

Inflammatory Networks in Renal Cell Carcinoma


 , ,
, , {kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
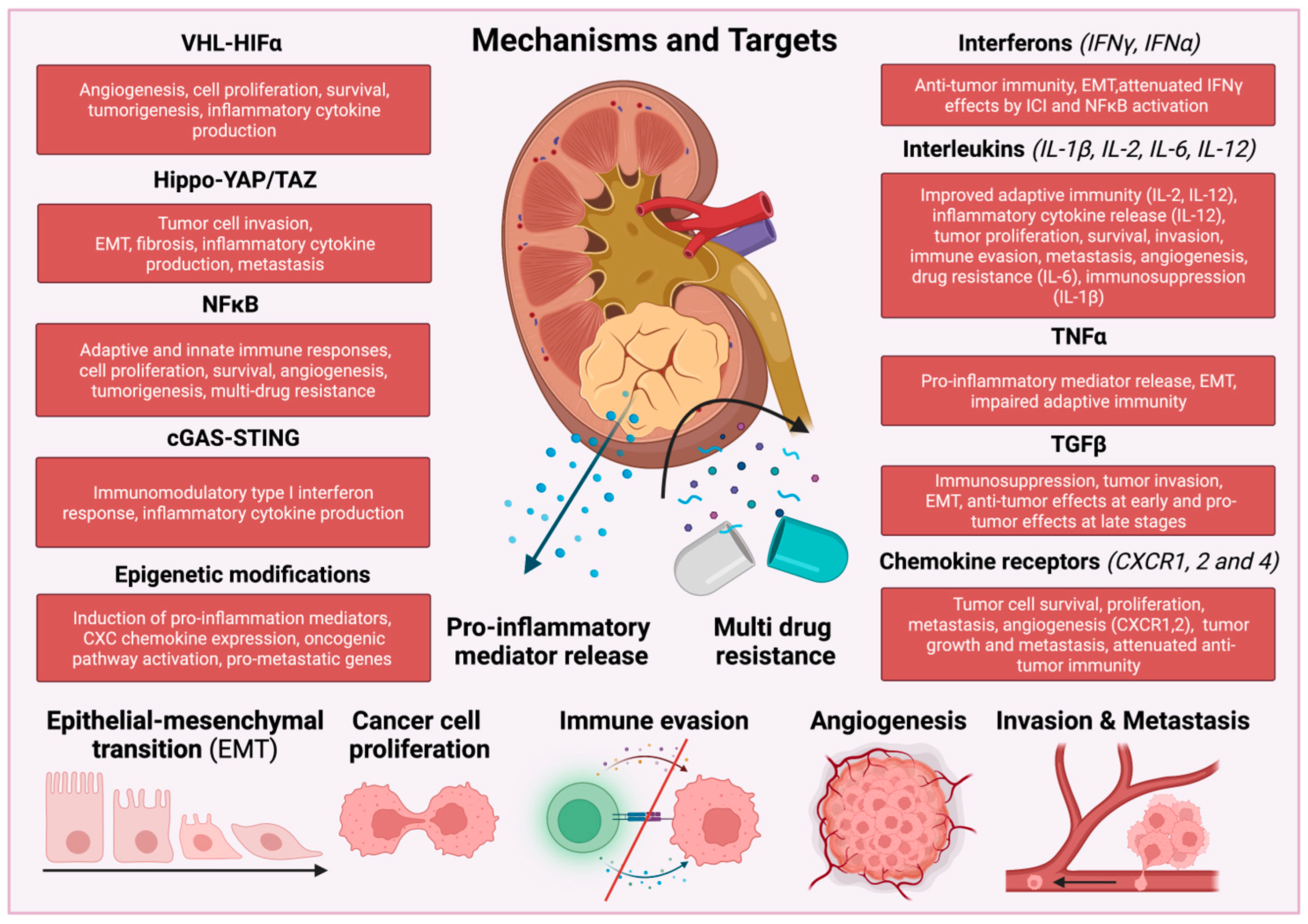
2. Inflammatory Signaling Pathways in RCC
3. vHL-HIFα
4. Hippo-YAP/TAZ
5. NF-κB Pathway
6. cGAS-STING and BAF Complex
7. Epigenetic Mechanisms in RCC
8. Inflammatory Immune Cell Landscape of RCC
9. Cytokine and Chemokine-Mediated Inflammatory Responses in RCC
9.1. IFNγ
9.2. IL-2
9.3. IL-12
9.4. IL-6/JAK/STAT3
9.5. IL-1β
9.6. TNFα
9.7. TGFβ
9.8. Chemokine Receptors
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Muglia, V.F.; Prando, A. Renal cell carcinoma: Histological classification and correlation with imaging findings. Radiol. Bras. 2015, 48, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Hansen, U.K.; Ramskov, S.; Bjerregaard, A.M.; Borch, A.; Andersen, R.; Draghi, A.; Donia, M.; Bentzen, A.K.; Marquard, A.M.; Szallasi, Z.; et al. Tumor-Infiltrating T Cells from Clear Cell Renal Cell Carcinoma Patients Recognize Neoepitopes Derived from Point and Frameshift Mutations. Front. Immunol. 2020, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Peired, A.J.; Lazzeri, E.; Guzzi, F.; Anders, H.J.; Romagnani, P. From kidney injury to kidney cancer. Kidney Int. 2021, 100, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Peired, A.J.; Antonelli, G.; Angelotti, M.L.; Allinovi, M.; Guzzi, F.; Sisti, A.; Semeraro, R.; Conte, C.; Mazzinghi, B.; Nardi, S.; et al. Acute kidney injury promotes development of papillary renal cell adenoma and carcinoma from renal progenitor cells. Sci. Transl. Med. 2020, 12, eaaw6003. [Google Scholar] [CrossRef]
- Qian, C.N.; Huang, D.; Wondergem, B.; Teh, B.T. Complexity of tumor vasculature in clear cell renal cell carcinoma. Cancer 2009, 115, 2282–2289. [Google Scholar] [CrossRef]
- Mertz, K.D.; Demichelis, F.; Kim, R.; Schraml, P.; Storz, M.; Diener, P.A.; Moch, H.; Rubin, M.A. Automated immunofluorescence analysis defines microvessel area as a prognostic parameter in clear cell renal cell cancer. Hum. Pathol. 2007, 38, 1454–1462. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Motzer, R.J. Systemic Therapy for Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda, S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta, E.; et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomised phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kadife, E.; Myers, M.; Kannourakis, G.; Prithviraj, P.; Ahmed, N. Determinants of resistance to VEGF-TKI and immune checkpoint inhibitors in metastatic renal cell carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 186. [Google Scholar] [CrossRef]
- Wu, Y.; Fu, X.; Zhu, X.; He, X.; Zou, C.; Han, Y.; Xu, M.; Huang, C.; Lu, X.; Zhao, Y. Prognostic role of systemic inflammatory response in renal cell carcinoma: A systematic review and meta-analysis. J. Cancer Res. Clin. Oncol. 2011, 137, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Koga, S.; Nishikido, M.; Noguchi, M.; Kanda, S.; Hayashi, T.; Saito, Y.; Kanetake, H. Predictive values of acute phase reactants, basic fetoprotein, and immunosuppressive acidic protein for staging and survival in renal cell carcinoma. Urology 2001, 58, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Roxburgh, C.S.; McMillan, D.C. Role of systemic inflammatory response in predicting survival in patients with primary operable cancer. Future Oncol. 2010, 6, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, S.; Lamb, G.W.; Aitchison, M.; McMillan, D.C. Prospective study of the relationship between the systemic inflammatory response, prognostic scoring systems and relapse-free and cancer-specific survival in patients undergoing potentially curative resection for renal cancer. BJU Int. 2008, 101, 959–963. [Google Scholar] [CrossRef]
- Gu, L.; Li, H.; Gao, Y.; Ma, X.; Chen, L.; Li, X.; Zhang, Y.; Fan, Y.; Zhang, X. The association of platelet count with clinicopathological significance and prognosis in renal cell carcinoma: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0125538. [Google Scholar] [CrossRef]
- Motzer, R.J.; Mazumdar, M.; Bacik, J.; Berg, W.; Amsterdam, A.; Ferrara, J. Survival and Prognostic Stratification of 670 Patients with Advanced Renal Cell Carcinoma. J. Clin. Oncol. 1999, 17, 2530. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Fox, P.; Hudson, M.; Brown, C.; Lord, S.; Gebski, V.; De Souza, P.; Lee, C.K. Markers of systemic inflammation predict survival in patients with advanced renal cell cancer. Br. J. Cancer 2013, 109, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.P.; Nayak, B.; Shanmugasundaram, K.; Friedrichs, W.; Sudarshan, S.; Eid, A.A.; DeNapoli, T.; Parekh, D.J.; Gorin, Y.; Block, K. Nox4 mediates renal cell carcinoma cell invasion through hypoxia-induced interleukin 6- and 8-production. PLoS ONE 2012, 7, e30712. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Montero, C.M.; Rini, B.I.; Finke, J.H. The immunology of renal cell carcinoma. Nat. Rev. Nephrol. 2020, 16, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Godwin, J.L.; Zibelman, M.; Plimack, E.R.; Geynisman, D.M. Immune checkpoint blockade as a novel immunotherapeutic strategy for renal cell carcinoma: A review of clinical trials. Discov Med 2014, 18, 341–350. [Google Scholar] [PubMed]
- Atkins, D.; Ferrone, S.; Schmahl, G.E.; Störkel, S.; Seliger, B. Down-regulation of HLA class I antigen processing molecules: An immune escape mechanism of renal cell carcinoma? J. Urol. 2004, 171, 885–889. [Google Scholar] [CrossRef]
- Dunker, K.; Schlaf, G.; Bukur, J.; Altermann, W.W.; Handke, D.; Seliger, B. Expression and regulation of non-classical HLA-G in renal cell carcinoma. Tissue Antigens 2008, 72, 137–148. [Google Scholar] [CrossRef]
- Seliger, B.; Atkins, D.; Bock, M.; Ritz, U.; Ferrone, S.; Huber, C.; Störkel, S. Characterization of human lymphocyte antigen class I antigen-processing machinery defects in renal cell carcinoma lesions with special emphasis on transporter-associated with antigen-processing down-regulation. Clin. Cancer Res. 2003, 9, 1721–1727. [Google Scholar]
- Wang, T.; Lu, R.; Kapur, P.; Jaiswal, B.S.; Hannan, R.; Zhang, Z.; Pedrosa, I.; Luke, J.J.; Zhang, H.; Goldstein, L.D.; et al. An Empirical Approach Leveraging Tumorgrafts to Dissect the Tumor Microenvironment in Renal Cell Carcinoma Identifies Missing Link to Prognostic Inflammatory Factors. Cancer Discov. 2018, 8, 1142–1155. [Google Scholar] [CrossRef]
- Donskov, F.; Maase, H.v.d. Impact of Immune Parameters on Long-Term Survival in Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2006, 24, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Tannir, N.M. Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat. Rev. 2018, 70, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Altmayr, F.; Jusek, G.; Holzmann, B. The neuropeptide calcitonin gene-related peptide causes repression of tumor necrosis factor-alpha transcription and suppression of ATF-2 promoter recruitment in Toll-like receptor-stimulated dendritic cells. J. Biol. Chem. 2010, 285, 3525–3531. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Xu, L.; Wang, Y.; Jiang, Q.; Liu, Z.; Zhang, J.; Zhou, Q.; Zeng, H.; Tong, S.; Wang, T.; et al. Tumor-associated Macrophage-derived Interleukin-23 Interlinks Kidney Cancer Glutamine Addiction with Immune Evasion. Eur. Urol. 2019, 75, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.W.; Ye, S.K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Kaelin, W.G., Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin. Cancer Biol. 2013, 23, 18–25. [Google Scholar] [CrossRef]
- Iliopoulos, O.; Levy, A.P.; Jiang, C.; Kaelin, W.G., Jr.; Goldberg, M.A. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10595–10599. [Google Scholar] [CrossRef]
- Labrousse-Arias, D.; Martinez-Alonso, E.; Corral-Escariz, M.; Bienes-Martinez, R.; Berridy, J.; Serrano-Oviedo, L.; Conde, E.; Garcia-Bermejo, M.L.; Gimenez-Bachs, J.M.; Salinas-Sanchez, A.S.; et al. VHL promotes immune response against renal cell carcinoma via NF-kappaB-dependent regulation of VCAM-1. J. Cell Biol. 2017, 216, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Staller, P.; Sulitkova, J.; Lisztwan, J.; Moch, H.; Oakeley, E.J.; Krek, W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature 2003, 425, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Galban, S.; Fan, J.; Martindale, J.L.; Cheadle, C.; Hoffman, B.; Woods, M.P.; Temeles, G.; Brieger, J.; Decker, J.; Gorospe, M. von Hippel-Lindau protein-mediated repression of tumor necrosis factor alpha translation revealed through use of cDNA arrays. Mol. Cell. Biol. 2003, 23, 2316–2328. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, S.; Higgins, P.J.; Samarakoon, R. Downstream Targets of VHL/HIF-alpha Signaling in Renal Clear Cell Carcinoma Progression: Mechanisms and Therapeutic Relevance. Cancers 2023, 15, 1316. [Google Scholar] [CrossRef]
- Monzon, F.A.; Alvarez, K.; Peterson, L.; Truong, L.; Amato, R.J.; Hernandez-McClain, J.; Tannir, N.; Parwani, A.V.; Jonasch, E. Chromosome 14q loss defines a molecular subtype of clear-cell renal cell carcinoma associated with poor prognosis. Mod. Pathol. 2011, 24, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Lal, P.; Dondeti, V.R.; Letrero, R.; Parekh, K.N.; Oquendo, C.E.; Greenberg, R.A.; Flaherty, K.T.; Rathmell, W.K.; Keith, B.; et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 2008, 14, 435–446. [Google Scholar] [CrossRef]
- Shen, C.; Beroukhim, R.; Schumacher, S.E.; Zhou, J.; Chang, M.; Signoretti, S.; Kaelin, W.G., Jr. Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov. 2011, 1, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kim, W.Y.; Lechpammer, M.; Kaelin, W.G., Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003, 1, e83. [Google Scholar] [CrossRef]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Cuvillier, O. The therapeutic potential of HIF-2 antagonism in renal cell carcinoma. Transl. Androl. Urol. 2017, 6, 131–133. [Google Scholar] [CrossRef]
- Hoefflin, R.; Harlander, S.; Schafer, S.; Metzger, P.; Kuo, F.; Schonenberger, D.; Adlesic, M.; Peighambari, A.; Seidel, P.; Chen, C.Y.; et al. HIF-1alpha and HIF-2alpha differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 2020, 11, 4111. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, T.L.; Bader, H.L.; Henderson, J.; Hsu, T. Conditional inactivation of the mouse von Hippel-Lindau tumor suppressor gene results in wide-spread hyperplastic, inflammatory and fibrotic lesions in the kidney. Oncogene 2015, 34, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.Y.; Lin, C.H.; Hsu, T. VHL Inactivation in Precancerous Kidney Cells Induces an Inflammatory Response via ER Stress-Activated IRE1α Signaling. Cancer Res. 2017, 77, 3406–3416. [Google Scholar] [CrossRef]
- Shan, B.; Wang, X.; Wu, Y.; Xu, C.; Xia, Z.; Dai, J.; Shao, M.; Zhao, F.; He, S.; Yang, L.; et al. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 2017, 18, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Nguyen-Tran, H.H.; Chen, C.Y.; Hsu, T. IL6 and CCL18 Mediate Cross-talk between VHL-Deficient Kidney Cells and Macrophages during Development of Renal Cell Carcinoma. Cancer Res. 2022, 82, 2716–2733. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Siemann, D.W. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers (Basel) 2020, 12, 1850. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhao, H.; Tian, L.; Nolley, R.; Diep, A.N.; Ernst, A.; Fuh, K.C.; Miao, Y.R.; von Eyben, R.; Leppert, J.T.; et al. S100A10 Is a Critical Mediator of GAS6/AXL-Induced Angiogenesis in Renal Cell Carcinoma. Cancer Res. 2019, 79, 5758–5768. [Google Scholar] [CrossRef]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Eisen, T.; Oudard, S.; Szczylik, C.; Gravis, G.; Heinzer, H.; Middleton, R.; Cihon, F.; Anderson, S.; Shah, S.; Bukowski, R.; et al. Sorafenib for older patients with renal cell carcinoma: Subset analysis from a randomized trial. J Natl Cancer Inst 2008, 100, 1454–1463. [Google Scholar] [CrossRef]
- Makhov, P.; Joshi, S.S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, X.D.; Sun, M.; Zhang, X.; German, P.; Bai, S.; Ding, Z.; Tannir, N.; Wood, C.G.; Matin, S.F.; et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.; Alekseev, B.; Rha, S.-Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Al-Salama, Z.T.; Syed, Y.Y.; Scott, L.J. Lenvatinib: A Review in Hepatocellular Carcinoma. Drugs 2019, 79, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, R.; Arduini, D.; Ciccarese, C.; Pierconti, F.; Strusi, A.; Piro, G.; Carbone, C.; Foschi, N.; Daniele, G.; Tortora, G. Targeting hypoxia-inducible factor pathways in sporadic and Von Hippel-Lindau syndrome-related kidney cancers. Crit. Rev. Oncol. Hematol. 2022, 176, 103750. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Belzutifan: First Approval. Drugs 2021, 81, 1921–1927. [Google Scholar] [CrossRef]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N. Engl. J. Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Fallah, J.; Brave, M.H.; Weinstock, C.; Mehta, G.U.; Bradford, D.; Gittleman, H.; Bloomquist, E.W.; Charlab, R.; Hamed, S.S.; Miller, C.P.; et al. FDA Approval Summary: Belzutifan for von Hippel-Lindau Disease-Associated Tumors. Clin. Cancer Res. 2022, 28, 4843–4848. [Google Scholar] [CrossRef]
- Patel, S.A.; Hirosue, S.; Rodrigues, P.; Vojtasova, E.; Richardson, E.K.; Ge, J.; Syafruddin, S.E.; Speed, A.; Papachristou, E.K.; Baker, D.; et al. The renal lineage factor PAX8 controls oncogenic signalling in kidney cancer. Nature 2022, 606, 999–1006. [Google Scholar] [CrossRef]
- Bleu, M.; Gaulis, S.; Lopes, R.; Sprouffske, K.; Apfel, V.; Holwerda, S.; Pregnolato, M.; Yildiz, U.; Cordoʹ, V.; Dost, A.F.M.; et al. PAX8 activates metabolic genes via enhancer elements in Renal Cell Carcinoma. Nat. Commun. 2019, 10, 3739. [Google Scholar] [CrossRef]
- Sun, N.; Petiwala, S.; Lu, C.; Hutti, J.E.; Hu, M.; Hu, M.; Domanus, M.H.; Mitra, D.; Addo, S.N.; Miller, C.P.; et al. VHL Synthetic Lethality Signatures Uncovered by Genotype-Specific CRISPR-Cas9 Screens. CRISPR J. 2019, 2, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Bakouny, Z.; Barbie, D.A. TBK1 Activation by VHL Loss in Renal Cell Carcinoma: A Novel HIF-Independent Vulnerability. Cancer Discov. 2020, 10, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xie, H.; Liu, X.; Potjewyd, F.; James, L.I.; Wilkerson, E.M.; Herring, L.E.; Xie, L.; Chen, X.; Cabrera, J.C.; et al. TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov. 2020, 10, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Nguyen, Q.H.; Singh, M.; Pavesic, M.W.; Nesterenko, I.; Nelson, L.J.; Liao, A.C.; Razorenova, O.V. Rho-associated kinase 1 inhibition is synthetically lethal with von Hippel-Lindau deficiency in clear cell renal cell carcinoma. Oncogene 2017, 36, 1080–1089. [Google Scholar] [CrossRef]
- Chakraborty, A.A.; Nakamura, E.; Qi, J.; Creech, A.; Jaffe, J.D.; Paulk, J.; Novak, J.S.; Nagulapalli, K.; McBrayer, S.K.; Cowley, G.S.; et al. HIF activation causes synthetic lethality between the VHL tumor suppressor and the EZH1 histone methyltransferase. Sci. Transl. Med. 2017, 9, eaal5272. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef]
- Nicholson, H.E.; Tariq, Z.; Housden, B.E.; Jennings, R.B.; Stransky, L.A.; Perrimon, N.; Signoretti, S.; Kaelin, W.G., Jr. HIF-independent synthetic lethality between CDK4/6 inhibition and VHL loss across species. Sci. Signal. 2019, 12, eaay0482. [Google Scholar] [CrossRef]
- Li, P.; Chen, T.; Kuang, P.; Liu, F.; Li, Z.; Liu, F.; Wang, Y.; Zhang, W.; Cai, X. Aurora-A/FOXO3A/SKP2 axis promotes tumor progression in clear cell renal cell carcinoma and dual-targeting Aurora-A/SKP2 shows synthetic lethality. Cell Death Dis. 2022, 13, 606. [Google Scholar] [CrossRef]
- Fu, M.; Hu, Y.; Lan, T.; Guan, K.L.; Luo, T.; Luo, M. The Hippo signalling pathway and its implications in human health and diseases. Signal Transduct. Target. Ther. 2022, 7, 376. [Google Scholar] [CrossRef]
- Xu, J.; Li, P.X.; Wu, J.; Gao, Y.J.; Yin, M.X.; Lin, Y.; Yang, M.; Chen, D.P.; Sun, H.P.; Liu, Z.B.; et al. Involvement of the Hippo pathway in regeneration and fibrogenesis after ischaemic acute kidney injury: YAP is the key effector. Clin. Sci. 2016, 130, 349–363. [Google Scholar] [CrossRef]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-beta-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [PubMed]
- McNeill, H.; Reginensi, A. Lats1/2 Regulate Yap/Taz to Control Nephron Progenitor Epithelialization and Inhibit Myofibroblast Formation. J. Am. Soc. Nephrol. 2017, 28, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Choi, H.I.; Park, J.S.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. Src-mediated crosstalk between FXR and YAP protects against renal fibrosis. FASEB J. 2019, 33, 11109–11122. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, D.; Zhang, Z.; Jin, D.; Xue, J.; Duan, L.; Zhang, Y.; Kang, X.; Lian, F. The critical role of the Hippo signaling pathway in kidney diseases. Front. Pharmacol. 2022, 13, 988175. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Liang, Y.; Zhu, X.; Wang, M.; Gui, Y.; Lu, Q.; Gu, M.; Xue, X.; Sun, X.; He, W.; et al. The signaling protein Wnt5a promotes TGFbeta1-mediated macrophage polarization and kidney fibrosis by inducing the transcriptional regulators Yap/Taz. J. Biol. Chem. 2018, 293, 19290–19302. [Google Scholar] [CrossRef]
- De Chiara, L.; Conte, C.; Semeraro, R.; Diaz-Bulnes, P.; Angelotti, M.L.; Mazzinghi, B.; Molli, A.; Antonelli, G.; Landini, S.; Melica, M.E.; et al. Tubular cell polyploidy protects from lethal acute kidney injury but promotes consequent chronic kidney disease. Nat. Commun. 2022, 13, 5805. [Google Scholar] [CrossRef]
- Chen, Y.B.; Xu, J.; Skanderup, A.J.; Dong, Y.; Brannon, A.R.; Wang, L.; Won, H.H.; Wang, P.I.; Nanjangud, G.J.; Jungbluth, A.A.; et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat. Commun. 2016, 7, 13131. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- White, S.M.; Avantaggiati, M.L.; Nemazanyy, I.; Di Poto, C.; Yang, Y.; Pende, M.; Gibney, G.T.; Ressom, H.W.; Field, J.; Atkins, M.B.; et al. YAP/TAZ Inhibition Induces Metabolic and Signaling Rewiring Resulting in Targetable Vulnerabilities in NF2-Deficient Tumor Cells. Dev. Cell 2019, 49, 425–443 e429. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; He, J.; Wang, D.L.; Cao, J.J.; Li, M.C.; Zhao, X.M.; Sheng, X.; Li, W.B.; Liu, W.J. Methylation-associated inactivation of LATS1 and its effect on demethylation or overexpression on YAP and cell biological function in human renal cell carcinoma. Int. J. Oncol. 2014, 45, 2511–2521. [Google Scholar] [CrossRef] [PubMed]
- Furth, N.; Aylon, Y. The LATS1 and LATS2 tumor suppressors: Beyond the Hippo pathway. Cell Death Differ. 2017, 24, 1488–1501. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Li, W.; Wang, G.; Shi, H.; Wang, K.; Yang, H.; Peng, B. NR1B2 suppress kidney renal clear cell carcinoma (KIRC) progression by regulation of LATS 1/2-YAP signaling. J. Exp. Clin. Cancer Res. 2019, 38, 343. [Google Scholar] [CrossRef]
- Ruan, H.; Bao, L.; Song, Z.; Wang, K.; Cao, Q.; Tong, J.; Cheng, G.; Xu, T.; Chen, X.; Liu, D.; et al. High expression of TAZ serves as a novel prognostic biomarker and drives cancer progression in renal cancer. Exp. Cell Res. 2019, 376, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Heng, C.; Zhou, Y.; Hu, Y.; Chen, S.; Wang, H.; Yang, H.; Jiang, Z.; Qian, S.; Wang, Y.; et al. Targeting mammalian serine/threonine-protein kinase 4 through Yes-associated protein/TEA domain transcription factor-mediated epithelial-mesenchymal transition ameliorates diabetic nephropathy orchestrated renal fibrosis. Metabolism 2020, 108, 154258. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Ahmad, R.; Giannico, G.A.; Zent, R.; Talmon, G.A.; Harris, R.C.; Clark, P.E.; Lokeshwar, V.; Dhawan, P.; Singh, A.B. Claudin-2 inhibits renal clear cell carcinoma progression by inhibiting YAP-activation. J. Exp. Clin. Cancer Res. 2021, 40, 77. [Google Scholar] [CrossRef] [PubMed]
- Harten, S.K.; Shukla, D.; Barod, R.; Hergovich, A.; Balda, M.S.; Matter, K.; Esteban, M.A.; Maxwell, P.H. Regulation of renal epithelial tight junctions by the von Hippel-Lindau tumor suppressor gene involves occludin and claudin 1 and is independent of E-cadherin. Mol. Biol. Cell 2009, 20, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.; Schiripo, T.A.; Haber, D.; Hariharan, I.K. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Nakada, C.; Mashio, M.; Narimatsu, T.; Yoshimoto, T.; Tanigawa, M.; Tsukamoto, Y.; Hijiya, N.; Takeuchi, I.; Nomura, T.; et al. Downregulation of SAV1 plays a role in pathogenesis of high-grade clear cell renal cell carcinoma. BMC Cancer 2011, 11, 523. [Google Scholar] [CrossRef]
- Mehra, R.; Vats, P.; Cieslik, M.; Cao, X.; Su, F.; Shukla, S.; Udager, A.M.; Wang, R.; Pan, J.; Kasaian, K.; et al. Biallelic Alteration and Dysregulation of the Hippo Pathway in Mucinous Tubular and Spindle Cell Carcinoma of the Kidney. Cancer Discov. 2016, 6, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Lv, S.; Wang, H.; Liu, C.; Zhai, Q.; Wang, S.; Cai, G.; Lu, D.; Sun, Z.; Wei, Q. Leukemia Inhibitory Factor Receptor Suppresses the Metastasis of Clear Cell Renal Cell Carcinoma Through Negative Regulation of the Yes-Associated Protein. DNA Cell Biol. 2018, 37, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Li, W.; Xu, A.; Shi, H.; Wang, K.; Yang, H.; Wang, R.; Peng, B. SH3BGRL2 inhibits growth and metastasis in clear cell renal cell carcinoma via activating hippo/TEAD1-Twist1 pathway. EBioMedicine 2020, 51, 102596. [Google Scholar] [CrossRef]
- Song, S.; Xie, M.; Scott, A.W.; Jin, J.; Ma, L.; Dong, X.; Skinner, H.D.; Johnson, R.L.; Ding, S.; Ajani, J.A. A Novel YAP1 Inhibitor Targets CSC-Enriched Radiation-Resistant Cells and Exerts Strong Antitumor Activity in Esophageal Adenocarcinoma. Mol. Cancer Ther. 2018, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Karin, M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert. Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Ohh, M. The von Hippel-Lindau tumor suppressor protein sensitizes renal cell carcinoma cells to tumor necrosis factor-induced cytotoxicity by suppressing the nuclear factor-kappaB-dependent antiapoptotic pathway. Cancer Res. 2003, 63, 7076–7080. [Google Scholar] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.L.; Yap, N.Y.; Rajandram, R.; Small, D.; Pailoor, J.; Ong, T.A.; Razack, A.H.; Wood, S.T.; Morais, C.; Gobe, G.C. Nuclear factor-kappa B subunits and their prognostic cancer-specific survival value in renal cell carcinoma patients. Pathology 2018, 50, 511–518. [Google Scholar] [CrossRef]
- Rebe, C.; Ghiringhelli, F. STAT3, a Master Regulator of Anti-Tumor Immune Response. Cancers 2019, 11, 1280. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, X.; Yang, P.; Zhang, X.; Peng, Y.; Li, D.; Yu, Y.; Wu, Y.; Wang, Y.; Zhang, J.; et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat. Commun. 2021, 12, 1394. [Google Scholar] [CrossRef] [PubMed]
- Evaristo, C.; Spranger, S.; Barnes, S.E.; Miller, M.L.; Molinero, L.L.; Locke, F.L.; Gajewski, T.F.; Alegre, M.-L. Cutting Edge: Engineering Active IKKβ in T Cells Drives Tumor Rejection. J. Immunol. 2016, 196, 2933–2938. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nag, S.A.; Zhang, R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr. Med. Chem. 2015, 22, 264–289. [Google Scholar] [CrossRef] [PubMed]
- Kondagunta, G.V.; Drucker, B.; Schwartz, L.; Bacik, J.; Marion, S.; Russo, P.; Mazumdar, M.; Motzer, R.J. Phase II trial of bortezomib for patients with advanced renal cell carcinoma. J. Clin. Oncol. 2004, 22, 3720–3725. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.D.; Jacobsen, K.M.; Li, W.; Shanker, A.; Sayers, T.J. Bortezomib Sensitizes Human Renal Cell Carcinomas to TRAIL Apoptosis through Increased Activation of Caspase-8 in the Death-Inducing Signaling Complex. Mol. Cancer Res. 2010, 8, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Erstad, D.J.; Cusack, J.C., Jr. Targeting the NF-κB pathway in cancer therapy. Surg. Oncol. Clin. N. Am. 2013, 22, 705–746. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Garbati, M.R. Inhibition of NF-κB signaling as a strategy in disease therapy. Curr. Top. Microbiol. Immunol. 2011, 349, 245–263. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Khoo, L.T.; Chen, L.Y. Role of the cGAS-STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep. 2018, 19, e46935. [Google Scholar] [CrossRef]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lin, Y.; Liu, L.-M.; Hou, Y.-L.; Qin, W.-T.; Zhang, L.; Jiang, S.-H.; Yang, Q.; Bai, Y.-R. Identification of Cytosolic DNA Sensor cGAS-STING as Immune-Related Risk Factor in Renal Carcinoma following Pan-Cancer Analysis. J. Immunol. Res. 2022, 2022, 7978042. [Google Scholar] [CrossRef] [PubMed]
- Mitrofanova, A.; Fontanella, A.; Tolerico, M.; Mallela, S.; Molina David, J.; Zuo, Y.; Boulina, M.; Kim, J.-J.; Santos, J.; Ge, M.; et al. Activation of Stimulator of IFN Genes (STING) Causes Proteinuria and Contributes to Glomerular Diseases. J. Am. Soc. Nephrol. 2022, 33, 2153–2173. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Lu, L.; Zhou, Y.; Liu, J.; Ma, H.; Fu, L.; Huang, S.; Zhang, Y.; Zhang, A.; Jia, Z. The novel STING antagonist H151 ameliorates cisplatin-induced acute kidney injury and mitochondrial dysfunction. Am. J. Physiol. -Ren. Physiol. 2021, 320, F608–F616. [Google Scholar] [CrossRef]
- Larkin, B.; Ilyukha, V.; Sorokin, M.; Buzdin, A.; Vannier, E.; Poltorak, A. Cutting edge: Activation of STING in T cells induces type I IFN responses and cell death. J. Immunol. 2017, 199, 397–402. [Google Scholar] [CrossRef]
- Alfert, A.; Moreno, N.; Kerl, K. The BAF complex in development and disease. Epigenetics Chromatin 2019, 12, 19. [Google Scholar] [CrossRef]
- Cooper, G.W.; Hong, A.L. SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers 2022, 14, 3645. [Google Scholar] [CrossRef]
- Msaouel, P.; Malouf, G.G.; Su, X.; Yao, H.; Tripathi, D.N.; Soeung, M.; Gao, J.; Rao, P.; Coarfa, C.; Creighton, C.J.; et al. Comprehensive Molecular Characterization Identifies Distinct Genomic and Immune Hallmarks of Renal Medullary Carcinoma. Cancer Cell 2020, 37, 720–734.e713. [Google Scholar] [CrossRef]
- Gao, W.; Li, W.; Xiao, T.; Liu, X.S.; Kaelin, W.G. Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL−/− clear cell renal carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 1027–1032. [Google Scholar] [CrossRef]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef]
- Burrows, A.E.; Smogorzewska, A.; Elledge, S.J. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc. Natl. Acad. Sci. USA 2010, 107, 14280–14285. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Morel, D.; Eychenne, T.; Colmet-Daage, L.; Bajrami, I.; Dorvault, N.; Garrido, M.; Meisenberg, C.; Lamb, A.; Ngo, C.; et al. PBRM1 Deficiency Confers Synthetic Lethality to DNA Repair Inhibitors in Cancer. Cancer Res. 2021, 81, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Schepisi, G.; Conteduca, V.; Casadei, C.; Gurioli, G.; Rossi, L.; Galla, V.; Cursano, M.C.; Brighi, N.; Lolli, C.; Menna, C.; et al. Potential Application of Chimeric Antigen Receptor (CAR)-T Cell Therapy in Renal Cell Tumors. Front. Oncol. 2020, 10, 565857. [Google Scholar] [CrossRef]
- Adam, P.J.; Terrett, J.A.; Steers, G.; Stockwin, L.; Loader, J.A.; Fletcher, G.C.; Lu, L.S.; Leach, B.I.; Mason, S.; Stamps, A.C.; et al. CD70 (TNFSF7) is expressed at high prevalence in renal cell carcinomas and is rapidly internalised on antibody binding. Br. J. Cancer 2006, 95, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Jilaveanu, L.B.; Sznol, J.; Aziz, S.A.; Duchen, D.; Kluger, H.M.; Camp, R.L. CD70 expression patterns in renal cell carcinoma. Hum. Pathol. 2012, 43, 1394–1399. [Google Scholar] [CrossRef]
- Pal, S.K.; Forero-Torres, A.; Thompson, J.A.; Morris, J.C.; Chhabra, S.; Hoimes, C.J.; Vogelzang, N.J.; Boyd, T.; Bergerot, P.G.; Adashek, J.J.; et al. A phase 1 trial of SGN-CD70A in patients with CD70-positive, metastatic renal cell carcinoma. Cancer 2019, 125, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.H.; Srinivasan, S.; Tan, N.; Tacheva-Grigorova, S.K.; Smith, B.; Mak, Y.S.L.; Ning, H.; Villanueva, J.; Wijewarnasuriya, D.; Lang, S.; et al. Preclinical Development and Evaluation of Allogeneic CAR T Cells Targeting CD70 for the Treatment of Renal Cell Carcinoma. Cancer Res. 2022, 82, 2610–2624. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Joosten, S.C.; Smits, K.M.; Aarts, M.J.; Melotte, V.; Koch, A.; Tjan-Heijnen, V.C.; van Engeland, M. Epigenetics in renal cell cancer: Mechanisms and clinical applications. Nat Rev Urol 2018, 15, 430–451. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, A.R.; O’Hagan, H.M. Interplay Between Inflammation and Epigenetic Changes in Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 69–117. [Google Scholar] [CrossRef]
- Karin, M.; Shalapour, S. Regulation of antitumor immunity by inflammation-induced epigenetic alterations. Cell. Mol. Immunol. 2022, 19, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Joosten, S.C.; Deckers, I.A.; Aarts, M.J.; Hoeben, A.; van Roermund, J.G.; Smits, K.M.; Melotte, V.; van Engeland, M.; Tjan-Heijnen, V.C. Prognostic DNA methylation markers for renal cell carcinoma: A systematic review. Epigenomics 2017, 9, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.R.; Latif, F. The epigenetic landscape of renal cancer. Nat. Rev. Nephrol. 2017, 13, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-H.; Haddad, A.; Wu, K.-J.; Zhao, H.-W.; Kapur, P.; Zhang, Z.-L.; Zhao, L.-Y.; Chen, Z.-H.; Zhou, Y.-Y.; Zhou, J.-C.; et al. A CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma. Nat. Commun. 2015, 6, 8699. [Google Scholar] [CrossRef]
- Ramankulov, A.; Lein, M.; Johannsen, M.; Schrader, M.; Miller, K.; Loening, S.A.; Jung, K. Serum amyloid A as indicator of distant metastases but not as early tumor marker in patients with renal cell carcinoma. Cancer Lett. 2008, 269, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Tomita, Y.; Imai, T.; Saito, T.; Katagiri, A.; Ohara-Mikami, Y.; Matsudo, T.; Takahashi, K. Significance of serum amyloid A on the prognosis in patients with renal cell carcinoma. Cancer 2001, 92, 2072–2075. [Google Scholar] [CrossRef] [PubMed]
- Nishida, J.; Momoi, Y.; Miyakuni, K.; Tamura, Y.; Takahashi, K.; Koinuma, D.; Miyazono, K.; Ehata, S. Epigenetic remodelling shapes inflammatory renal cancer and neutrophil-dependent metastasis. Nat. Cell Biol. 2020, 22, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.; Patel, S.A.; Harewood, L.; Olan, I.; Vojtasova, E.; Syafruddin, S.E.; Zaini, M.N.; Richardson, E.K.; Burge, J.; Warren, A.Y.; et al. NF-κB–Dependent Lymphoid Enhancer Co-option Promotes Renal Carcinoma Metastasis. Cancer Discov. 2018, 8, 850–865. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, B.C.; Jeong, S.H.; Jeong, C.W.; Ku, J.H.; Kwak, C.; Kim, H.H. Histone Demethylase LSD1 Regulates Kidney Cancer Progression by Modulating Androgen Receptor Activity. Int. J. Mol. Sci. 2020, 21, 6089. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, J.; Kong, W.; Huang, J.; Dong, B.; Huang, Y.; Xue, W.; Zhang, J. LSD1 inhibition suppresses the growth of clear cell renal cell carcinoma via upregulating P21 signaling. Acta Pharm. Sin. B 2019, 9, 324–334. [Google Scholar] [CrossRef]
- Ma, Y.; Zheng, Y.; Ji, Y.; Wang, X.; Ye, B. Raloxifene, identified as a novel LSD1 inhibitor, suppresses the migration of renal cell carcinoma. Future Med. Chem. 2021, 13, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.C.; Ma, Y.C.; Qin, W.P.; Ding, L.N.; Zheng, Y.C.; Zhu, Y.L.; Zhai, X.Y.; Yang, J.; Ma, C.Y.; Guan, Y.Y. Design and synthesis of tranylcypromine derivatives as novel LSD1/HDACs dual inhibitors for cancer treatment. Eur. J. Med. Chem. 2017, 140, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33, 495–511.e412. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tan, J.; Lim, K.J.; Koh, J.; Ooi, W.F.; Li, Z.; Huang, D.; Xing, M.; Chan, Y.S.; Qu, J.Z.; et al. VHL Deficiency Drives Enhancer Activation of Oncogenes in Clear Cell Renal Cell Carcinoma. Cancer Discov. 2017, 7, 1284–1305. [Google Scholar] [CrossRef] [PubMed]
- Hutt, D.M.; Roth, D.M.; Vignaud, H.; Cullin, C.; Bouchecareilh, M. The Histone Deacetylase Inhibitor, Vorinostat, Represses Hypoxia Inducible Factor 1 Alpha Expression through Translational Inhibition. PLoS ONE 2014, 9, e106224. [Google Scholar] [CrossRef]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef]
- Amin, A.; Hammers, H. The Evolving Landscape of Immunotherapy-Based Combinations for Frontline Treatment of Advanced Renal Cell Carcinoma. Front. Immunol. 2018, 9, 3120. [Google Scholar] [CrossRef] [PubMed]
- Raghubar, A.M.; Roberts, M.J.; Wood, S.; Healy, H.G.; Kassianos, A.J.; Mallett, A.J. Cellular milieu in clear cell renal cell carcinoma. Front. Oncol. 2022, 12, 943583. [Google Scholar] [CrossRef]
- Nakano, O.; Sato, M.; Naito, Y.; Suzuki, K.; Orikasa, S.; Aizawa, M.; Suzuki, Y.; Shintaku, I.; Nagura, H.; Ohtani, H. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: Clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001, 61, 5132–5136. [Google Scholar]
- Zhang, S.; Zhang, E.; Long, J.; Hu, Z.; Peng, J.; Liu, L.; Tang, F.; Li, L.; Ouyang, Y.; Zeng, Z. Immune infiltration in renal cell carcinoma. Cancer Sci. 2019, 110, 1564–1572. [Google Scholar] [CrossRef]
- Yao, J.; Xi, W.; Zhu, Y.; Wang, H.; Hu, X.; Guo, J. Checkpoint molecule PD-1-assisted CD8(+) T lymphocyte count in tumor microenvironment predicts overall survival of patients with metastatic renal cell carcinoma treated with tyrosine kinase inhibitors. Cancer Manag. Res. 2018, 10, 3419–3431. [Google Scholar] [CrossRef]
- Remark, R.; Alifano, M.; Cremer, I.; Lupo, A.; Dieu-Nosjean, M.C.; Riquet, M.; Crozet, L.; Ouakrim, H.; Goc, J.; Cazes, A.; et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: Influence of tumor origin. Clin. Cancer Res. 2013, 19, 4079–4091. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749 e718. [Google Scholar] [CrossRef] [PubMed]
- Varn, F.S.; Wang, Y.; Mullins, D.W.; Fiering, S.; Cheng, C. Systematic Pan-Cancer Analysis Reveals Immune Cell Interactions in the Tumor Microenvironment. Cancer Res. 2017, 77, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Kanazawa, T.; Kidani, Y.; Yoshida, T.; Hirata, M.; Nishida, K.; Nojima, S.; Yamamoto, Y.; Kato, T.; Hatano, K.; et al. Tumour grade significantly correlates with total dysfunction of tumour tissue-infiltrating lymphocytes in renal cell carcinoma. Sci. Rep. 2020, 10, 6220. [Google Scholar] [CrossRef] [PubMed]
- Senbabaoglu, Y.; Gejman, R.S.; Winer, A.G.; Liu, M.; Van Allen, E.M.; de Velasco, G.; Miao, D.; Ostrovnaya, I.; Drill, E.; Luna, A.; et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016, 17, 231. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Bakouny, Z.; Hirsch, L.; Flippot, R.; Van Allen, E.M.; Wu, C.J.; Choueiri, T.K. Beyond conventional immune-checkpoint inhibition - novel immunotherapies for renal cell carcinoma. Nat Rev Clin Oncol 2021, 18, 199–214. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Becht, E.; Pagès, F.; Skliris, G.; Verkarre, V.; Vano, Y.; Mejean, A.; Saint-Aubert, N.; Lacroix, L.; Natario, I.; et al. Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clin. Cancer Res. 2015, 21, 3031–3040. [Google Scholar] [CrossRef]
- Thompson, R.H.; Dong, H.; Lohse, C.M.; Leibovich, B.C.; Blute, M.L.; Cheville, J.C.; Kwon, E.D. PD-1 is expressed by tumor-infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin. Cancer Res. 2007, 13, 1757–1761. [Google Scholar] [CrossRef]
- Beuselinck, B.; Job, S.; Becht, E.; Karadimou, A.; Verkarre, V.; Couchy, G.; Giraldo, N.; Rioux-Leclercq, N.; Molinie, V.; Sibony, M.; et al. Molecular subtypes of clear cell renal cell carcinoma are associated with sunitinib response in the metastatic setting. Clin. Cancer Res. 2015, 21, 1329–1339. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-Infiltrating and Peripheral Blood T-cell Immunophenotypes Predict Early Relapse in Localized Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef] [PubMed]
- Piedra-Quintero, Z.L.; Wilson, Z.; Nava, P.; Guerau-de-Arellano, M. CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Front. Immunol. 2020, 11, 597959. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, A.; Chowdhury, N.; Haake, S.M.; Ager, C.; Wang, V.; Vlahos, L.; Guo, X.V.; Aggen, D.H.; Rathmell, W.K.; Jonasch, E.; et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 2021, 184, 2988–3005.e2916. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- Bi, K.; He, M.X.; Bakouny, Z.; Kanodia, A.; Napolitano, S.; Wu, J.; Grimaldi, G.; Braun, D.A.; Cuoco, M.S.; Mayorga, A.; et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 2021, 39, 649–661 e645. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Street, K.; Burke, K.P.; Cookmeyer, D.L.; Denize, T.; Pedersen, C.B.; Gohil, S.H.; Schindler, N.; Pomerance, L.; Hirsch, L.; et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 2021, 39, 632–648.e638. [Google Scholar] [CrossRef]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-γ: Teammate or opponent in the tumour microenvironment? Nat. Rev. Immunol. 2022, 22, 158–172. [Google Scholar] [CrossRef]
- Mo, X.; Zhang, H.; Preston, S.; Martin, K.; Zhou, B.; Vadalia, N.; Gamero, A.M.; Soboloff, J.; Tempera, I.; Zaidi, M.R. Interferon-γ Signaling in Melanocytes and Melanoma Cells Regulates Expression of CTLA-4. Cancer Res. 2018, 78, 436–450. [Google Scholar] [CrossRef]
- Hänze, J.; Wegner, M.; Noessner, E.; Hofmann, R.; Hegele, A. Co-Regulation of Immune Checkpoint PD-L1 with Interferon-Gamma Signaling is Associated with a Survival Benefit in Renal Cell Cancer. Target. Oncol. 2020, 15, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Tao, J.; Zhu, W.; Liu, W.; Anwaier, A.; Tian, X.; Su, J.; Shi, G.; Huang, H.; Wei, G.; et al. Comprehensive Multi-Omics Identification of Interferon-gamma Response Characteristics Reveals That RBCK1 Regulates the Immunosuppressive Microenvironment of Renal Cell Carcinoma. Front. Immunol. 2021, 12, 734646. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Adams, G.P. Interferon-γ-induced necrosis: An antitumor biotherapeutic perspective. J. Interferon Cytokine Res. 2013, 33, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Nogusa, S.; Thapa, R.J.; Shaller, C.; Simmons, H.; Peri, S.; Adams, G.P.; Balachandran, S. Anti-CD70 Immunocytokines for Exploitation of Interferon-γ-Induced RIP1-Dependent Necrosis in Renal Cell Carcinoma. PLoS ONE 2013, 8, e61446. [Google Scholar] [CrossRef] [PubMed]
- Tsimafeyeu, I.; Demidov, L.; Stepanova, E.; Wynn, N.; Ta, H. Overexpression of fibroblast growth factor receptors FGFR1 and FGFR2 in renal cell carcinoma. Scand. J. Urol. Nephrol. 2011, 45, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Kamiyama, H.; Ichikawa, K.; Fukushima, S.; Ozawa, Y.; Yamaguchi, S.; Goda, S.; Kimura, T.; Kodama, K.; Matsuki, M.; et al. Inhibition of FGFR Reactivates IFNγ Signaling in Tumor Cells to Enhance the Combined Antitumor Activity of Lenvatinib with Anti-PD-1 Antibodies. Cancer Res. 2022, 82, 292–306. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Matsui, J.; Matsushima, T.; Obaishi, H.; Miyazaki, K.; Nakamura, K.; Tohyama, O.; Semba, T.; Yamaguchi, A.; Hoshi, S.S.; et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc. Cell 2014, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Varo, N.; Alegre, E.; Díaz, A.; Melero, I. Immunosuppression routed via the kynurenine pathway: A biochemical and pathophysiologic approach. Adv. Clin. Chem. 2008, 45, 155–197. [Google Scholar] [CrossRef] [PubMed]
- Trott, J.F.; Kim, J.; Aboud, O.A.; Wettersten, H.; Stewart, B.; Berryhill, G.; Uzal, F.; Hovey, R.C.; Chen, C.-H.; Anderson, K.; et al. Inhibiting tryptophan metabolism enhances interferon therapy in kidney cancer. Oncotarget 2016, 7, 66540–66557. [Google Scholar] [CrossRef]
- Kim, K.; Taylor, S.L.; Ganti, S.; Guo, L.; Osier, M.V.; Weiss, R.H. Urine metabolomic analysis identifies potential biomarkers and pathogenic pathways in kidney cancer. Omics 2011, 15, 293–303. [Google Scholar] [CrossRef]
- Lo, U.G.; Bao, J.; Cen, J.; Yeh, H.C.; Luo, J.; Tan, W.; Hsieh, J.T. Interferon-induced IFIT5 promotes epithelial-to-mesenchymal transition leading to renal cancer invasion. Am. J. Clin. Exp. Urol. 2019, 7, 31–45. [Google Scholar] [PubMed]
- Au-Yeung, N.; Mandhana, R.; Horvath, C.M. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. Jakstat 2013, 2, e23931. [Google Scholar] [CrossRef] [PubMed]
- Lo, U.G.; Pong, R.-C.; Yang, D.; Gandee, L.; Hernandez, E.; Dang, A.; Lin, C.-J.; Santoyo, J.; Ma, S.; Sonavane, R.; et al. IFNγ-Induced IFIT5 Promotes Epithelial-to-Mesenchymal Transition in Prostate Cancer via miRNA Processing. Cancer Res. 2019, 79, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Charych, D.H.; Hoch, U.; Langowski, J.L.; Lee, S.R.; Addepalli, M.K.; Kirk, P.B.; Sheng, D.; Liu, X.; Sims, P.W.; VanderVeen, L.A.; et al. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clin. Cancer Res. 2016, 22, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Park, J.C.; Pyo, K.H.; Song, Y.J.; Lee, W.J.; Kim, J.; Ji, H.N.; Park, S.S.; Koh, Y.J.; Lee, K.; Cho, B.; et al. GI101, a novel triple-targeting bispecific CD80-IgG4-IL2variant fusion protein, elicits synergistic anti-tumour effects in preclinical models. Ann. Oncol. 2019, 30, v500. [Google Scholar] [CrossRef]
- Pyo, K.-H.; Koh, Y.J.; Synn, C.-B.; Kim, J.H.; Byeon, Y.; Jo, H.N.; Kim, Y.S.; Lee, W.; Kim, D.H.; Lee, S.; et al. Abstract 1826: Comprehensive preclinical study on GI-101, a novel CD80-IgG4-IL2 variant protein, as a therapeutic antibody candidate with bispecific immuno-oncology target. Cancer Res. 2021, 81, 1826. [Google Scholar] [CrossRef]
- Grohmann, U.; Belladonna, M.L.; Bianchi, R.; Orabona, C.; Ayroldi, E.; Fioretti, M.C.; Puccetti, P. IL-12 acts directly on DC to promote nuclear localization of NF-κB and primes DC for IL-12 production. Immunity 1998, 9, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Aste-Amezaga, M.; D’Andrea, A.; Kubin, M.; Trinchieri, G. Cooperation of natural killer cell stimulatory factor/interleukin-12 with other stimuli in the induction of cytokines and cytotoxic cell-associated molecules in human T and NK cells. Cell. Immunol. 1994, 156, 480–492. [Google Scholar] [CrossRef]
- Cheever, M.A. Twelve immunotherapy drugs that could cure cancers. Immunol. Rev. 2008, 222, 357–368. [Google Scholar] [CrossRef]
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef]
- Greiner, J.W.; Morillon, Y.M., 2nd; Schlom, J. NHS-IL12, a Tumor-Targeting Immunocytokine. Immunotargets Ther. 2021, 10, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Rudman, S.M.; Jameson, M.B.; McKeage, M.J.; Savage, P.; Jodrell, D.I.; Harries, M.; Acton, G.; Erlandsson, F.; Spicer, J.F. A Phase 1 Study of AS1409, a Novel Antibody-Cytokine Fusion Protein, in Patients with Malignant Melanoma or Renal Cell Carcinoma. Clin. Cancer Res. 2011, 17, 1998–2005. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, X.; Wang, J.; Gao, D.; Li, Y.; Li, H.; Chu, Y.; Zhang, Z.; Liu, H.; Jiang, G.; et al. Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat. Commun. 2017, 8, 1395. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, K.; Czarnecka, A.M.; Escudier, B.; Lian, F.; Szczylik, C. Interleukin-6 as an emerging regulator of renal cell cancer. Urol. Oncol. 2015, 33, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Horiguchi, A.; Oya, M.; Marumo, K.; Murai, M. STAT3, but not ERKs, mediates the IL-6-induced proliferation of renal cancer cells, ACHN and 769P. Kidney Int. 2002, 61, 926–938. [Google Scholar] [CrossRef]
- Chang, W.H.; Lai, A.G. An immunoevasive strategy through clinically-relevant pan-cancer genomic and transcriptomic alterations of JAK-STAT signaling components. Mol. Med. 2019, 25, 46. [Google Scholar] [CrossRef] [PubMed]
- Linker-Israeli, M.; Deans, R.J.; Wallace, D.J.; Prehn, J.; Ozeri-Chen, T.; Klinenberg, J.R. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J. Immunol. 1991, 147, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lei, C.T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Pecoits-Filho, R.; Heimburger, O.; Barany, P.; Suliman, M.; Fehrman-Ekholm, I.; Lindholm, B.; Stenvinkel, P. Associations between circulating inflammatory markers and residual renal function in CRF patients. Am. J. Kidney Dis. 2003, 41, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Dalla Vestra, M.; Mussap, M.; Gallina, P.; Bruseghin, M.; Cernigoi, A.M.; Saller, A.; Plebani, M.; Fioretto, P. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J. Am. Soc. Nephrol. 2005, 16 (Suppl. S1), S78–S82. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Masaki, T.; Hirai, T.; Doi, S.; Kuratsune, M.; Arihiro, K.; Kohno, N.; Yorioka, N. Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrol. Dial. Transplant. 2008, 23, 3418–3426. [Google Scholar] [CrossRef] [PubMed]
- Horii, Y.; Iwano, M.; Hirata, E.; Shiiki, M.; Fujii, Y.; Dohi, K.; Ishikawa, H. Role of interleukin-6 in the progression of mesangial proliferative glomerulonephritis. Kidney Int. Suppl. 1993, 39, S71–S75. [Google Scholar] [PubMed]
- Zhang, J.; Li, Y.; Shan, K.; Wang, L.; Qiu, W.; Lu, Y.; Zhao, D.; Zhu, G.; He, F.; Wang, Y. Sublytic C5b-9 induces IL-6 and TGF-beta1 production by glomerular mesangial cells in rat Thy-1 nephritis through p300-mediated C/EBPbeta acetylation. FASEB J. 2014, 28, 1511–1525. [Google Scholar] [CrossRef] [PubMed]
- Roofeh, D.; Lin, C.J.F.; Goldin, J.; Kim, G.H.; Furst, D.E.; Denton, C.P.; Huang, S.; Khanna, D.; the focuSSced Investigators. Tocilizumab Prevents Progression of Early Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2021, 73, 1301–1310. [Google Scholar] [CrossRef]
- Gomez-Guerrero, C.; Lopez-Armada, M.J.; Gonzalez, E.; Egido, J. Soluble IgA and IgG aggregates are catabolized by cultured rat mesangial cells and induce production of TNF-alpha and IL-6, and proliferation. J. Immunol. 1994, 153, 5247–5255. [Google Scholar] [CrossRef]
- Kielar, M.L.; John, R.; Bennett, M.; Richardson, J.A.; Shelton, J.M.; Chen, L.; Jeyarajah, D.R.; Zhou, X.J.; Zhou, H.; Chiquett, B.; et al. Maladaptive role of IL-6 in ischemic acute renal failure. J. Am. Soc. Nephrol. 2005, 16, 3315–3325. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Jayakumar, C.; Ramesh, G. Proximal tubule-specific overexpression of netrin-1 suppresses acute kidney injury-induced interstitial fibrosis and glomerulosclerosis through suppression of IL-6/STAT3 signaling. Am. J. Physiol. Renal Physiol. 2013, 304, F1054–F1065. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.Y.; Negrier, S.; Combaret, V.; Attali, S.; Goillot, E.; Merrouche, Y.; Mercatello, A.; Ravault, A.; Tourani, J.M.; Moskovtchenko, J.F.; et al. Serum level of interleukin 6 as a prognosis factor in metastatic renal cell carcinoma. Cancer Res. 1992, 52, 3317–3322. [Google Scholar] [PubMed]
- Ljungberg, B.; Grankvist, K.; Rasmuson, T. Serum interleukin-6 in relation to acute-phase reactants and survival in patients with renal cell carcinoma. Eur. J. Cancer 1997, 33, 1794–1798. [Google Scholar] [CrossRef] [PubMed]
- Negrier, S.; Perol, D.; Menetrier-Caux, C.; Escudier, B.; Pallardy, M.; Ravaud, A.; Douillard, J.Y.; Chevreau, C.; Lasset, C.; Blay, J.Y.; et al. Interleukin-6, interleukin-10, and vascular endothelial growth factor in metastatic renal cell carcinoma: Prognostic value of interleukin-6--from the Groupe Francais d’Immunotherapie. J. Clin. Oncol. 2004, 22, 2371–2378. [Google Scholar] [CrossRef]
- Hrab, M.; Olek-Hrab, K.; Antczak, A.; Kwias, Z.; Milecki, T. Interleukin-6 (IL-6) and C-reactive protein (CRP) concentration prior to total nephrectomy are prognostic factors in localized renal cell carcinoma (RCC). Rep. Pract. Oncol. Radiother. 2013, 18, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Gudbrandsdottir, G.; Aarstad, H.H.; Bostad, L.; Hjelle, K.M.; Aarstad, H.J.; Bruserud, O.; Tvedt, T.H.A.; Beisland, C. Serum levels of the IL-6 family of cytokines predict prognosis in renal cell carcinoma (RCC). Cancer Immunol. Immunother. 2021, 70, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Polimeno, M.; Napolitano, M.; Costantini, S.; Portella, L.; Esposito, A.; Capone, F.; Guerriero, E.; Trotta, A.; Zanotta, S.; Pucci, L.; et al. Regulatory T cells, interleukin (IL)-6, IL-8, vascular endothelial growth factor (VEGF), CXCL10, CXCL11, epidermal growth factor (EGF) and hepatocyte growth factor (HGF) as surrogate markers of host immunity in patients with renal cell carcinoma. BJU Int. 2013, 112, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Imarisio, I.; Ganini, C.; Sacchi, L.; Quaglini, S.; Giunta, V.; De Amici, M. Changes in circulating pro-angiogenic cytokines, other than VEGF, before progression to sunitinib therapy in advanced renal cell carcinoma patients. Oncology 2013, 84, 115–122. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, D.; Zong, H.; Zhu, L.; Wang, L.; Wang, X.; Zhu, X.; Song, X.; Wang, J. Growth-induced stress enhances epithelial-mesenchymal transition induced by IL-6 in clear cell renal cell carcinoma via the Akt/GSK-3beta/beta-catenin signaling pathway. Oncogenesis 2017, 6, e375. [Google Scholar] [CrossRef]
- Esteban, E.; Exposito, F.; Crespo, G.; Lambea, J.; Pinto, A.; Puente, J.; Arranz, J.A.; Redrado, M.; Rodriguez-Antona, C.; de Andrea, C.; et al. Circulating Levels of the Interferon-gamma-Regulated Chemokines CXCL10/CXCL11, IL-6 and HGF Predict Outcome in Metastatic Renal Cell Carcinoma Patients Treated with Antiangiogenic Therapy. Cancers 2021, 13, 2849. [Google Scholar] [CrossRef]
- Ishibashi, K.; Haber, T.; Breuksch, I.; Gebhard, S.; Sugino, T.; Kubo, H.; Hata, J.; Koguchi, T.; Yabe, M.; Kataoka, M.; et al. Overriding TKI resistance of renal cell carcinoma by combination therapy with IL-6 receptor blockade. Oncotarget 2017, 8, 55230–55245. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, H.; Xu, L.; An, H.; Liu, W.; Liu, Y.; Lin, Z.; Xu, J. p21-activated kinase 1 determines stem-like phenotype and sunitinib resistance via NF-kappaB/IL-6 activation in renal cell carcinoma. Cell Death Dis. 2015, 6, e1637. [Google Scholar] [CrossRef] [PubMed]
- Quan, Z.; He, Y.; Luo, C.; Xia, Y.; Zhao, Y.; Liu, N.; Wu, X. Interleukin 6 induces cell proliferation of clear cell renal cell carcinoma by suppressing hepaCAM via the STAT3-dependent up-regulation of DNMT1 or DNMT3b. Cell Signal. 2017, 32, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yang, R.; Wei, R.; Du, Y.; He, P.; Liu, X. Pan-cancer analysis reveals RIPK2 predicts prognosis and promotes immune therapy resistance via triggering cytotoxic T lymphocytes dysfunction. Mol. Med. 2022, 28, 47. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhou, B.; Qian, C.; Vasquez, A.; Kamra, M.; Chatterjee, A.; Lee, Y.J.; Yuan, X.; Ellis, L.; Di Vizio, D.; et al. Receptor-interacting protein kinase 2 (RIPK2) stabilizes c-Myc and is a therapeutic target in prostate cancer metastasis. Nat. Commun. 2022, 13, 669. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.F.; Negrier, S.; James, N.D.; Kocak, I.; Hawkins, R.; Davis, H.; Prabhakar, U.; Qin, X.; Mulders, P.; Berns, B. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. Br. J. Cancer 2010, 103, 1154–1162. [Google Scholar] [CrossRef]
- Schafer, J.A.; Kjesbo, N.K.; Gleason, P.P. Formulary review of 2 new biologic agents: Tocilizumab for rheumatoid arthritis and ustekinumab for plaque psoriasis. J. Manag. Care Pharm. 2010, 16, 402–416. [Google Scholar] [CrossRef]
- Ruperto, N.; Brunner, H.I.; Ramanan, A.V.; Horneff, G.; Cuttica, R.; Henrickson, M.; Anton, J.; Boteanu, A.L.; Penades, I.C.; Minden, K.; et al. Subcutaneous dosing regimens of tocilizumab in children with systemic or polyarticular juvenile idiopathic arthritis. Rheumatology 2021, 60, 4568–4580. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.I.; Ruperto, N.; Zuber, Z.; Cuttica, R.; Keltsev, V.; Xavier, R.M.; Burgos-Vargas, R.; Penades, I.C.; Silverman, E.D.; Espada, G.; et al. Efficacy and Safety of Tocilizumab for Polyarticular-Course Juvenile Idiopathic Arthritis in the Open-Label Two-Year Extension of a Phase III Trial. Arthritis Rheumatol. 2021, 73, 530–541. [Google Scholar] [CrossRef]
- De Benedetti, F.; Brunner, H.I.; Ruperto, N.; Kenwright, A.; Wright, S.; Calvo, I.; Cuttica, R.; Ravelli, A.; Schneider, R.; Woo, P.; et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N. Engl. J. Med. 2012, 367, 2385–2395. [Google Scholar] [CrossRef]
- Stone, J.H.; Klearman, M.; Collinson, N. Trial of Tocilizumab in Giant-Cell Arteritis. N. Engl. J. Med. 2017, 377, 1494–1495. [Google Scholar] [CrossRef] [PubMed]
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018, 23, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): A phase 2, randomised, controlled trial. Lancet 2016, 387, 2630–2640. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Goldin, J.; Kim, G.; Kuwana, M.; Allanore, Y.; Matucci-Cerinic, M.; Distler, O.; Shima, Y.; et al. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2020, 8, 963–974. [Google Scholar] [CrossRef]
- Angus, D.C.; Berry, S.; Lewis, R.J.; Al-Beidh, F.; Arabi, Y.; van Bentum-Puijk, W.; Bhimani, Z.; Bonten, M.; Broglio, K.; Brunkhorst, F.; et al. The REMAP-CAP (Randomized Embedded Multifactorial Adaptive Platform for Community-acquired Pneumonia) Study. Rationale and Design. Ann. Am. Thorac. Soc. 2020, 17, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Group, R.C. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 1637–1645. [Google Scholar] [CrossRef]
- Ishibashi, K.; Koguchi, T.; Matsuoka, K.; Onagi, A.; Tanji, R.; Takinami-Honda, R.; Hoshi, S.; Onoda, M.; Kurimura, Y.; Hata, J.; et al. Interleukin-6 induces drug resistance in renal cell carcinoma. Fukushima J. Med. Sci. 2018, 64, 103–110. [Google Scholar] [CrossRef]
- Oguro, T.; Ishibashi, K.; Sugino, T.; Hashimoto, K.; Tomita, S.; Takahashi, N.; Yanagida, T.; Haga, N.; Aikawa, K.; Suzutani, T.; et al. Humanised antihuman IL-6R antibody with interferon inhibits renal cell carcinoma cell growth in vitro and in vivo through suppressed SOCS3 expression. Eur. J. Cancer 2013, 49, 1715–1724. [Google Scholar] [CrossRef]
- Anders, H.J. Of Inflammasomes and Alarmins: IL-1beta and IL-1alpha in Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 2564–2575. [Google Scholar] [CrossRef]
- Chittezhath, M.; Dhillon, M.K.; Lim, J.Y.; Laoui, D.; Shalova, I.N.; Teo, Y.L.; Chen, J.; Kamaraj, R.; Raman, L.; Lum, J.; et al. Molecular profiling reveals a tumor-promoting phenotype of monocytes and macrophages in human cancer progression. Immunity 2014, 41, 815–829. [Google Scholar] [CrossRef]
- Krelin, Y.; Voronov, E.; Dotan, S.; Elkabets, M.; Reich, E.; Fogel, M.; Huszar, M.; Iwakura, Y.; Segal, S.; Dinarello, C.A.; et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007, 67, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Najjar, Y.G.; Rayman, P.; Jia, X.; Pavicic, P.G., Jr.; Rini, B.I.; Tannenbaum, C.; Ko, J.; Haywood, S.; Cohen, P.; Hamilton, T.; et al. Myeloid-Derived Suppressor Cell Subset Accumulation in Renal Cell Carcinoma Parenchyma Is Associated with Intratumoral Expression of IL1β, IL8, CXCL5, and Mip-1α. Clin. Cancer Res. 2017, 23, 2346–2355. [Google Scholar] [CrossRef] [PubMed]
- Aggen, D.H.; Ager, C.R.; Obradovic, A.Z.; Chowdhury, N.; Ghasemzadeh, A.; Mao, W.; Chaimowitz, M.G.; Lopez-Bujanda, Z.A.; Spina, C.S.; Hawley, J.E.; et al. Blocking IL1 Beta Promotes Tumor Regression and Remodeling of the Myeloid Compartment in a Renal Cell Carcinoma Model: Multidimensional Analyses. Clin. Cancer Res. 2021, 27, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. Landmark 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Ikemoto, S.; Narita, K.; Sugimura, K.; Wada, S.; Yasumoto, R.; Kishimoto, T.; Nakatani, T. Interleukin-6, tumour necrosis factor α and interleukin-1β in patients with renal cell carcinoma. Br. J. Cancer 2002, 86, 1396–1400. [Google Scholar] [CrossRef]
- Mikami, S.; Mizuno, R.; Kosaka, T.; Saya, H.; Oya, M.; Okada, Y. Expression of TNF-α and CD 44 is implicated in poor prognosis, cancer cell invasion, metastasis and resistance to the sunitinib treatment in clear cell renal cell carcinomas. Int. J. Cancer 2015, 136, 1504–1514. [Google Scholar] [CrossRef]
- Ho, M.-Y.; Tang, S.-J.; Chuang, M.-J.; Cha, T.-L.; Li, J.-Y.; Sun, G.-H.; Sun, K.-H. TNF-α Induces Epithelial–Mesenchymal Transition of Renal Cell Carcinoma Cells via a GSK3β-Dependent MechanismGSK-3β Inactivation Enhances Tumorigenicity of RCCs. Mol. Cancer Res. 2012, 10, 1109–1119. [Google Scholar] [CrossRef]
- Wu, S.-T.; Sun, G.-H.; Hsu, C.-Y.; Huang, C.-S.; Wu, Y.-H.; Wang, H.-H.; Sun, K.-H. Tumor necrosis factor-α induces epithelial–mesenchymal transition of renal cell carcinoma cells via a nuclear factor kappa B-independent mechanism. Exp. Biol. Med. 2011, 236, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiao, M.; Wu, K.; Li, L.; Zhu, G.; Wang, X.; He, D.; Wu, D. TNF-α induced epithelial mesenchymal transition increases stemness properties in renal cell carcinoma cells. Int. J. Clin. Exp. Med. 2014, 7, 4951. [Google Scholar] [PubMed]
- Chuang, M.J.; Sun, K.H.; Tang, S.J.; Deng, M.W.; Wu, Y.H.; Sung, J.S.; Cha, T.L.; Sun, G.H. Tumor-derived tumor necrosis factor-alpha promotes progression and epithelial-mesenchymal transition in renal cell carcinoma cells. Cancer Sci. 2008, 99, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.H.; Sun, G.H.; Wu, Y.C.; Ko, B.J.; Hsu, H.T.; Wu, S.T. TNF-α augments CXCR2 and CXCR3 to promote progression of renal cell carcinoma. J. Cell. Mol. Med. 2016, 20, 2020–2028. [Google Scholar] [CrossRef]
- Zhong, M.; Zhu, M.; Liu, Y.; Lin, Y.; Wang, L.; Ye, Y.; Chen, H.; Yang, Y.; Zhuang, G.; Huang, J. TNFAIP8 promotes the migration of clear cell renal cell carcinoma by regulating the EMT. J. Cancer 2020, 11, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gracia, J.L.; Prior, C.; Guillén-Grima, F.; Segura, V.; Gonzalez, A.; Panizo, A.; Melero, I.; Grande-Pulido, E.; Gurpide, A.; Gil-Bazo, I.; et al. Identification of TNF-alpha and MMP-9 as potential baseline predictive serum markers of sunitinib activity in patients with renal cell carcinoma using a human cytokine array. Br. J. Cancer 2009, 101, 1876–1883. [Google Scholar] [CrossRef]
- Raval, G.; Biswas, S.; Rayman, P.; Biswas, K.; Sa, G.; Ghosh, S.; Thornton, M.; Hilston, C.; Das, T.; Bukowski, R.; et al. TNF-α Induction of GM2 Expression on Renal Cell Carcinomas Promotes T Cell Dysfunction1. J. Immunol. 2007, 178, 6642–6652. [Google Scholar] [CrossRef]
- Hillman, G.G.; Puri, R.K.; Kukuruga, M.A.; Pontes, J.E.; Haas, G.P. Growth and major histocompatibility antigen expression regulation by IL-4, interferon-gamma (IFN-gamma) and tumour necrosis factor-alpha (TNF-alpha) on human renal cell carcinoma. Clin. Exp. Immunol. 1994, 96, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Falkensammer, C.; Jöhrer, K.; Gander, H.; Ramoner, R.; Putz, T.; Rahm, A.; Greil, R.; Bartsch, G.; Thurnher, M. IL-4 inhibits the TNF-alpha induced proliferation of renal cell carcinoma (RCC) and cooperates with TNF-alpha to induce apoptotic and cytokine responses by RCC: Implications for antitumor immune responses. Cancer Immunol. Immunother. 2006, 55, 1228–1237. [Google Scholar] [CrossRef]
- Bauer, S.; Oosterwijk-Wakka, J.C.; Adrian, N.; Oosterwijk, E.; Fischer, E.; Wüest, T.; Stenner, F.; Perani, A.; Cohen, L.S.; Knuth, A.K.; et al. Targeted therapy of renal cell carcinoma: Synergistic activity of cG250-TNF and IFNg. Int. J. Cancer 2009, 125, 115–123. [Google Scholar] [CrossRef]
- Harrison, M.L.; Obermueller, E.; Maisey, N.R.; Hoare, S.; Edmonds, K.; Li, N.F.; Chao, D.; Hall, K.; Lee, C.; Timotheadou, E.; et al. Tumor necrosis factor alpha as a new target for renal cell carcinoma: Two sequential phase II trials of infliximab at standard and high dose. J. Clin. Oncol. 2007, 25, 4542–4549. [Google Scholar] [CrossRef]
- Larkin, J.M.; Ferguson, T.R.; Pickering, L.M.; Edmonds, K.; James, M.G.; Thomas, K.; Banerji, U.; Berns, B.; de Boer, C.; Gore, M.E. A phase I/II trial of sorafenib and infliximab in advanced renal cell carcinoma. Br. J. Cancer 2010, 103, 1149–1153. [Google Scholar] [CrossRef]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta 2008, 1782, 197–228. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. The regulation of TGFbeta signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef]
- Bostrom, A.K.; Lindgren, D.; Johansson, M.E.; Axelson, H. Effects of TGF-beta signaling in clear cell renal cell carcinoma cells. Biochem. Biophys. Res. Commun. 2013, 435, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Y.; Liu, X.S.; Huang, X.R.; Yu, X.Q.; Lan, H.Y. Diverse Role of TGF-beta in Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Takemura, T.; Murakami, K.; Okada, M.; Hino, S.; Miyamoto, H.; Maki, S. Transforming growth factor-beta protein and mRNA in glomeruli in normal and diseased human kidneys. Lab. Investig. 1993, 68, 154–163. [Google Scholar]
- Yamamoto, T.; Watanabe, T.; Ikegaya, N.; Fujigaki, Y.; Matsui, K.; Masaoka, H.; Nagase, M.; Hishida, A. Expression of types I, II, and III TGF-beta receptors in human glomerulonephritis. J. Am. Soc. Nephrol. 1998, 9, 2253–2261. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakamura, T.; Noble, N.A.; Ruoslahti, E.; Border, W.A. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1993, 90, 1814–1818. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Ziyadeh, F.N.; Alzahabi, B.; McGowan, T.A.; Kapoor, S.; Kurnik, B.R.; Kurnik, P.B.; Weisberg, L.S. Increased renal production of transforming growth factor-beta1 in patients with type II diabetes. Diabetes 1997, 46, 854–859. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Languino, L.R.; Ruoslahti, E.; Border, W.A. Elevated expression of transforming growth factor-beta and proteoglycan production in experimental glomerulonephritis. Possible role in expansion of the mesangial extracellular matrix. J. Clin. Investig. 1990, 86, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y. Targeting TGF-beta Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [PubMed]
- Tomooka, S.; Border, W.A.; Marshall, B.C.; Noble, N.A. Glomerular matrix accumulation is linked to inhibition of the plasmin protease system. Kidney Int. 1992, 42, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Huang, X.R.; Xiao, J.; Chen, H.Y.; Zhong, X.; Chung, A.C.; Lan, H.Y. Diverse roles of TGF-beta receptor II in renal fibrosis and inflammation in vivo and in vitro. J. Pathol. 2012, 227, 175–188. [Google Scholar] [CrossRef]
- Hegele, A.; Varga, Z.; von Knobloch, R.; Heidenreich, A.; Kropf, J.; Hofmann, R. TGF-beta1 in patients with renal cell carcinoma. Urol. Res. 2002, 30, 126–129. [Google Scholar] [CrossRef]
- Mitropoulos, D.; Kiroudi, A.; Christelli, E.; Serafetinidis, E.; Zervas, A.; Anastasiou, I.; Dimopoulos, C. Expression of transforming growth factor beta in renal cell carcinoma and matched non-involved renal tissue. Urol. Res. 2004, 32, 317–322. [Google Scholar] [CrossRef]
- Sitaram, R.T.; Mallikarjuna, P.; Landstrom, M.; Ljungberg, B. Transforming growth factor-beta promotes aggressiveness and invasion of clear cell renal cell carcinoma. Oncotarget 2016, 7, 35917–35931. [Google Scholar] [CrossRef] [PubMed]
- Copland, J.A.; Luxon, B.A.; Ajani, L.; Maity, T.; Campagnaro, E.; Guo, H.; LeGrand, S.N.; Tamboli, P.; Wood, C.G. Genomic profiling identifies alterations in TGFbeta signaling through loss of TGFbeta receptor expression in human renal cell carcinogenesis and progression. Oncogene 2003, 22, 8053–8062. [Google Scholar] [CrossRef] [PubMed]
- Nishida, J.; Miyazono, K.; Ehata, S. Decreased TGFBR3/betaglycan expression enhances the metastatic abilities of renal cell carcinoma cells through TGF-beta-dependent and -independent mechanisms. Oncogene 2018, 37, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, H.; Yamamoto, T.; Fujigaki, Y.; Misaki, T.; Ohashi, N.; Takayama, T.; Suzuki, S.; Mugiya, S.; Oda, T.; Uchida, C.; et al. Reduction of transforming growth factor-beta type II receptor is caused by the enhanced ubiquitin-dependent degradation in human renal cell carcinoma. Int. J. Cancer 2010, 127, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Margulis, V.; Maity, T.; Zhang, X.Y.; Cooper, S.J.; Copland, J.A.; Wood, C.G. Type III transforming growth factor-beta (TGF-beta) receptor mediates apoptosis in renal cell carcinoma independent of the canonical TGF-beta signaling pathway. Clin. Cancer Res. 2008, 14, 5722–5730. [Google Scholar] [CrossRef] [PubMed]
- Tretbar, S.; Krausbeck, P.; Muller, A.; Friedrich, M.; Vaxevanis, C.; Bukur, J.; Jasinski-Bergner, S.; Seliger, B. TGF-beta inducible epithelial-to-mesenchymal transition in renal cell carcinoma. Oncotarget 2019, 10, 1507–1524. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Feng, H.; Yang, J.; Yin, A.; Qian, A.; Sugiyama, H. Landscape of immune cell infiltration in clear cell renal cell carcinoma to aid immunotherapy. Cancer Sci. 2021, 112, 2126–2139. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Li, C.G.; Chantry, A.; Stayner, C.; Horsfield, J.; Eccles, M.R. SMAD proteins directly suppress PAX2 transcription downstream of transforming growth factor-beta 1 (TGF-beta1) signalling in renal cell carcinoma. Oncotarget 2018, 9, 26852–26867. [Google Scholar] [CrossRef] [PubMed]
- Feldkoren, B.; Hutchinson, R.; Rapoport, Y.; Mahajan, A.; Margulis, V. Integrin signaling potentiates transforming growth factor-beta 1 (TGF-beta1) dependent down-regulation of E-Cadherin expression—Important implications for epithelial to mesenchymal transition (EMT) in renal cell carcinoma. Exp. Cell Res. 2017, 355, 57–66. [Google Scholar] [CrossRef]
- Tian, T.; Fu, X.; Hu, L.; Yang, X.; Sun, P.; Sun, F. FAST1 Predicts Poor Survival of Renal Carcinoma and Promotes Its Progression Through the TGF-beta/Smad Pathway. Onco Targets Ther. 2021, 14, 1487–1499. [Google Scholar] [CrossRef]
- Ananth, S.; Knebelmann, B.; Gruning, W.; Dhanabal, M.; Walz, G.; Stillman, I.E.; Sukhatme, V.P. Transforming growth factor beta1 is a target for the von Hippel-Lindau tumor suppressor and a critical growth factor for clear cell renal carcinoma. Cancer Res. 1999, 59, 2210–2216. [Google Scholar]
- Meng, F.; Li, Y.; Tian, X.; Fu, L.; Yin, Y.; Sui, C.; Ma, P.; Jiang, Y. Identification of TGF-beta-activated kinase 1 as a possible novel target for renal cell carcinoma intervention. Biochem. Biophys. Res. Commun. 2014, 453, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Busse, A.; Asemissen, A.; Nonnenmacher, A.; Ochsenreither, S.; Fusi, A.; Braun, F.; Stather, D.; Schmittel, A.; Miller, K.; Thiel, E.; et al. Systemic immune tuning in renal cell carcinoma: Favorable prognostic impact of TGF-beta1 mRNA expression in peripheral blood mononuclear cells. J. Immunother. 2011, 34, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhang, Q.; Zhen, Q.; Zhao, Y.; Liu, N.; Li, T.; Hao, Y.; Zhang, Y.; Luo, C.; Wu, X. Negative regulation of tumor-infiltrating NK cell in clear cell renal cell carcinoma patients through the exosomal pathway. Oncotarget 2017, 8, 37783–37795. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wen, W.; Yuan, J.; Helfand, B.; Li, Y.; Shi, C.; Tian, F.; Zheng, J.; Wang, F.; Chen, L.; et al. Immunotherapy for human renal cell carcinoma by adoptive transfer of autologous transforming growth factor beta-insensitive CD8+ T cells. Clin. Cancer Res. 2010, 16, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-beta1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.E.; Smith, D.C.; Branton, M.H.; Penzak, S.R.; Kopp, J.B. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2007, 2, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.D.; Ding, M.; Tong, P.; Chong, Y.Y.; Gu, W.Y.; Li, Y.; Fang, X.J.; Li, N. Synergistic effects of low-dose chemotherapy and T cells in renal cell carcinoma. Oncol. Rep. 2020, 44, 897–908. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Q.; Mao, J.; Qin, T.; Sun, Y.; Yang, J.; Han, Y.; Li, L.; Li, Q. Salinomycin suppresses cancer cell stemness and attenuates TGF-beta-induced epithelial-mesenchymal transition of renal cell carcinoma cells. Chem. Biol. Interact. 2018, 296, 145–153. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, X.; Guo, Z.; Huang, N.; Li, J.; Lv, Y.; Han, L.; Zheng, W.; Xu, D.; Chai, D.; et al. The Anti-fibrosis drug Pirfenidone modifies the immunosuppressive tumor microenvironment and prevents the progression of renal cell carcinoma by inhibiting tumor autocrine TGF-β. Cancer Biol. Ther. 2022, 23, 150–162. [Google Scholar] [CrossRef]
- Heidegger, I.; Pircher, A.; Pichler, R. Targeting the Tumor Microenvironment in Renal Cell Cancer Biology and Therapy. Front Oncol 2019, 9, 490. [Google Scholar] [CrossRef]
- Song, W.; He, D.; Chen, Y.; Yeh, C.R.; Hsu, I.; Huang, Q.; Zhang, X.; Chang, L.S.; Zuo, L.; Chen, J.; et al. Targeting newly identified ERβ/TGF-β1/SMAD3 signals with the FDA-approved anti-estrogen Faslodex or an ERβ selective antagonist in renal cell carcinoma. Mol. Oncol. 2018, 12, 2055–2071. [Google Scholar] [CrossRef] [PubMed]
- Gurujeyalakshmi, G.; Hollinger, M.A.; Giri, S.N. Pirfenidone inhibits PDGF isoforms in bleomycin hamster model of lung fibrosis at the translational level. Am. J. Physiol. 1999, 276, L311–L318. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999, 291, 367–373. [Google Scholar] [PubMed]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of pirfenidone on procollagen gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999, 289, 211–218. [Google Scholar] [PubMed]
- Chan, M.K.; Chung, J.Y.; Tang, P.C.; Chan, A.S.; Ho, J.Y.; Lin, T.P.; Chen, J.; Leung, K.T.; To, K.F.; Lan, H.Y.; et al. TGF-beta signaling networks in the tumor microenvironment. Cancer Lett 2022, 550, 215925. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients with Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.J. On-Target Anti-TGF-beta Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef]
- Dodagatta-Marri, E.; Meyer, D.S.; Reeves, M.Q.; Paniagua, R.; To, M.D.; Binnewies, M.; Broz, M.L.; Mori, H.; Wu, D.; Adoumie, M.; et al. α-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α-TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J. Immunother. Cancer 2019, 7, 62. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008, 267, 226–244. [Google Scholar] [CrossRef]
- Schott, A.F.; Goldstein, L.J.; Cristofanilli, M.; Ruffini, P.A.; McCanna, S.; Reuben, J.M.; Perez, R.P.; Kato, G.; Wicha, M. Phase Ib Pilot Study to Evaluate Reparixin in Combination with Weekly Paclitaxel in Patients with HER-2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5358–5365. [Google Scholar] [CrossRef] [PubMed]
- Dufies, M.; Grytsai, O.; Ronco, C.; Camara, O.; Ambrosetti, D.; Hagege, A.; Parola, J.; Mateo, L.; Ayrault, M.; Giuliano, S.; et al. New CXCR1/CXCR2 inhibitors represent an effective treatment for kidney or head and neck cancers sensitive or refractory to reference treatments. Theranostics 2019, 9, 5332–5346. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; Mezzapelle, R. The Chemokine Receptor CXCR4 in Cell Proliferation and Tissue Regeneration. Front. Immunol. 2020, 11, 2109. [Google Scholar] [CrossRef] [PubMed]
- Romoli, S.; Angelotti, M.L.; Antonelli, G.; Kumar Vr, S.; Mulay, S.R.; Desai, J.; Anguiano Gomez, L.; Thomasova, D.; Eulberg, D.; Klussmann, S.; et al. CXCL12 blockade preferentially regenerates lost podocytes in cortical nephrons by targeting an intrinsic podocyte-progenitor feedback mechanism. Kidney Int. 2018, 94, 1111–1126. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Long, Q.; Guan, B.; Mu, L. Prognostic Value of High CXCR4 Expression in Renal Cell Carcinoma: A System Review and Meta-Analysis. Dis. Markers 2015, 2015, 568980. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gassenmaier, M.; Maruschke, M.; Riesenberg, R.; Pohla, H.; Stief, C.G.; Zimmermann, W.; Buchner, A. Expression and Prognostic Significance of a Comprehensive Epithelial-Mesenchymal Transition Gene Set in Renal Cell Carcinoma. J. Urol. 2014, 191, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.M.; Ji, S.; Li, Y.; Fu, L.Y.; Jiang, T.; Meng, F.D. β-Catenin promotes cell proliferation, migration, and invasion but induces apoptosis in renal cell carcinoma. Onco Targets Ther. 2017, 10, 711–724. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Vaishampayan, U.; Matrana, M.; Rha, S.Y.; Saavedra, A.Z.; Ho, T.; Keam, B.; Lee, J.L.; Joseph, R.; Ali, S.; et al. 1186PD—Safety and efficacy of the oral CXCR4 inhibitor X4P-001 + axitinib in advanced renal cell carcinoma patients: An analysis of subgroup responses by prior treatment. Ann. Oncol. 2019, 30, v482–v483. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Atkins, M.B.; Rose, T.L.; Alter, R.S.; Ju, Y.; Niland, K.; Wang, Y.; Arbeit, R.; Parasuraman, S.; Gan, L.; et al. A phase 1b trial of the CXCR4 inhibitor mavorixafor and nivolumab in advanced renal cell carcinoma patients with no prior response to nivolumab monotherapy. Investig. New Drugs 2021, 39, 1019–1027. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruk, L.; Mamtimin, M.; Braun, A.; Anders, H.-J.; Andrassy, J.; Gudermann, T.; Mammadova-Bach, E. Inflammatory Networks in Renal Cell Carcinoma. Cancers 2023, 15, 2212. https://doi.org/10.3390/cancers15082212
Kruk L, Mamtimin M, Braun A, Anders H-J, Andrassy J, Gudermann T, Mammadova-Bach E. Inflammatory Networks in Renal Cell Carcinoma. Cancers. 2023; 15(8):2212. https://doi.org/10.3390/cancers15082212
Chicago/Turabian StyleKruk, Linus, Medina Mamtimin, Attila Braun, Hans-Joachim Anders, Joachim Andrassy, Thomas Gudermann, and Elmina Mammadova-Bach. 2023. "Inflammatory Networks in Renal Cell Carcinoma" Cancers 15, no. 8: 2212. https://doi.org/10.3390/cancers15082212
APA StyleKruk, L., Mamtimin, M., Braun, A., Anders, H.-J., Andrassy, J., Gudermann, T., & Mammadova-Bach, E. (2023). Inflammatory Networks in Renal Cell Carcinoma. Cancers, 15(8), 2212. https://doi.org/10.3390/cancers15082212