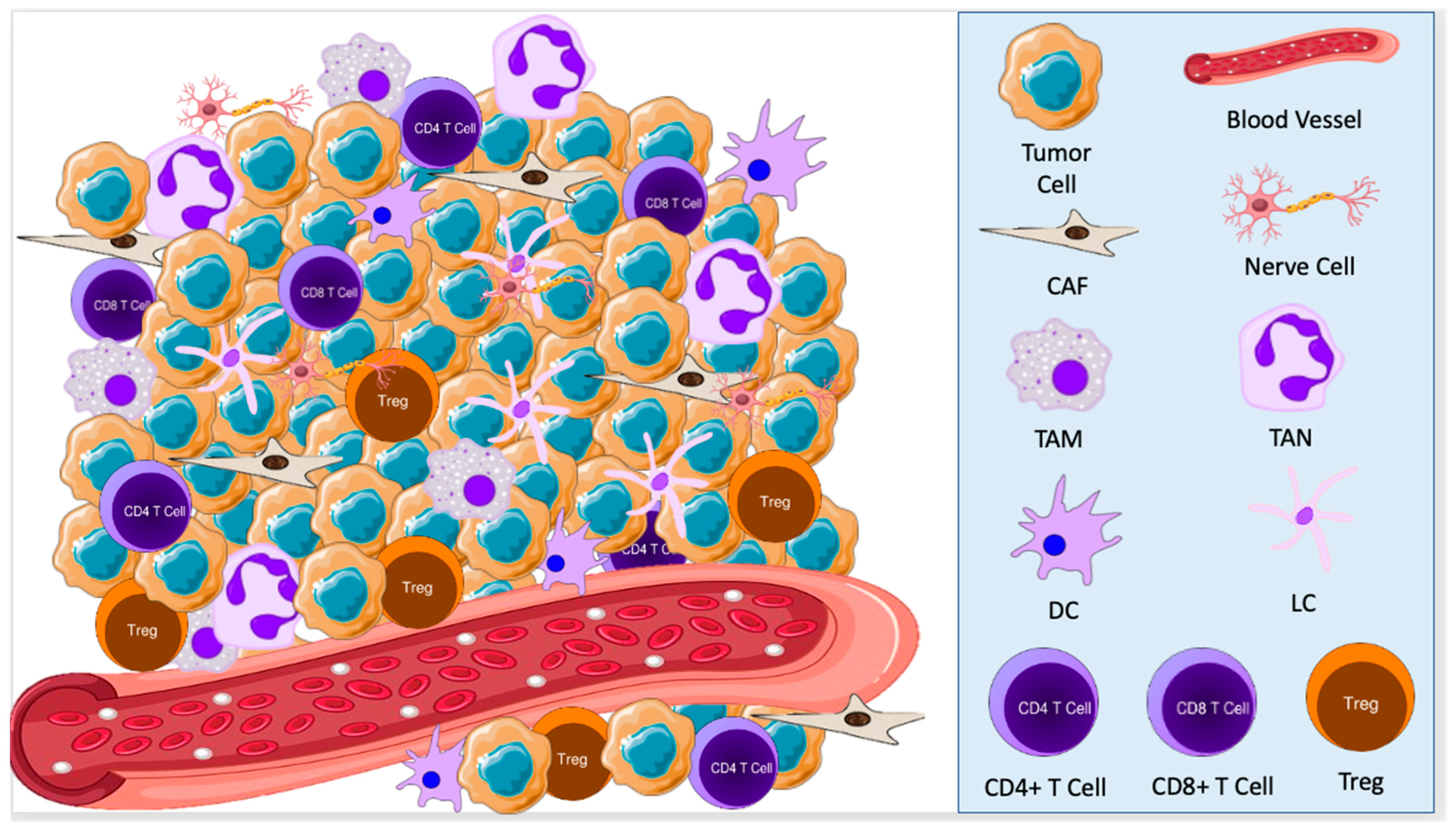
2.1. Cellular Components of the TME in BCC
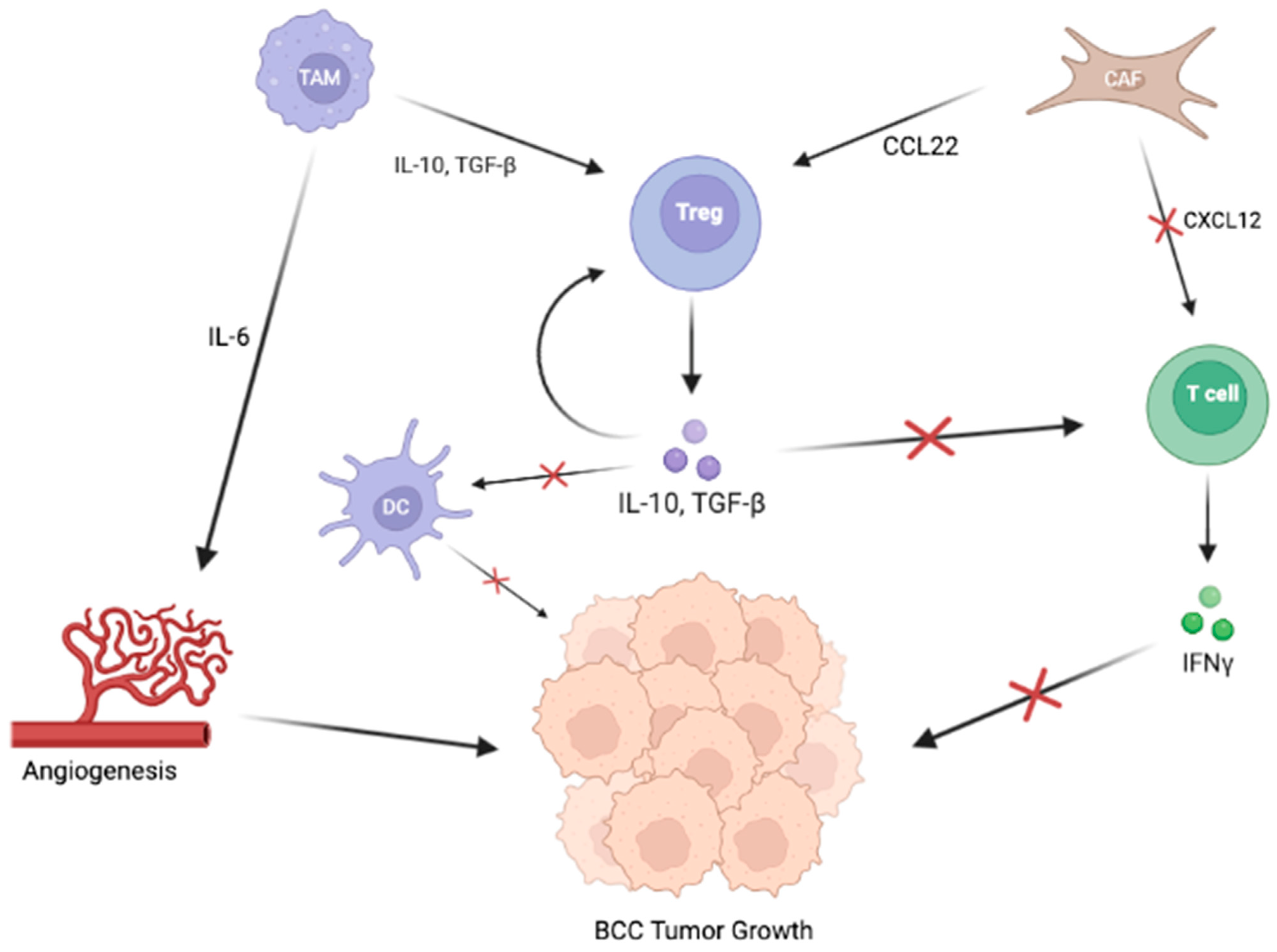
2.2. Signaling Pathways in the TME of BCC
3. Squamous Cell Carcinoma
3.1. Cellular Components of the TME in SCC
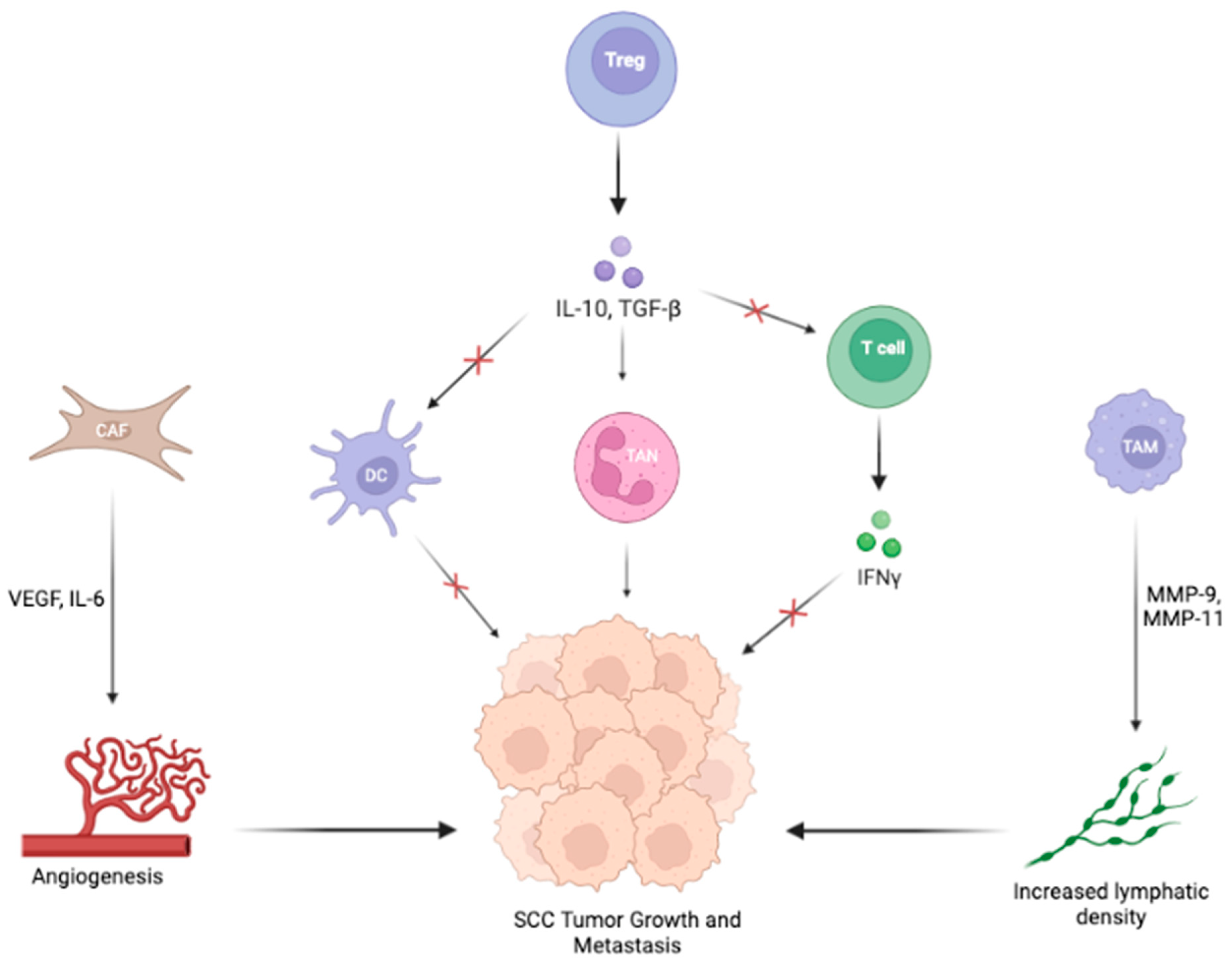
3.2. Signaling Pathways in the TME of SCC
4. Therapeutic Considerations for Skin Cancer
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gruber, P.; Zito, P.M. Skin Cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Leiter, U.; Eigentler, T.; Garbe, C. Epidemiology of Skin Cancer. In Sunlight, Vitamin D and Skin Cancer; Springer: New York, NY, USA, 2014; pp. 120–140. ISBN 978-1-4939-0436-5. [Google Scholar]
- Bashline, B. Skin Cancer: Squamous and Basal Cell Carcinomas. FP Essent 2019, 481, 17–22. [Google Scholar] [PubMed]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Capasso, A.; Viggiano, D.; Lee, M.W.; Palladino, G.; Bilancio, G.; Simeoni, M.; Capolongo, G.; Secondulfo, C.; Ronchi, A.; Caputo, A.; et al. Kidney Transplant Modifies the Architecture and Microenvironment of Basal Cell Carcinomas. Kidney Blood Press Res. 2020, 45, 368–377. [Google Scholar] [CrossRef]
- Amôr, N.G.; Santos, P.S.d.S.; Campanelli, A.P. The Tumor Microenvironment in SCC: Mechanisms and Therapeutic Opportunities. Front. Cell Dev. Biol. 2021, 9, 636544. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e22. [Google Scholar] [CrossRef]
- Amit, M.; Takahashi, H.; Dragomir, M.P.; Lindemann, A.; Gleber-Netto, F.O.; Pickering, C.R.; Anfossi, S.; Osman, A.A.; Cai, Y.; Wang, R.; et al. Loss of P53 Drives Neuron Reprogramming in Head and Neck Cancer. Nature 2020, 578, 449–454. [Google Scholar] [CrossRef]
- Moisejenko-Golubovica, J.; Volkov, O.; Ivanova, A.; Groma, V. Analysis of the Occurrence and Distribution of Primary and Recurrent Basal Cell Carcinoma of Head and Neck Coupled to the Assessment of Tumor Microenvironment and Sonic Hedgehog Signaling. Rom. J. Morphol. Embryol. 2020, 61, 821–831. [Google Scholar] [CrossRef]
- Cahoon, E.K.; Kitahara, C.M.; Ntowe, E.; Bowen, E.M.; Doody, M.M.; Alexander, B.H.; Lee, T.; Little, M.P.; Linet, M.S.; Freedman, D.M. Female Estrogen-Related Factors and Incidence of Basal Cell Carcinoma in a Nationwide US Cohort. J. Clin. Oncol. 2015, 33, 4058–4065. [Google Scholar] [CrossRef]
- Kuklinski, L.F.; Zens, M.S.; Perry, A.E.; Gossai, A.; Nelson, H.H.; Karagas, M.R. Sex Hormones and the Risk of Keratinocyte Cancers among Women in the United States: A Population-Based Case-Control Study. Int. J. Cancer 2016, 139, 300–309. [Google Scholar] [CrossRef]
- Grund-Gröschke, S.; Ortner, D.; Szenes-Nagy, A.B.; Zaborsky, N.; Weiss, R.; Neureiter, D.; Wipplinger, M.; Risch, A.; Hammerl, P.; Greil, R.; et al. Epidermal Activation of Hedgehog Signaling Establishes an Immunosuppressive Microenvironment in Basal Cell Carcinoma by Modulating Skin Immunity. Mol. Oncol. 2020, 14, 1930–1946. [Google Scholar] [CrossRef]
- Lefrançois, P.; Xie, P.; Gunn, S.; Gantchev, J.; Villarreal, A.M.; Sasseville, D.; Litvinov, I.V. In Silico Analyses of the Tumor Microenvironment Highlight Tumoral Inflammation, a Th2 Cytokine Shift and a Mesenchymal Stem Cell-like Phenotype in Advanced in Basal Cell Carcinomas. J. Cell Commun. Signal. 2020, 14, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Ressler, J.M.; Zila, N.; Korosec, A.; Yu, J.; Silmbrod, R.; Bachmayr, V.; Tittes, J.; Strobl, J.; Lichtenberger, B.M.; Hoeller, C.; et al. Myofibroblast Stroma Differentiation in Infiltrative Basal Cell Carcinoma Is Accompanied by Regulatory T-cells. J. Cutan. Pathol. 2023, cup.14381. [Google Scholar] [CrossRef]
- Beksaç, B.; İlter, N.; Erdem, Ö.; Çakmak, P.; Çenetoğlu, S.; Yapar, D. Sparsity of Dendritic Cells and Cytotoxic T Cells in Tumor Microenvironment May Lead to Recurrence in Basal Cell Carcinoma. Int. J. Dermatol. 2020, 59, 1258–1263. [Google Scholar] [CrossRef]
- Omland, S.H.; Wettergren, E.E.; Mollerup, S.; Asplund, M.; Mourier, T.; Hansen, A.J.; Gniadecki, R. Cancer Associated Fibroblasts (CAFs) Are Activated in Cutaneous Basal Cell Carcinoma and in the Peritumoural Skin. BMC Cancer 2017, 17, 675. [Google Scholar] [CrossRef] [PubMed]
- Kaporis, H.G.; Guttman-Yassky, E.; Lowes, M.A.; Haider, A.S.; Fuentes-Duculan, J.; Darabi, K.; Whynot-Ertelt, J.; Khatcherian, A.; Cardinale, I.; Novitskaya, I.; et al. Human Basal Cell Carcinoma Is Associated with Foxp3+ T Cells in a Th2 Dominant Microenvironment. J. Investig. Dermatol. 2007, 127, 2391–2398. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Juarez, C.F.; Lee, G.H.; Liu, Y.; Wang, S.; Karikomi, M.; Sha, Y.; Chow, R.Y.; Nguyen, T.T.L.; Iglesias, V.S.; Aasi, S.; et al. Single-Cell Analysis of Human Basal Cell Carcinoma Reveals Novel Regulators of Tumor Growth and the Tumor Microenvironment. Sci. Adv. 2022, 8, eabm7981. [Google Scholar] [CrossRef] [PubMed]
- Lacina, L.; Smetana, K.; Dvoránková, B.; Pytlík, R.; Kideryová, L.; Kucerová, L.; Plzáková, Z.; Stork, J.; Gabius, H.-J.; André, S. Stromal Fibroblasts from Basal Cell Carcinoma Affect Phenotype of Normal Keratinocytes. Br. J. Dermatol. 2007, 156, 819–829. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A Peek into Cancer-Associated Fibroblasts: Origins, Functions and Translational Impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Sasaki, K.; Sugai, T.; Ishida, K.; Osakabe, M.; Amano, H.; Kimura, H.; Sakuraba, M.; Kashiwa, K.; Kobayashi, S. Analysis of Cancer-Associated Fibroblasts and the Epithelial-Mesenchymal Transition in Cutaneous Basal Cell Carcinoma, Squamous Cell Carcinoma, and Malignant Melanoma. Hum. Pathol. 2018, 79, 1–8. [Google Scholar] [CrossRef]
- König, S.; Nitzki, F.; Uhmann, A.; Dittmann, K.; Theiss-Suennemann, J.; Herrmann, M.; Reichardt, H.M.; Schwendener, R.; Pukrop, T.; Schulz-Schaeffer, W.; et al. Depletion of Cutaneous Macrophages and Dendritic Cells Promotes Growth of Basal Cell Carcinoma in Mice. PLoS ONE 2014, 9, e93555. [Google Scholar] [CrossRef]
- Tjiu, J.-W.; Chen, J.-S.; Shun, C.-T.; Lin, S.-J.; Liao, Y.-H.; Chu, C.-Y.; Tsai, T.-F.; Chiu, H.-C.; Dai, Y.-S.; Inoue, H.; et al. Tumor-Associated Macrophage-Induced Invasion and Angiogenesis of Human Basal Cell Carcinoma Cells by Cyclooxygenase-2 Induction. J. Investig. Dermatol. 2009, 129, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Omland, S.H.; Nielsen, P.S.; Gjerdrum, L.M.R.; Gniadecki, R. Immunosuppressive Environment in Basal Cell Carcinoma: The Role of Regulatory T Cells. Acta Derm. Venereol. 2016, 96, 917–921. [Google Scholar] [CrossRef]
- Elamin, I.; Zecević, R.D.; Vojvodić, D.; Medenica, L.; Pavlović, M.D. Cytokine Concentrations in Basal Cell Carcinomas of Different Histological Types and Localization. Acta Dermatovenerol. Alp. Pannonica Adriat. 2008, 17, 55–59. [Google Scholar]
- Weber, F.; Byrne, S.N.; Le, S.; Brown, D.A.; Breit, S.N.; Scolyer, R.A.; Halliday, G.M. Transforming Growth Factor-Beta1 Immobilises Dendritic Cells within Skin Tumours and Facilitates Tumour Escape from the Immune System. Cancer Immunol. Immunother. 2005, 54, 898–906. [Google Scholar] [CrossRef]
- Cernadas, M.; Lu, J.; Watts, G.; Brenner, M.B. CD1a Expression Defines an Interleukin-12 Producing Population of Human Dendritic Cells. Clin. Exp. Immunol. 2009, 155, 523–533. [Google Scholar] [CrossRef]
- Dimovska Nilsson, K.; Neittaanmäki, N.; Zaar, O.; Angerer, T.B.; Paoli, J.; Fletcher, J.S. TOF-SIMS Imaging Reveals Tumor Heterogeneity and Inflammatory Response Markers in the Microenvironment of Basal Cell Carcinoma. Biointerphases 2020, 15, 041012. [Google Scholar] [CrossRef]
- Pellegrini, C.; Orlandi, A.; Costanza, G.; Di Stefani, A.; Piccioni, A.; Di Cesare, A.; Chiricozzi, A.; Ferlosio, A.; Peris, K.; Fargnoli, M.C. Expression of IL-23/Th17-Related Cytokines in Basal Cell Carcinoma and in the Response to Medical Treatments. PLoS ONE 2017, 12, e0183415. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, M.; Modlin, R.L.; Ohmen, J.D.; Moy, R.L. Local Expression of Antiinflammatory Cytokines in Cancer. J. Clin. Investig. 1993, 91, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Westin, A.T.; Gardinassi, L.G.; Soares, E.G.; Da Silva, J.S.; Donadi, E.A.; Da Silva Souza, C. HLA-G, Cytokines, and Cytokine Receptors in the Non-Aggressive Basal Cell Carcinoma Microenvironment. Arch. Dermatol. Res 2022, 314, 247–256. [Google Scholar] [CrossRef]
- Fania, L.; Didona, D.; Di Pietro, F.R.; Verkhovskaia, S.; Morese, R.; Paolino, G.; Donati, M.; Ricci, F.; Coco, V.; Ricci, F.; et al. Cutaneous Squamous Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2021, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Quadri, M.; Marconi, A.; Sandhu, S.K.; Kiss, A.; Efimova, T.; Palazzo, E. Investigating Cutaneous Squamous Cell Carcinoma in Vitro and in Vivo: Novel 3D Tools and Animal Models. Front. Med. 2022, 9, 875517. [Google Scholar] [CrossRef] [PubMed]
- Flemming, J.P.; Hill, B.L.; Haque, M.W.; Raad, J.; Bonder, C.S.; Harshyne, L.A.; Rodeck, U.; Luginbuhl, A.; Wahl, J.K.; Tsai, K.Y.; et al. MiRNA- and Cytokine-associated Extracellular Vesicles Mediate Squamous Cell Carcinomas. J. Extracell. Vesicles 2020, 9, 1790159. [Google Scholar] [CrossRef] [PubMed]
- Nappi, A.; Miro, C.; Pezone, A.; Tramontano, A.; Di Cicco, E.; Sagliocchi, S.; Cicatiello, A.G.; Murolo, M.; Torabinejad, S.; Abbotto, E.; et al. Loss of P53 Activates Thyroid Hormone via Type 2 Deiodinase and Enhances DNA Damage. Nat. Commun. 2023, 14, 1244. [Google Scholar] [CrossRef] [PubMed]
- Nappi, A.; Di Cicco, E.; Miro, C.; Cicatiello, A.G.; Sagliocchi, S.; Mancino, G.; Ambrosio, R.; Luongo, C.; Di Girolamo, D.; De Stefano, M.A.; et al. The NANOG Transcription Factor Induces Type 2 Deiodinase Expression and Regulates the Intracellular Activation of Thyroid Hormone in Keratinocyte Carcinomas. Cancers 2020, 12, 715. [Google Scholar] [CrossRef]
- Miro, C.; Nappi, A.; Cicatiello, A.G.; Di Cicco, E.; Sagliocchi, S.; Murolo, M.; Belli, V.; Troiani, T.; Albanese, S.; Amiranda, S.; et al. Thyroid Hormone Enhances Angiogenesis and the Warburg Effect in Squamous Cell Carcinomas. Cancers 2021, 13, 2743. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-KappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.; Xiao, M.; Wang, C.; Qin, Z. FSP1+ Fibroblasts Promote Skin Carcinogenesis by Maintaining MCP-1-Mediated Macrophage Infiltration and Chronic Inflammation. Am. J. Pathol. 2011, 178, 382–390. [Google Scholar] [CrossRef]
- Santana, A.L.; Felsen, D.; Carucci, J.A. Interleukin-22 and Cyclosporine in Aggressive Cutaneous Squamous Cell Carcinoma. Dermatol. Clin. 2017, 35, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, J.S.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Pierson, K.C.; Pitts-Kiefer, A.; Fan, L.; Belkin, D.A.; Wang, C.Q.F.; Bhuvanendran, S.; Johnson-Huang, L.M.; et al. Tumor-Associated Macrophages in the Cutaneous SCC Microenvironment Are Heterogeneously Activated. J. Investig. Dermatol. 2011, 131, 1322–1330. [Google Scholar] [CrossRef]
- Kambayashi, Y.; Fujimura, T.; Aiba, S. Comparison of Immunosuppressive and Immunomodulatory Cells in Keratoacanthoma and Cutaneous Squamous Cell Carcinoma. Acta Derm. Venereol. 2013, 93, 663–668. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.; Liu, Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef]
- Nasti, T.H.; Iqbal, O.; Tamimi, I.A.; Geise, J.T.; Katiyar, S.K.; Yusuf, N. Differential Roles of T-Cell Subsets in Regulation of Ultraviolet Radiation Induced Cutaneous Photocarcinogenesis. Photochem. Photobiol. 2011, 87, 387–398. [Google Scholar] [CrossRef]
- Halliday, G.M.; Patel, A.; Hunt, M.J.; Tefany, F.J.; Barnetson, R.S. Spontaneous Regression of Human Melanoma/Nonmelanoma Skin Cancer: Association with Infiltrating CD4+ T Cells. World J. Surg. 1995, 19, 352–358. [Google Scholar] [CrossRef]
- Patel, A.; Halliday, G.M.; Barnetson, R.S. CD4+ T Lymphocyte Infiltration Correlates with Regression of a UV-Induced Squamous Cell Carcinoma. J. Dermatol. Sci. 1995, 9, 12–19. [Google Scholar] [CrossRef]
- Freeman, A.; Bridge, J.A.; Maruthayanar, P.; Overgaard, N.H.; Jung, J.-W.; Simpson, F.; Prow, T.W.; Soyer, H.P.; Frazer, I.H.; Freeman, M.; et al. Comparative Immune Phenotypic Analysis of Cutaneous Squamous Cell Carcinoma and Intraepidermal Carcinoma in Immune-Competent Individuals: Proportional Representation of CD8+ T-Cells but Not FoxP3+ Regulatory T-Cells Is Associated with Disease Stage. PLoS ONE 2014, 9, e110928. [Google Scholar] [CrossRef]
- de Oliveira, C.E.; Gasparoto, T.H.; Pinheiro, C.R.; Amôr, N.G.; Nogueira, M.R.S.; Kaneno, R.; Garlet, G.P.; Lara, V.S.; Silva, J.S.; Cavassani, K.A.; et al. CCR5-Dependent Homing of T Regulatory Cells to the Tumor Microenvironment Contributes to Skin Squamous Cell Carcinoma Development. Mol. Cancer Ther. 2017, 16, 2871–2880. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of Natural Killer Cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Khou, S.; Popa, A.; Luci, C.; Bihl, F.; Meghraoui-Kheddar, A.; Bourdely, P.; Salavagione, E.; Cosson, E.; Rubod, A.; Cazareth, J.; et al. Tumor-Associated Neutrophils Dampen Adaptive Immunity and Promote Cutaneous Squamous Cell Carcinoma Development. Cancers 2020, 12, 1860. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Seddon, A.; Hock, B.; Miller, A.; Frei, L.; Pearson, J.; McKenzie, J.; Simcock, J.; Currie, M. Cutaneous Squamous Cell Carcinomas with Markers of Increased Metastatic Risk Are Associated with Elevated Numbers of Neutrophils and/or Granulocytic Myeloid Derived Suppressor Cells. J. Dermatol. Sci. 2016, 83, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Puig, S.; Berrocal, A. Management of High-Risk and Advanced Basal Cell Carcinoma. Clin. Transl. Oncol. 2015, 17, 497–503. [Google Scholar] [CrossRef]
- Work Group; Invited Reviewers; Kim, J.Y.S.; Kozlow, J.H.; Mittal, B.; Moyer, J.; Olenecki, T.; Rodgers, P. Guidelines of Care for the Management of Cutaneous Squamous Cell Carcinoma. J. Am. Acad. Dermatol. 2018, 78, 560–578. [Google Scholar] [CrossRef] [PubMed]
- Shalhout, S.Z.; Emerick, K.S.; Kaufman, H.L.; Miller, D.M. Immunotherapy for Non-Melanoma Skin Cancer. Curr. Oncol. Rep. 2021, 23, 125. [Google Scholar] [CrossRef]
- Lipson, E.J.; Lilo, M.T.; Ogurtsova, A.; Esandrio, J.; Xu, H.; Brothers, P.; Schollenberger, M.; Sharfman, W.H.; Taube, J.M. Basal Cell Carcinoma: PD-L1/PD-1 Checkpoint Expression and Tumor Regression after PD-1 Blockade. J. Immunother. Cancer 2017, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Ansary, T.M.; Hossain, M.D.R.; Komine, M.; Ohtsuki, M. Immunotherapy for the Treatment of Squamous Cell Carcinoma: Potential Benefits and Challenges. Int. J. Mol. Sci. 2022, 23, 8530. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and Safety of Vismodegib in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- Jin, H.-T.; Ahmed, R.; Okazaki, T. Role of PD-1 in Regulating T-Cell Immunity. Curr. Top. Microbiol. Immunol. 2011, 350, 17–37. [Google Scholar] [CrossRef]
- Gross, N.D.; Miller, D.M.; Khushalani, N.I.; Divi, V.; Ruiz, E.S.; Lipson, E.J.; Meier, F.; Su, Y.B.; Swiecicki, P.L.; Atlas, J.; et al. Neoadjuvant Cemiplimab for Stage II to IV Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2022, 387, 1557–1568. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Lu, Q.; Qu, Y.; Ding, Y.; Kang, X. P75NTR/ProBDNF Modulates Basal Cell Carcinoma (BCC) Immune Microenvironment via Necroptosis Signaling Pathway. J. Immunol. Res. 2021, 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.-R.; Ghezelbash, S.; Xie, P.; Fotovati, M.; Litvinov, I.V.; Lefrançois, P. Comparison of the Basal Cell Carcinoma (BCC) Tumour Microenvironment to Other Solid Malignancies. Cancers 2023, 15, 305. [Google Scholar] [CrossRef] [PubMed]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene Laherparepvec: First in Class Oncolytic Virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, E.; Stafford, H.; Buell, J.; Ramesh, U.; Amit, M.; Nagarajan, P.; Migden, M.; Yaniv, D. Review of the Tumor Microenvironment in Basal and Squamous Cell Carcinoma. Cancers 2023, 15, 2453. https://doi.org/10.3390/cancers15092453
Chiang E, Stafford H, Buell J, Ramesh U, Amit M, Nagarajan P, Migden M, Yaniv D. Review of the Tumor Microenvironment in Basal and Squamous Cell Carcinoma. Cancers. 2023; 15(9):2453. https://doi.org/10.3390/cancers15092453
Chicago/Turabian StyleChiang, Elizabeth, Haleigh Stafford, Jane Buell, Uma Ramesh, Moran Amit, Priyadharsini Nagarajan, Michael Migden, and Dan Yaniv. 2023. "Review of the Tumor Microenvironment in Basal and Squamous Cell Carcinoma" Cancers 15, no. 9: 2453. https://doi.org/10.3390/cancers15092453
APA StyleChiang, E., Stafford, H., Buell, J., Ramesh, U., Amit, M., Nagarajan, P., Migden, M., & Yaniv, D. (2023). Review of the Tumor Microenvironment in Basal and Squamous Cell Carcinoma. Cancers, 15(9), 2453. https://doi.org/10.3390/cancers15092453