
Adefovir Dipivoxil as a Therapeutic Candidate for Medullary Thyroid Carcinoma: Targeting RET and STAT3 Proto-Oncogenes


 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture & Media
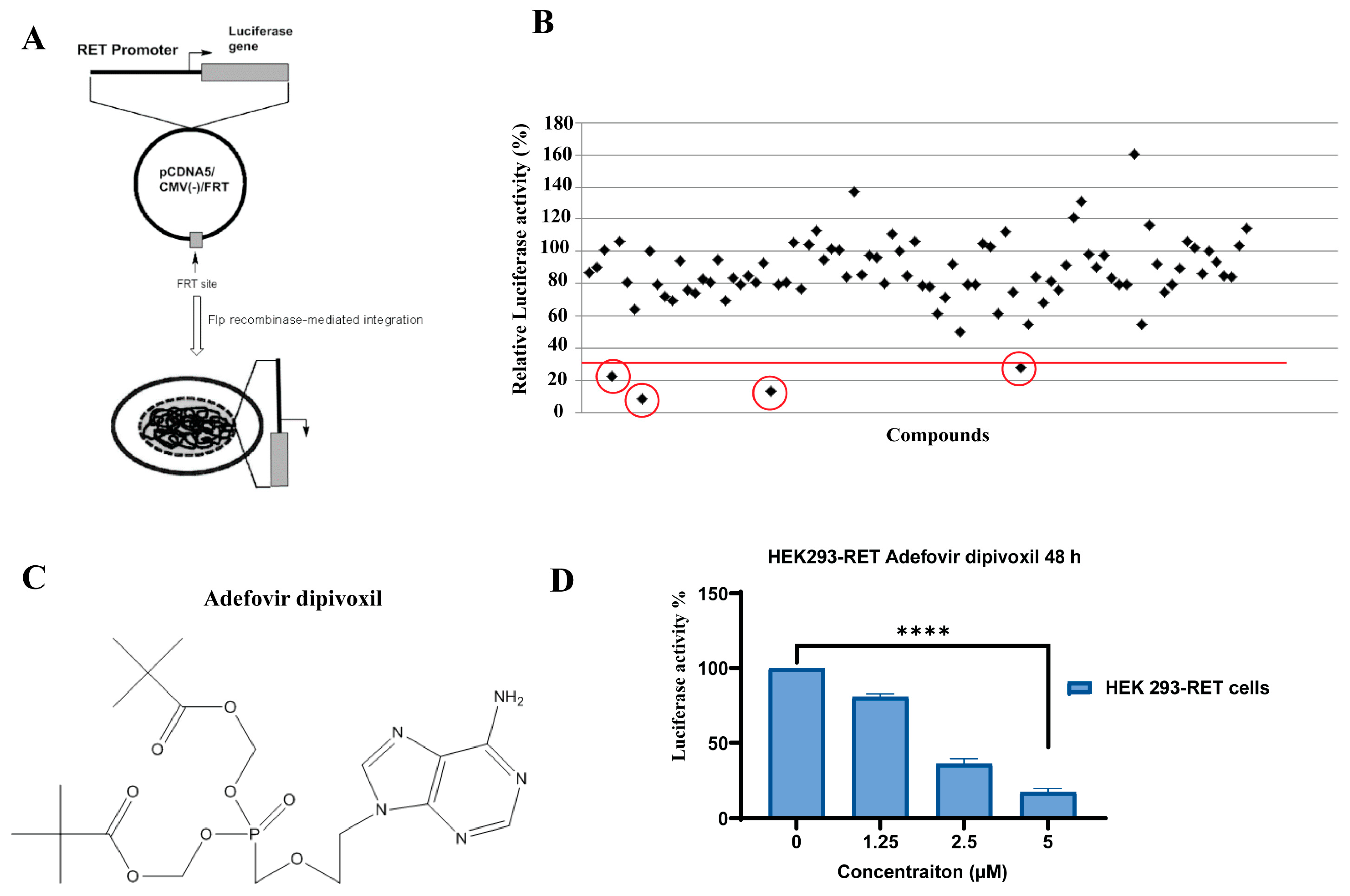
2.2. Cell Based Screening Using Luciferase Assay
2.3. Reverse Transcription PCR Analysis of the RET mRNA Synthesis in TT Cells
2.4. Western Blotting
2.5. Cell Viability Assay
2.6. Flow Cytometry
2.7. Protein Preparation
2.8. Ligand Preparation
2.9. Ligand-Protein Molecular Docking and Prime MM-GBSA Calculations
2.10. Biotinylation of 5′-Phosphate Group of Adefovir
2.11. Spheroid Formation
2.12. Xenograft Model
2.13. Statistical Analysis
3. Results
3.1. Adefovir Dipivoxil Is Identified as a Novel RET Transcription Inhibitor
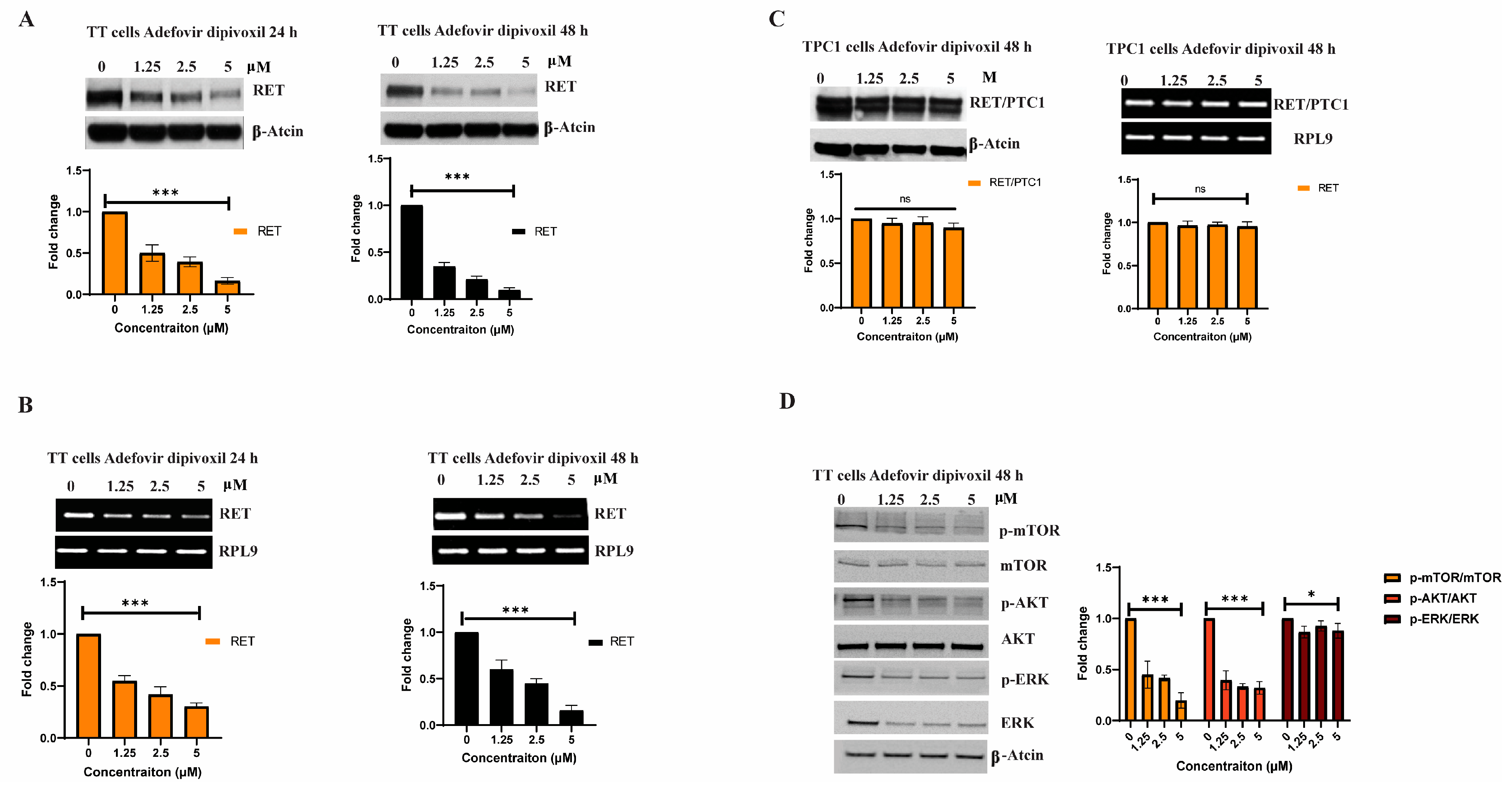
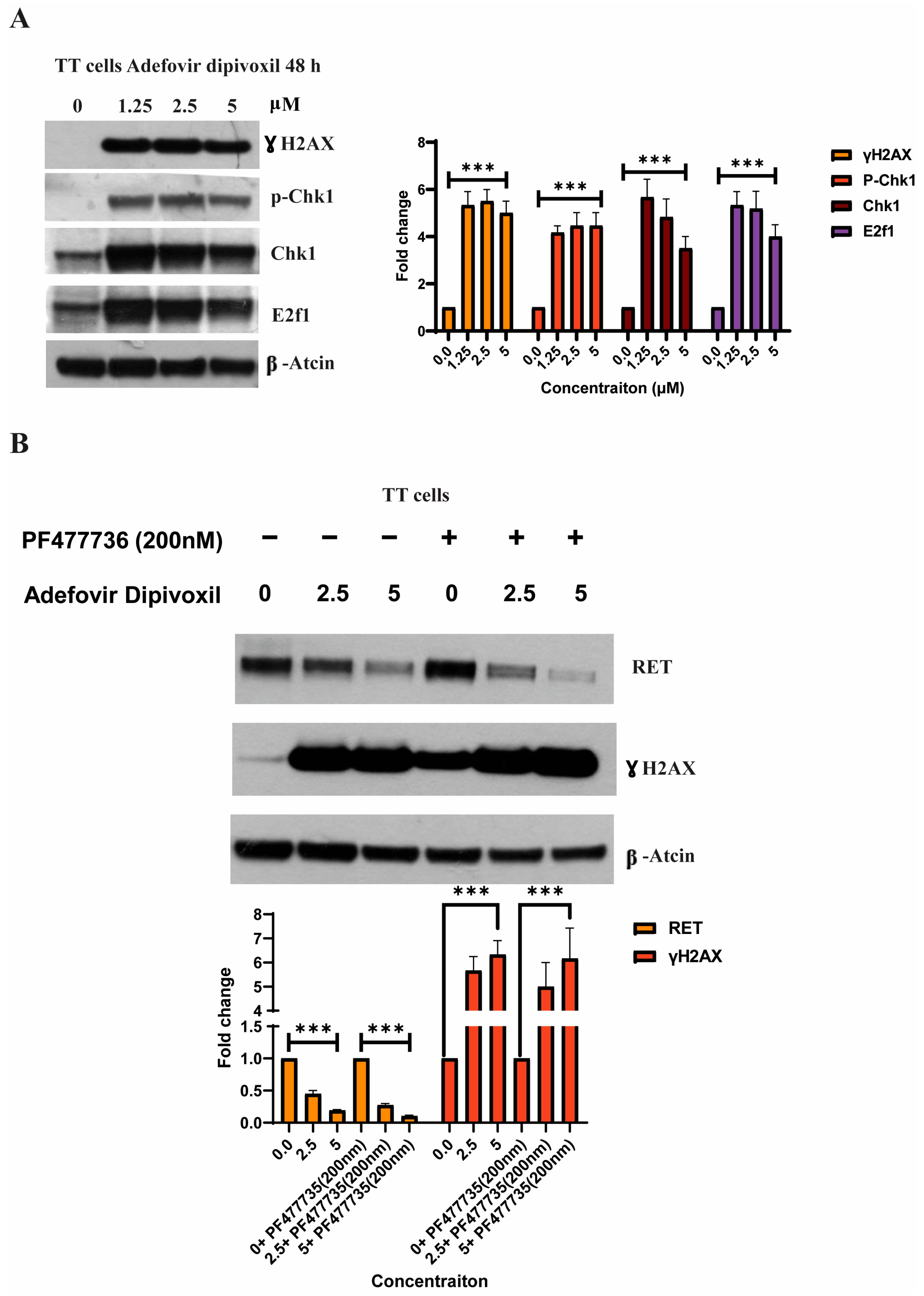
3.2. Adfovivir Dipivoxil Affects RET Expression and RET Signaling Pathway
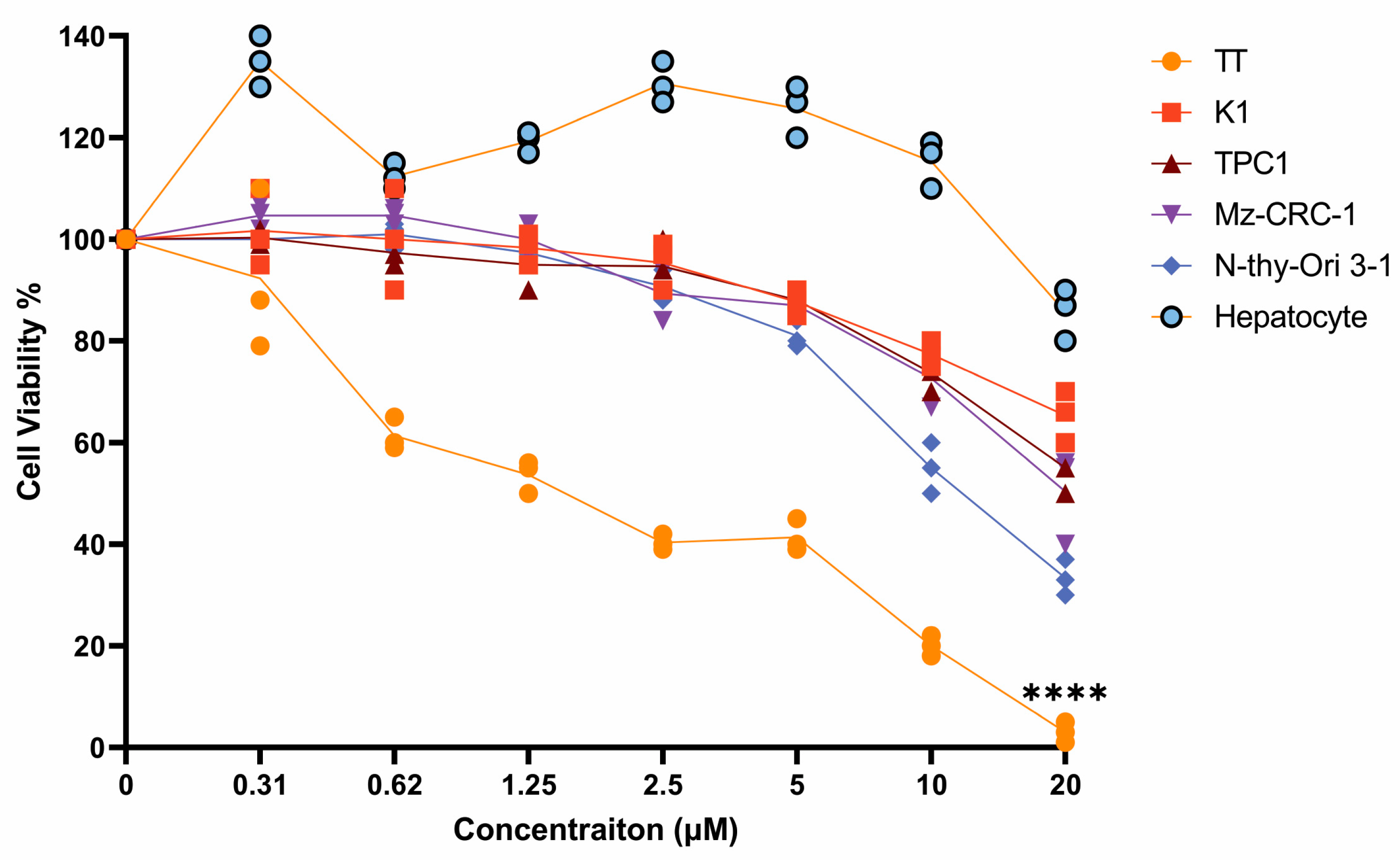
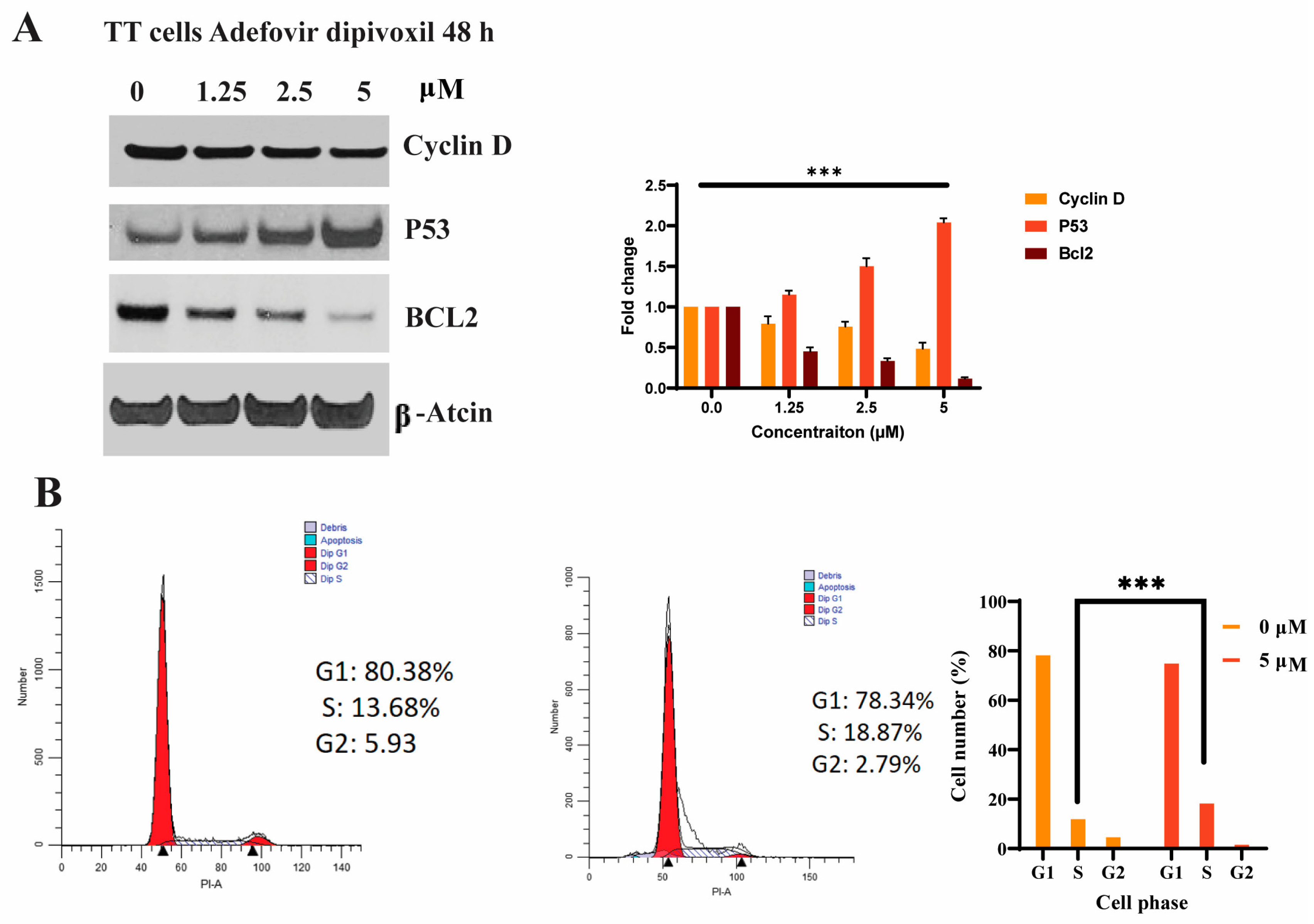
3.3. Effects of Adefovir Dipivoxil in Cell Proliferation
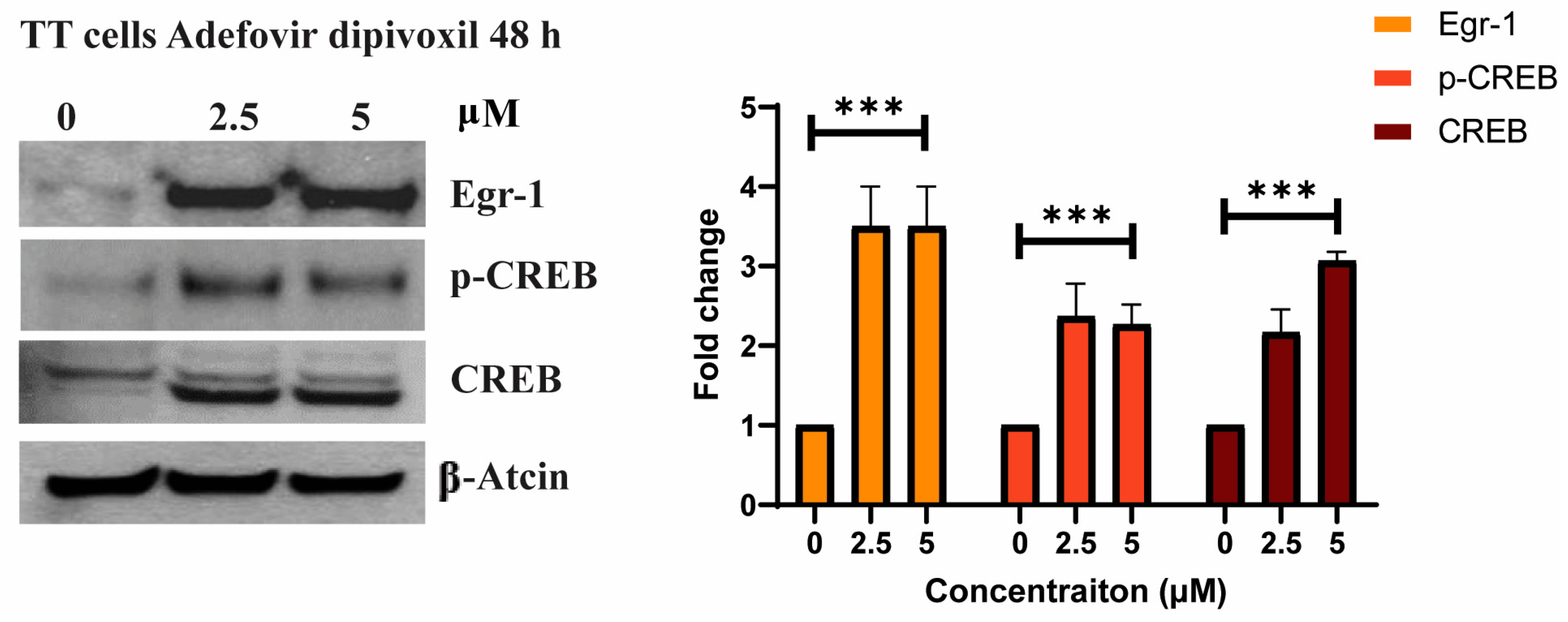
3.4. The Effects of Adefovir on Egr-1 and CREB
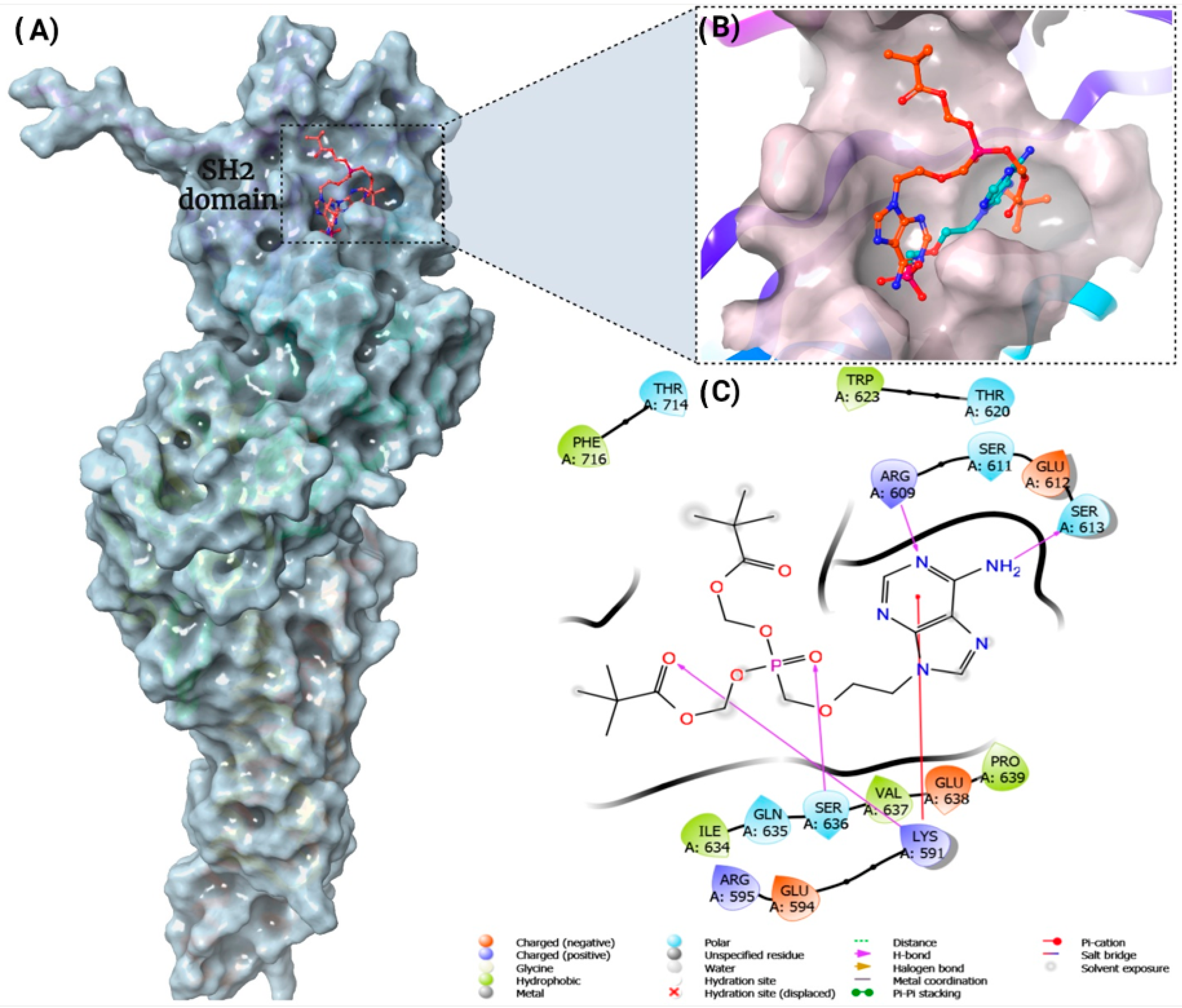
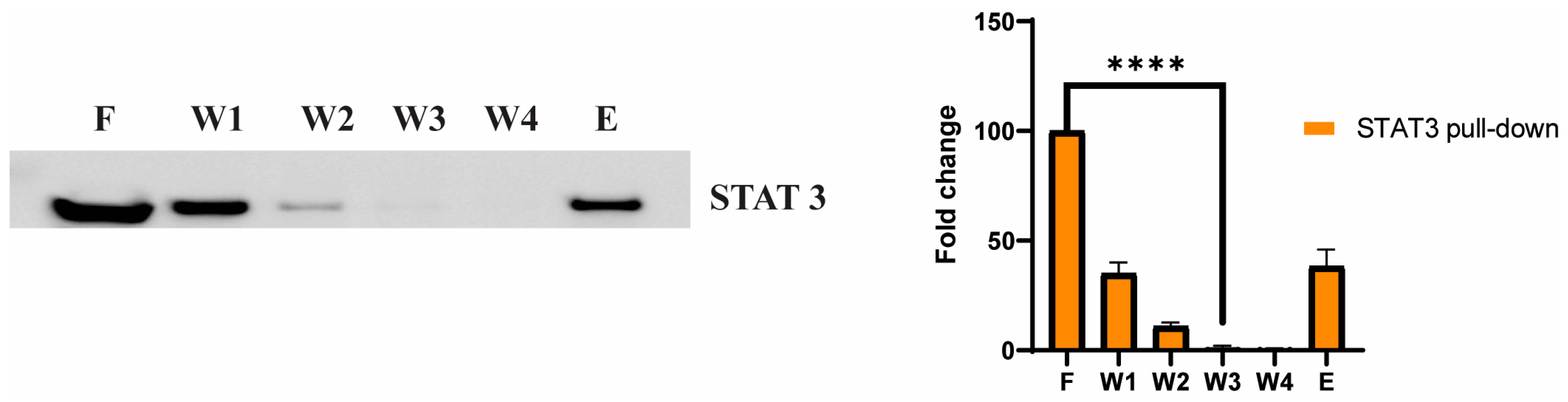
3.5. STAT3 as a Potential Cellular Target for Adefovir
3.6. Adefovir Dipivoxil Effects on STAT3 and pSTAT3 in MTC Cell Line
3.7. Cell Cycle Effects of Adefovir Dipivoxil
3.8. Characterizing DNA Damage Checkpoints Activated by Adefovir
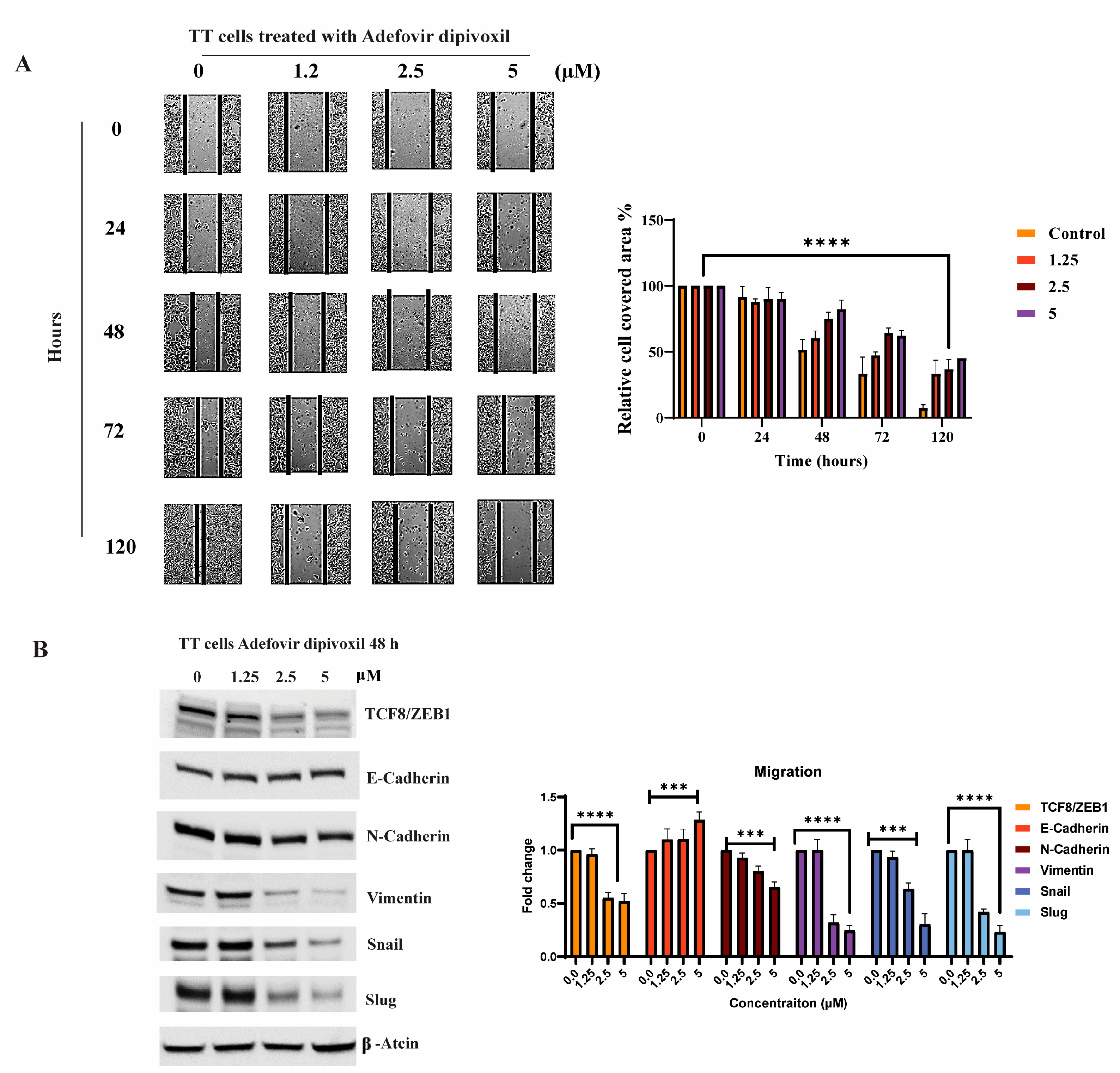
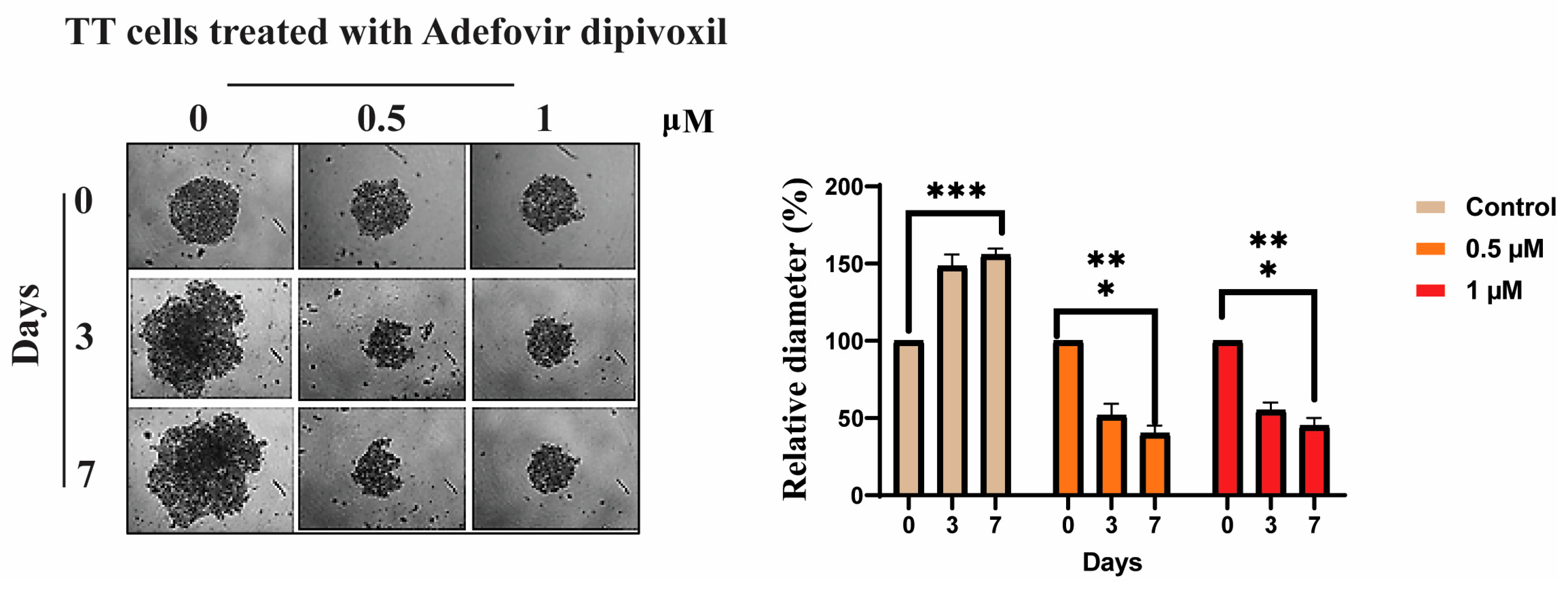
3.9. Signaling Pathways Involved in the Migration, Invasion, and Resistance of TT Cells and Spheroid Formation
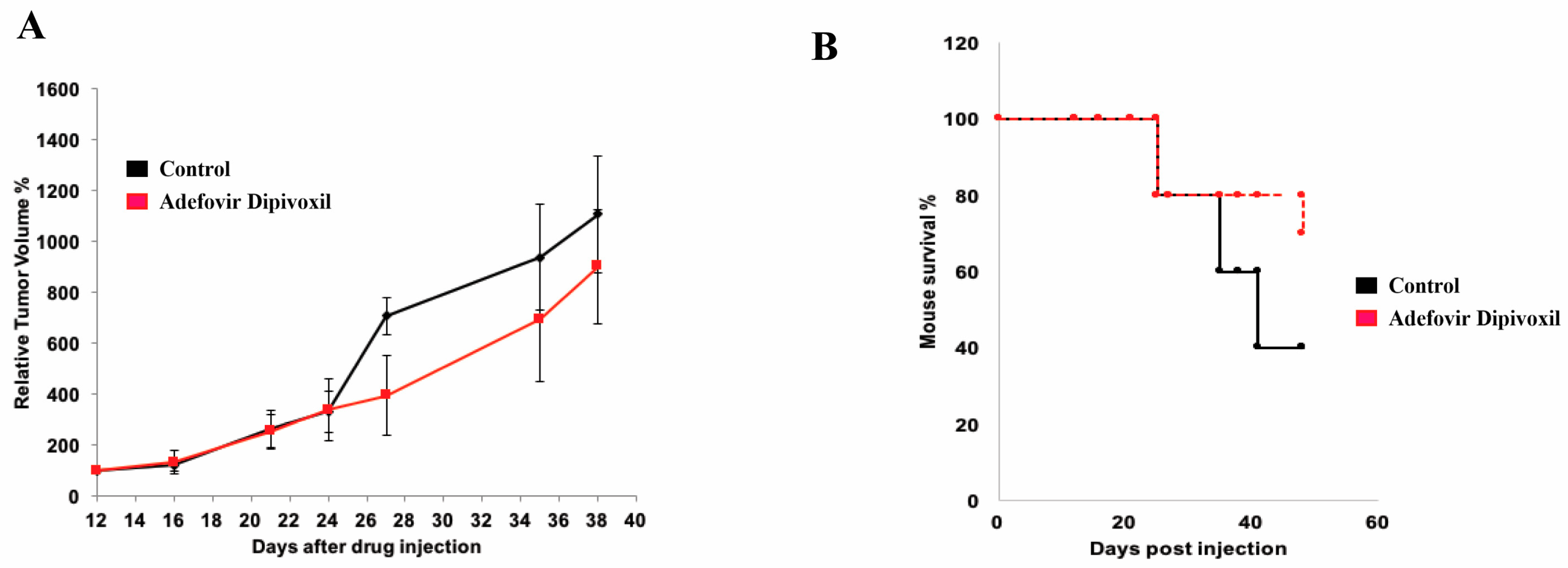
3.10. In Vivo Antitumor Effect of Adefovir Dipivoxil in Xenograft Models of Human Medullary Thyroid Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Priya, S.R.; Dravid, C.S.; Digumarti, R.; Dandekar, M. Targeted Therapy for Medullary Thyroid Cancer: A Review. Front. Oncol. 2017, 7, 238. [Google Scholar] [CrossRef] [PubMed]
- Gimm, O.; Dralle, H. Therapy for medullary thyroid cancer. In Thyroid Cancer, 2nd ed.; NHS: London, UK, 2005. [Google Scholar]
- Viola, D.; Elisei, R. Management of Medullary Thyroid Cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Nozhat, Z.; Hedayati, M. Medullary thyroid carcinoma: A review on ethical considerations in treatment of children. J. Pediatr. Endocrinol. Metab. 2016, 29, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Accardo, G.; Conzo, G.; Esposito, D.; Gambardella, C.; Mazzella, M.; Castaldo, F.; Di Donna, C.; Polistena, A.; Avenia, N.; Colantuoni, V.; et al. Genetics of medullary thyroid cancer: An overview. Int. J. Surg. 2017, 41, S2–S6. [Google Scholar] [CrossRef]
- Ishizaka, Y.; Itoh, F.; Tahira, T.; Ikeda, I.; Sugimura, T.; Tucker, J.; Fertitta, A.; Carrano, A.V.; Nagao, M. Human ret proto-oncogene mapped to chromosome 10q11.2. Oncogene 1989, 4, 1519–1521. [Google Scholar]
- Manfredi, G.I.; Dicitore, A.; Gaudenzi, G.; Caraglia, M.; Persani, L.; Vitale, G. PI3K/Akt/mTOR signaling in medullary thyroid cancer: A promising molecular target for cancer therapy. Endocrine 2015, 48, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Kumarasamy, V.M.; Sun, D. Demonstration of a potent RET transcriptional inhibitor for the treatment of medullary thyroid carcinoma based on an ellipticine derivative. Int. J. Oncol. 2017, 51, 145–157. [Google Scholar] [CrossRef]
- Santoro, M.; Carlomagno, F. Central role of RET in thyroid cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a009233. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Hu, Z.I.; Lai, G.G.Y.; Tan, D.S.W. Targeting ret-driven cancers: Lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol. 2018, 15, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized double-blind phase III, trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef]
- Grabowski, P.; Briest, F.; Baum, R.P.; Zaknun, J.J.; Kulkarni, H.R.; Zeitz, M.; Hörsch, D. Vandetanib therapy in medullary thyroid cancer. Drugs Today 2012, 48, 723. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Schlumberger, M.J.; Müller, S.P.; Schöffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, J.N.; Fancher, K.M. Cabozantinib: A Multitargeted Oral Tyrosine Kinase Inhibitor. Pharmacotherapy 2018, 38, 357–369. [Google Scholar] [CrossRef]
- Markham, A. Selpercatinib: First Approval. Drugs 2020, 80, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Belli, C.; Penault-Llorca, F.; Ladanyi, M.; Normanno, N.; Scoazec, J.Y.; Lacroix, L.; Reis-Filho, J.S.; Subbiah, V.; Gainor, J.F.; Endris, V.; et al. ESMO recommendations on the standard methods to detect RET fusions and mutations in daily practice and clinical research. Ann. Oncol. 2021, 32, 337–350. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Fallahi, P.; Elia, G.; Ragusa, F.; Ruffilli, I.; Paparo, S.R.; Antonelli, A. Thyroid autoimmune disorders and cancer. Semin. Cancer Biol. 2020, 64, 135–146. [Google Scholar] [CrossRef]
- Subbiah, V.; Shen, T.; Terzyan, S.S.; Liu, X.; Hu, X.; Patel, K.P.; Hu, M.; Cabanillas, M.; Behrang, A.; Meric-Bernstam, F.; et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann. Oncol 2021, 32, 261–268. [Google Scholar] [CrossRef]
- Hartmann, J.T.; Haap, M.; Kopp, H.G.; Lipp, H.P. Tyrosine kinase inhibitors—A review on pharmacology, metabolism and side effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming Cancer Therapeutic Bottleneck By Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Meheus, L.; Huys, I.; Pantziarka, P. Repurposing Drugs in Oncology: Next Steps. Trends Cancer 2017, 3, 543–546. [Google Scholar] [CrossRef]
- Frantzi, M.; Latosinska, A.; Mokou, M.; Mischak, H.; Vlahou, A. Drug repurposing in oncology. Lancet Oncol. 2020, 21, e543. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.J.; Kumarasamy, V.; Camacho, D.; Sun, D. Involvement of G-quadruplex structures in regulation of human RET gene expression by small molecules in human medullary thyroid carcinoma TT cells. Oncogene 2015, 34, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Seto, W.K.; Lai, C.L.; Yuen, M.F. Adefovir dipivoxil. In Kucers the Use of Antibiotics: A Clinical Review of Antibacterial, Antifungal, Antiparasitic, and Antiviral Drugs, 7th ed.; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Dando, T.M.; Plosker, G.L. Adefovir Dipivoxil: A Review of its Use in Chronic Hepatitis B. Drugs 2003, 63, 2215–2234. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, D.W. Antiviral Agents. In Principles and Practice of Pediatric Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Gu, J.; Sun, R.; Shen, S.; Yu, Z. The curative effect of adefovir dipivoxil treating HBeAg negative chronic hepatitis B and treating HBeAg positive chronic hepatitis B combining interferon α-2b. Pak. J. Pharm. Sci. 2015, 28, 1493–1498. [Google Scholar]
- Alqahtani, T.; Kumarasamy, V.M.; Huczyński, A.; Sun, D. Salinomycin and its derivatives as potent RET transcriptional inhibitors for the treatment of medullary thyroid carcinoma. Int. J. Oncol. 2020, 56, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R.; Sadeghi-Nejad, A. Role of RET protein-tyrosine kinase inhibitors in the treatment RET-driven thyroid and lung cancers. Pharmacol. Res. 2018, 128, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Desterke, C.; Féraud, O.; Guibert, M.; De Rose, R.F.; Opolon, P.; Divers, D.; Gobbo, E.; Griscelli, F.; Schlumberger, M.; et al. Transcriptional landscape of a RETC634Y-mutated iPSC and its CRISPR-corrected isogenic control reveals the putative role of EGR1 transcriptional program in the development of multiple endocrine neoplasia type 2A-associated cancers. Stem Cell Res. 2018, 26, 8–16. [Google Scholar] [CrossRef]
- Mulligan, L.M. RET revisited: Expanding the oncogenic portfolio. Nat. Rev. Cancer 2014, 14, 173–186. [Google Scholar] [CrossRef]
- Andrew, S.D.; Capes-Davis, A.; Delhanty, P.J.D.; Marsh, D.J.; Mulligan, L.M.; Robinson, B.G. Transcriptional repression of the RET proto-oncogene by a mitogen activated protein kinase-dependent signalling pathway. Gene 2002, 298, 9–19. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Lee, S.H.; Kim, J.S.; Wimalasena, J.; Kitajima, S.; Baek, S.J. Activating transcription factor 3 and early growth response 1 are the novel targets of LY294002 in a phosphatidylinositol 3-kinase-independent pathway. Cancer Res. 2006, 66, 2376–2384. [Google Scholar] [CrossRef]
- Szelag, M.; Sikorski, K.; Czerwoniec, A.; Szatkowska, K.; Wesoly, J.; Bluyssen, H.A.R. In silico simulations of STAT1 and STAT3 inhibitors predict SH2 domain cross-binding specificity. Eur. J. Pharm. 2013, 720, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Schuringa, J.J.; Wojtachnio, K.; Hagens, W.; Vellenga, E.; Buys, C.H.; Hofstra, R.; Kruijer, W. MEN2A-RET-induced cellular transformation by activation of STAT3. Oncogene 2001, 20, 5350–5358. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Chen, M.C.; Chiu, C.Y.; Song, Y.M.; Lin, S.Y. Cdk5 regulates STAT3 activation and cell proliferation in medullary thyroid carcinoma cells. J. Biol. Chem. 2007, 282, 2776–2784. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Rapa, I.; Saggiorato, E.; Giachino, D.; Palestini, N.; Orlandi, F.; Papotti, M.; Volante, M. Mammalian target of rapamycin pathway activation is associated to RET mutation status in medullary thyroid, carcinoma. J. Clin. Endocrinol. Metab. 2011, 96, 2146–2153. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Averous, J.; Fonseca, B.D.; Proud, C.G. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene 2008, 27, 1106–1113. [Google Scholar] [CrossRef]
- Patel, A.; Seraia, E.; Ebner, D.; Ryan, A.J. Adefovir dipivoxil induces DNA replication stress and augments ATR inhibitor-related cytotoxicity. Int. J. Cancer 2020, 147, 1474–1484. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Bassett, D.; Urrabaz, R.; Sun, D. Cellular response and molecular mechanism of antitumor activity by leinamycin in MiaPaCa human pancreatic cancer cells. Anticancer. Drugs 2004, 15, 689–697. [Google Scholar] [CrossRef]
- Giardino, E.; Catalano, R.; Barbieri, A.M.; Treppiedi, D.; Mangili, F.; Spada, A.; Arosio, M.; Mantovani, G.; Peverelli, E. Cofilin is a mediator of RET-promoted medullary thyroid carcinoma cell migration, invasion and proliferation. Mol. Cell. Endocrinol. 2019, 495, 110519. [Google Scholar] [CrossRef]
- Rodriguez, L.G.; Wu, X.; Guan, J.L. Wound-healing assay. In Cell Migration; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Shakib, H.; Rajabi, S.; Dehghan, M.H.; Mashayekhi, F.J.; Safari-Alighiarloo, N.; Hedayati, M. Epithelial-to-mesenchymal transition in thyroid cancer: A comprehensive review. Endocrine 2019, 66, 435–455. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Yoon, C.-H.; Kim, R.-K.; Lim, E.-J.; Oh, Y.S.; Hwang, S.-G.; An, S.; Yoon, G.; Gye, M.C.; Yi, J.-M.; et al. Claudin-1 induces epithelial-mesenchymal transition through activation of the c-Abl-ERK signaling pathway in human liver cells. Oncogene 2013, 32, 4873–4882. [Google Scholar] [CrossRef] [PubMed]
- Garg, M. Epithelial-mesenchymal transition—Activating transcription factors—Multifunctional regulators in cancer. World J. Stem Cells 2013, 5, 188. [Google Scholar] [CrossRef]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Da, C.; Wu, K.; Yue, C.; Bai, P.; Wang, R.; Wang, G.; Zhao, M.; Lv, Y.; Hou, P. N-cadherin promotes thyroid tumorigenesis through modulating major signaling pathways. Oncotarget 2017, 8, 8131–8142. [Google Scholar] [CrossRef]
- Alqahtani, T.; Alswied, A.; Sun, D. Selective antitumor activity of datelliptium toward medullary thyroid carcinoma by downregulating ret transcriptional activity. Cancers 2021, 13, 3288. [Google Scholar] [CrossRef]
- Haque, S.K.; Ariceta, G.; Batlle, D. Proximal renal tubular acidosis: A not so rare disorder of multiple etiologies. Nephrol. Dial. Transplant. 2012, 27, 4273–4287. [Google Scholar] [CrossRef]
- Lee, J.G.; Lee, J.; Lee, J.J.; Song, S.H.; Ju, M.K.; Choi, G.H.; Kim, M.S.; Choi, J.S.; Kim, S.I.; Joo, D.J. Adefovir- or lamivudine-induced renal tubular dysfunction after liver transplantation. Medicine 2015, 94, e1569. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.J.; Chaloner, K.; Cohn, D.L.; Grant, L.B.; Alston, B.; Brosgart, C.L.; Schmetter, B.; El-Sadr, W.M.; Sampson, J.; Terry Beirn Community Programs for Clinical Research on AIDS. The safety and efficacy of adefovir dipivoxil in patients with advanced HIV disease: A randomized, placebo-controlled trial. AIDS 2001, 15, 1695–1700. [Google Scholar] [CrossRef]
- Drosten, M.; Hilken, G.; Böckmann, M.; Rödicker, F.; Mise, N.; Cranston, A.N.; Dahmen, U.; Ponder, B.A.J.; Pützer, B.M. Role of MEN2A-Derived RET in Maintenance and Proliferation of Medullary Thyroid Carcinoma. JNCI J. Natl. Cancer Inst. 2004, 96, 1231–1239. [Google Scholar] [CrossRef]
- Sastry, R.; Williams, E.A.; Koffie, R.; Fehnel, K.; Wirth, L.; Nahed, B. Metastatic medullary thyroid cancer to the brain: A case report and review of the literature. Int. J. Endocr. Oncol. 2018, 5, IJE11. [Google Scholar] [CrossRef]
- Niafar, M.; Dabiri, S.; Bozorgi, F.; Niafar, F.; Gholami, N. Metastatic medullary thyroid carcinoma: A case report. J. Res. Med. Sci. 2011, 16, 568. [Google Scholar] [PubMed]
- Kanteti, A.P.; Atiya, S.; Hein, A.; Cox, J.L.; Duarte, E.M. Medullary Thyroid Carcinoma Presenting as Metastatic Disease to the Breast. Case Rep. Pathol. 2020, 2020, 6138409. [Google Scholar] [CrossRef] [PubMed]
- Joly, M.M.; Williams, M.M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Young, C.D.; Sarbassov, D.D.; Muller, W.J.; Brantley-Sieders, D.; Cook, R.S. Two distinct mTORC2-dependent pathways converge on Rac1 to drive breast cancer metastasis. Breast Cancer Res. 2017, 19, 74. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Guo, H.; Yu, H.; Chen, Y.; Xu, H.; Zhao, G. The Role of the Transcription Factor EGR1 in Cancer. Front. Oncol. 2021, 11, 642547. [Google Scholar] [CrossRef]
- Wong, K.M.; Song, J.; Wong, Y.H. CTCF and EGR1 suppress breast cancer cell migration through transcriptional control of Nm23-H1. Sci. Rep. 2021, 11, 491. [Google Scholar] [CrossRef]
- Inoue, K.; Fry, E.A. Tumor suppression by the EGR1, DMP1, ARF, p53, and PTEN Network. Cancer Investig. 2018, 36, 520–536. [Google Scholar] [CrossRef]
- Horibata, S.; Rice, E.J.; Zheng, H.; Mukai, C.; Chu, T.; Marks, B.A.; Coonrod, S.A.; Danko, C.G. A bi-stable feedback loop between GDNF, EGR1, and ERα contribute to endocrine resistant breast cancer. PLoS ONE 2018, 13, e0194522. [Google Scholar] [CrossRef]
- Pritchard, M.T.; Malinak, R.N.; Nagy, L.E. Early growth response (EGR)-1 is required for timely cell-cycle entry and progression in hepatocytes after acute carbon tetrachloride exposure in mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 300, G1124–G1131. [Google Scholar] [CrossRef]
- Wang, C.; Husain, K.; Zhang, A.; Centeno, B.A.; Chen, D.-T.; Tong, Z.; Sebti, S.M.; Malafa, M.P. EGR-1/Bax pathway plays a role in vitamin E d-tocotrienol-induced apoptosis in pancreatic cancer cells. J. Nutr. Biochem. 2015, 26, 797–807. [Google Scholar] [CrossRef]
- Wang, X.; Crowe, P.J.; Goldstein, D.; Yang, J.-L. STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers. Int. J. Oncol. 2012, 41, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Beebe, J.D.; Liu, J.Y.; Zhang, J.T. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol. Ther 2018, 191, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Melillo, R.M.; Carlomagno, F.; Fusco, A.; Vecchio, G. Molecular mechanisms of RET activation in human cancer. In Annals of the New York Academy of Sciences; Wiley-Blackwell: Hoboken, NJ, USA, 2002. [Google Scholar]
- Pützer, B.M.; Drosten, M. The RET proto-oncogene: A potential target for molecular cancer therapy. Trends Mol. Med. 2004, 10, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Kim, D.W.; Suh, J.M.; Kim, H.; Song, J.H.; Hwang, E.S.; Park, K.C.; Chung, H.K.; Kim, J.M.; Lee, T.-H.; et al. Activation of signal transducer and activator of transcription 3 by oncogenic RET/PTC (rearranged in transformation/papillary thyroid carcinoma) tyrosine kinase: Roles in specific gene regulation and cellular transformation. Mol. Endocrinol. 2003, 17, 1155–1166. [Google Scholar] [CrossRef]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis. JAK-STAT 2012, 1, 65–72. [Google Scholar] [CrossRef]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—Using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- El-Tanani, M.; Al Khatib, A.O.; Aladwan, S.M.; Abuelhana, A.; McCarron, P.A.; Tambuwala, M.M. Importance of STAT3 signalling in cancer, metastasis and therapeutic interventions. Cell Signal 2022, 92, 110275. [Google Scholar] [CrossRef]
- Park, I.H.; Li, C. Characterization of molecular recognition of STAT3 SH2 domain inhibitors through molecular simulation. J. Mol. Recognit. 2011, 24, 254–265. [Google Scholar] [CrossRef]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases-the lessons from the mouse models: Inhibition = deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef]
- Xu, C.; Hu, Y.; Chen, B.; Li, D.; Liang, R.; Shen, M.; Wu, M.; Tao, M. Metastasis-associated gene 1 (MTA1) enhances cisplatin resistance of malignant pleural mesothelioma by ATR-Chk1-mediated DNA repair. Ann. Transl. Med. 2021, 9, 670. [Google Scholar] [CrossRef]
- Wikiniyadhanee, R.; Lerksuthirat, T.; Stitchantrakul, W.; Chitphuk, S.; Takeda, S.; Dejsuphong, D. ATR Inhibitor Synergizes PARP Inhibitor Cytotoxicity in Homologous Recombination Repair Deficiency TK6 Cell Lines. Biomed Res. Int. 2023, 2023, 7891753. [Google Scholar] [CrossRef]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, L.M. GDNF and the RET receptor in cancer: New insights and therapeutic potential. Front. Physiol. 2019, 9, 1873. [Google Scholar] [CrossRef]
- Valiquette, M.; Parent, S.; Loisel, T.; Bouvier, M. Mutation of tyrosine-141 inhibits insulin-promoted tyrosine phosphorylation and increased responsiveness of the human beta 2-adrenergic receptor. EMBO J. 1995, 14, 5542–5549. [Google Scholar] [CrossRef]
- Lian, E.Y.; Hyndman, B.D.; Moodley, S.; Maritan, S.M.; Mulligan, L.M. RET isoforms contribute differentially to invasive processes in pancreatic ductal adenocarcinoma. Oncogene 2020, 39, 6493–6510. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Corum, L.; Meng, Q.; Blenis, J.; Zheng, J.Z.; Shi, X.; Flynn, D.C.; Jiang, B.-H. PI3K induced actin filament remodeling through Akt p70S6K1: Implication of essential role in cell migration. Am. J. Physiol. Cell Physiol. 2004, 286, C153–C163. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Song, Y.; Su, D.; Liu, Y.; Shi, X.; Lu, S. Early Growth Response Protein-1 Involves in Transforming Growth factor-b1 Induced Epithelial-Mesenchymal Transition and Inhibits Migration of Non-Small-Cell Lung Cancer Cells. Asian Pac. J. Cancer Prev. APJCP 2015, 16, 4137–4142. [Google Scholar] [CrossRef]
- Kim, J.; Kang, S.; Lee, H.; Choi, S.; Hong, S. Oxytocin inhibits head and neck squamous cell carcinoma cell migration by early growth response-1 upregulation. Anti-Cancer Drugs 2017, 28, 613–622. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, J.; Jeong, Y.; Kim, G.; Kim, N.; Choi, Y. Induction of Egr-1 is associated with anti-metastatic and anti-invasive ability of beta-lapachone in human hepatocarcinoma cells. Biosci. Biotechnol. Biochem. 2007, 71, 2169–2176. [Google Scholar] [CrossRef]
- Liu, P.; Li, J.; Lu, H.; Xu, B. Thalidomide inhibits leukemia cell invasion and migration by upregulation of early growth response gene 1. Leuk Lymphoma 2009, 50, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xu, B.; Li, J.; Lu, L.H. LY294002 inhibits leukemia cell invasion and migration through early growth response gene 1 induction independent of phosphatidylinositol 3-kinase-Akt pathway. Biochem. Biophys. Res. Commun. 2008, 377, 187–190. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Standard-Precision (SP) Scoring Function | Extra Precision (XP) Scoring Function | ||||
|---|---|---|---|---|---|---|
| Glide Score | Glide Energy (Kcal/mol) | MMGBSA Binding Free Energy (Kcal/mol) | Glide Score | Glide Energy (Kcal/mol) | MMGBSA Binding Free Energy (Kcal/mol) | |
| Adefovir dipivoxil | −3.760 | −47.268 | −61.39 | −4.037 | −48.464 | −61.23 |
| Adefovir | −6.173 | −41.017 | −32.11 | −5.680 | −39.024 | −28.30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqahtani, T.; Kumarasamy, V.; Alghamdi, S.S.; Suliman, R.S.; Bin Saleh, K.; Alrashed, M.A.; Aldhaeefi, M.; Sun, D. Adefovir Dipivoxil as a Therapeutic Candidate for Medullary Thyroid Carcinoma: Targeting RET and STAT3 Proto-Oncogenes. Cancers 2023, 15, 2163. https://doi.org/10.3390/cancers15072163
Alqahtani T, Kumarasamy V, Alghamdi SS, Suliman RS, Bin Saleh K, Alrashed MA, Aldhaeefi M, Sun D. Adefovir Dipivoxil as a Therapeutic Candidate for Medullary Thyroid Carcinoma: Targeting RET and STAT3 Proto-Oncogenes. Cancers. 2023; 15(7):2163. https://doi.org/10.3390/cancers15072163
Chicago/Turabian StyleAlqahtani, Tariq, Vishnu Kumarasamy, Sahar Saleh Alghamdi, Rasha Saad Suliman, Khalid Bin Saleh, Mohammed A. Alrashed, Mohammed Aldhaeefi, and Daekyu Sun. 2023. "Adefovir Dipivoxil as a Therapeutic Candidate for Medullary Thyroid Carcinoma: Targeting RET and STAT3 Proto-Oncogenes" Cancers 15, no. 7: 2163. https://doi.org/10.3390/cancers15072163
APA StyleAlqahtani, T., Kumarasamy, V., Alghamdi, S. S., Suliman, R. S., Bin Saleh, K., Alrashed, M. A., Aldhaeefi, M., & Sun, D. (2023). Adefovir Dipivoxil as a Therapeutic Candidate for Medullary Thyroid Carcinoma: Targeting RET and STAT3 Proto-Oncogenes. Cancers, 15(7), 2163. https://doi.org/10.3390/cancers15072163