



The Role of Mammalian STK38 in DNA Damage Response and Targeting for Radio-Sensitization
 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Structure, Function, and Regulation of STK38
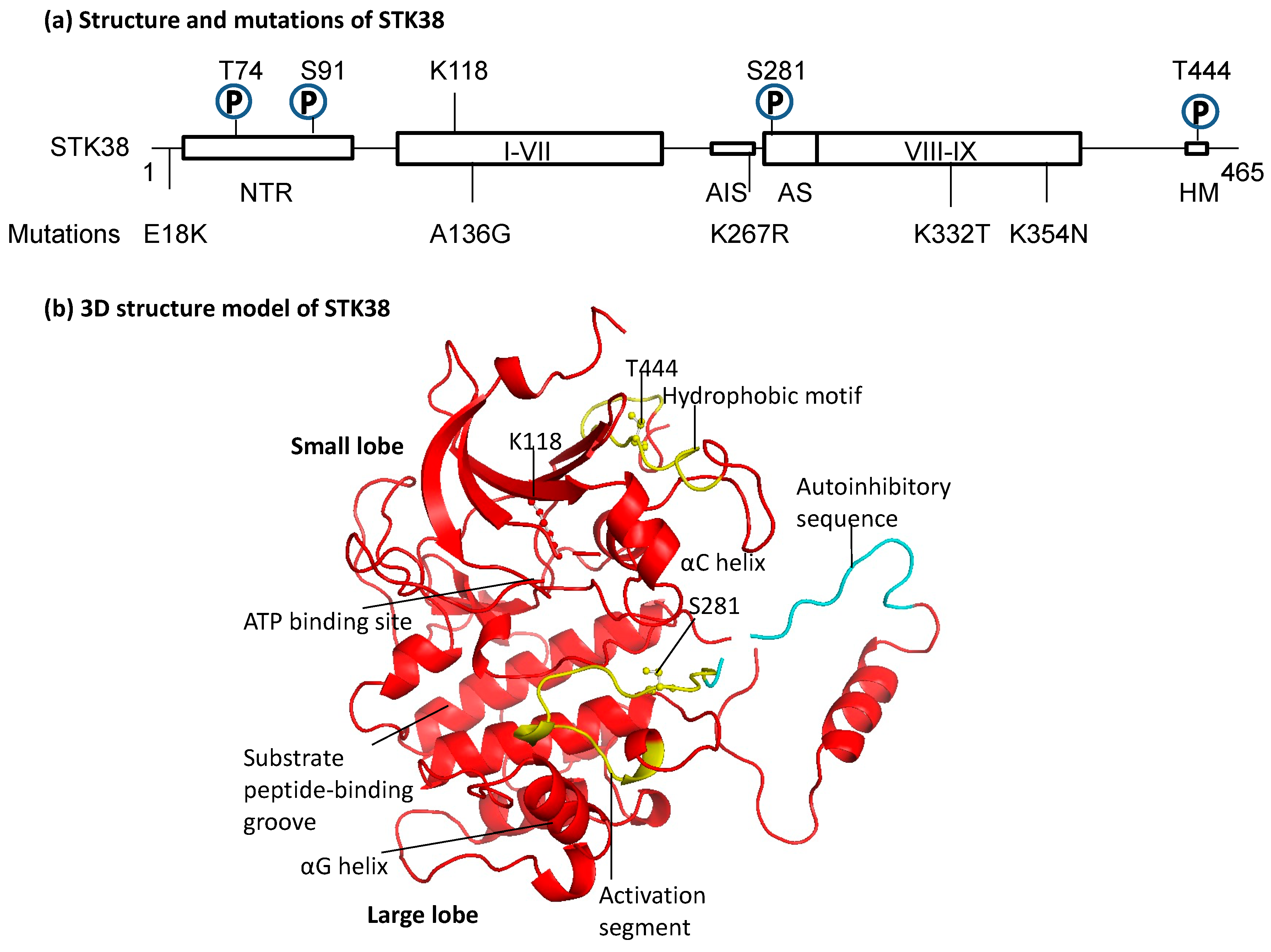
2.1. Structure of STK38
2.2. Regulation of Expression and Activity of STK38
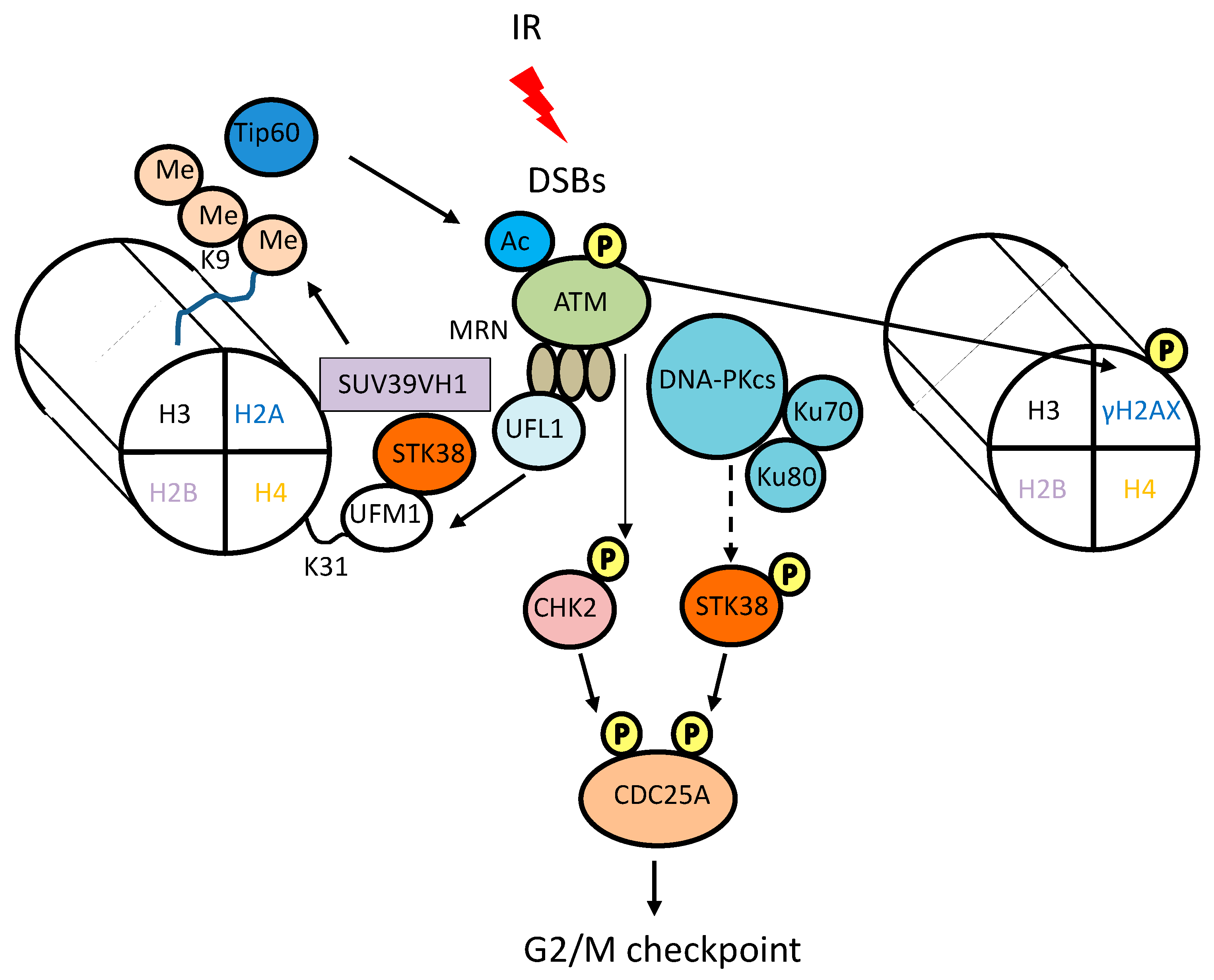
3. Involvement of STK38 in DNA Damage Signaling
4. Role of STK38 in DNA Damage-Induced Cell Cycle Checkpoint
5. Role of STK38 in Cell Proliferation, Cell Survival, and Autophagy
6. Involvement of STK38 in Malignancy
7. Targeting of STK38 for Radio-Sensitization
8. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tamaskovic, R.; Bichsel, S.J.; Hemmings, B.A. NDR Family of AGC Kinases--Essential Regulators of the Cell Cycle and Morphogenesis. FEBS Lett. 2003, 546, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Bidlingmaier, S.; Weiss, E.L.; Seidel, C.; Drubin, D.G.; Snyder, M. The Cbk1p Pathway Is Important for Polarized Cell Growth and Cell Separation in Saccharomyces Cerevisiae. Mol. Cell. Biol. 2001, 21, 2449. [Google Scholar] [CrossRef] [PubMed]
- Verde, F.; Wiley, D.J.; Nurse, P. Fission Yeast Orb6, a Ser/Thr Protein Kinase Related to Mammalian Rho Kinase and Myotonic Dystrophy Kinase, Is Required for Maintenance of Cell Polarity and Coordinates Cell Morphogenesis with the Cell Cycle. Proc. Natl. Acad. Sci. USA 1998, 95, 7526–7531. [Google Scholar] [CrossRef] [PubMed]
- Toyn, J.H.; Johnston, L.H. The Dbf2 and Dbf20 Protein Kinases of Budding Yeast Are Activated after the Metaphase to Anaphase Cell Cycle Transition. EMBO J. 1994, 13, 1103. [Google Scholar] [CrossRef]
- Zallen, J.A.; Peckol, E.L.; Tobin, D.M.; Bargmann, C.I. Neuronal Cell Shape and Neurite Initiation Are Regulated by the Ndr Kinase SAX-1, a Member of the Orb6/COT-1/Warts Serine/Threonine Kinase Family. Mol. Biol. Cell 2000, 11, 3177–3190. [Google Scholar] [CrossRef] [PubMed]
- Hergovich, A.; Stegert, M.R.; Schmitz, D.; Hemmings, B.A. NDR kinases regulate essential cell processes from yeast to humans. Nat. Rev. Mol. Cell Biol. 2006, 7, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Hergovich, A.; Lamla, S.; Nigg, E.A.; Hemmings, B.A. Centrosome-Associated NDR Kinase Regulates Centrosome Duplication. Mol. Cell 2007, 25, 625–634. [Google Scholar] [CrossRef]
- Chiba, S.; Ikeda, M.; Katsunuma, K.; Ohashi, K.; Mizuno, K. MST2- and Furry-Mediated Activation of NDR1 Kinase Is Critical for Precise Alignment of Mitotic Chromosomes. Curr. Biol. 2009, 19, 675–681. [Google Scholar] [CrossRef]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The Nuts and Bolts of AGC Protein Kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Komander, D.; Garg, R.; Wan, P.T.C.; Ridley, A.J.; Barford, D. Mechanism of Multi-Site Phosphorylation from a ROCK-I:RhoE Complex Structure. EMBO J. 2008, 27, 3175–3185. [Google Scholar] [CrossRef]
- Devroe, E.; Erdjument-Bromage, H.; Tempst, P.; Silver, P.A. Human Mob Proteins Regulate the NDR1 and NDR2 Serine-Threonine Kinases. J. Biol. Chem. 2004, 279, 24444–24451. [Google Scholar] [CrossRef] [PubMed]
- Bichsel, S.J.; Tamaskovic, R.; Stegert, M.R.; Hemmings, B.A. Mechanism of Activation of NDR (Nuclear Dbf2-Related) Protein Kinase by the HMOB1 Protein. J. Biol. Chem. 2004, 279, 35228–35235. [Google Scholar] [CrossRef] [PubMed]
- Millward, T.A.; Hess, D.; Hemmings, B.A. Ndr Protein Kinase Is Regulated by Phosphorylation on Two Conserved Sequence Motifs. J. Biol. Chem. 1999, 274, 33847–33850. [Google Scholar] [CrossRef] [PubMed]
- Tamaskovic, R.; Bichsel, S.J.; Rogniaux, H.; Stegert, M.R.; Hemmings, B.A. Mechanism of Ca2+-Mediated Regulation of NDR Protein Kinase through Autophosphorylation and Phosphorylation by an Upstream Kinase. J. Biol. Chem. 2003, 278, 6710–6718. [Google Scholar] [CrossRef]
- Enomoto, A.; Kido, N.; Ito, M.; Takamatsu, N.; Miyagawa, K. Serine-Threonine Kinase 38 Is Regulated by Glycogen Synthase Kinase-3 and Modulates Oxidative Stress-Induced Cell Death. Free Radic. Biol. Med. 2012, 52, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Fukasawa, T.; Tsumoto, H.; Karube, M.; Nakagawa, K.; Yoshizaki, A.; Sato, S.; Miura, Y.; Miyagawa, K. Prevention of Calpain-Dependent Degradation of STK38 by MEKK2-Mediated Phosphorylation. Sci. Rep. 2019, 9, 16010. [Google Scholar] [CrossRef]
- Cornils, H.; Stegert, M.R.; Hergovich, A.; Hynx, D.; Schmitz, D.; Dirnhofer, S.; Hemmings, B.A. Ablation of the Kinase NDR1 Predisposes Mice to the Development of T Cell Lymphoma. Sci. Signal. 2010, 3, ra47. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL Workspace: A Web-Based Environment for Protein Structure Homology Modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An Automated Protein Homology-Modeling Server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Stegert, M.R.; Tamaskovic, R.; Bichsel, S.J.; Hergovich, A.; Hemmings, B.A. Regulation of NDR2 Protein Kinase by Multi-Site Phosphorylation and the S100B Calcium-Binding Protein. J. Biol. Chem. 2004, 279, 23806–23812. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Fukasawa, T.; Takamatsu, N.; Ito, M.; Morita, A.; Hosoi, Y.; Miyagawa, K. The HSP90 Inhibitor 17-Allylamino-17-Demethoxygeldanamycin Modulates Radiosensitivity by Downregulating Serine/Threonine Kinase 38 via Sp1 Inhibition. Eur. J. Cancer 2013, 49, 3547–3558. [Google Scholar] [CrossRef] [PubMed]
- Devroe, E.; Silver, P.A.; Engelman, A. HIV-1 Incorporates and Proteolytically Processes Human NDR1 and NDR2 Serine-Threonine Kinases. Virology 2005, 331, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Stork, O.; Zhdanov, A.; Kudersky, A.; Yoshikawa, T.; Obata, K.; Pape, H.C. Neuronal Functions of the Novel Serine/Threonine Kinase Ndr2. J. Biol. Chem. 2004, 279, 45773–45781. [Google Scholar] [CrossRef]
- Martin, A.P.; Jacquemyn, M.; Lipecka, J.; Chhuon, C.; Aushev, V.N.; Meunier, B.; Singh, M.K.; Carpi, N.; Piel, M.; Codogno, P.; et al. STK38 Kinase Acts as XPO1 Gatekeeper Regulating the Nuclear Export of Autophagy Proteins and Other Cargoes. EMBO Rep. 2019, 20, e48150. [Google Scholar] [CrossRef] [PubMed]
- Paul, I.; Batth, T.S.; Iglesias-Gato, D.; Al-Araimi, A.; Al-Haddabi, I.; Alkharusi, A.; Norstedt, G.; Olsen, J.V.; Zadjali, F.; Flores-Morales, A. The ubiquitin ligase Cullin5SOCS2 regulates NDR1/STK38 stability and NF-κB transactivation. Sci. Rep. 2017, 7, 2800. [Google Scholar] [CrossRef]
- Millward, T.; Cron, P.; Hemmings, B.A. Molecular Cloning and Characterization of a Conserved Nuclear Serine(Threonine) Protein Kinase. Proc. Natl. Acad. Sci. USA 1995, 92, 5022–5026. [Google Scholar] [CrossRef]
- Hergovich, A.; Bichsel, S.J.; Hemmings, B.A. Human NDR Kinases Are Rapidly Activated by MOB Proteins through Recruitment to the Plasma Membrane and Phosphorylation. Mol. Cell. Biol. 2005, 25, 8259–8272. [Google Scholar] [CrossRef]
- Stegert, M.R.; Hergovich, A.; Tamaskovic, R.; Bichsel, S.J.; Hemmings, B.A. Regulation of NDR Protein Kinase by Hydrophobic Motif Phosphorylation Mediated by the Mammalian Ste20-like Kinase MST3. Mol. Cell. Biol. 2005, 25, 11019–11029. [Google Scholar] [CrossRef]
- Steel, G.G. Basic Clinical Radiobiology; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Yu, J.; Nowsheen, S.; Wang, M.; Tu, X.; Liu, T.; Li, H.; Wang, L.; Lou, Z. UFL1 promotes histone H4 ufmylation and ATM activation. Nat. Commun. 2019, 18, 1242. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Qin, B.; Yu, J.; Nowsheen, S.; Nowsheen, S.; Zhao, F.; Wang, L.; Lou, Z. STK38 Promotes ATM Activation by Acting as a Reader of Histone H4 Ufmylation. Sci. Adv. 2020, 6, eaax8214. [Google Scholar] [CrossRef]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Shiga, S.; Murata, Y.; Hashimoto, T.; Urushihara, Y.; Fujishima, Y.; Kudo, K.; Sonohara, Y.; Kurusu, M.; Takeda, K.; Jingu, K.; et al. DNA-PKcs Is Activated under Nutrient Starvation and Activates Akt, MST1, FoxO3a, and NDR1. Biochem. Biophys. Res. Commun. 2020, 521, 668–673. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Wassing, I.E.; Esashi, F. RAD51: Beyond the break. Semin. Cell Dev. Biol. 2021, 113, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, R.; Erdogan, M.K.; Ditsiou, A.; Spanswick, V.; Garcia-Gomez, J.J.; Hartley, J.A.; Esashi, F.; Hergovich, A.; Gomez, V. hMOB2 deficiency impairs homologous recombination-mediated DNA repair and sensitises cancer cells to PARP inhibitors. Cell Signal. 2021, 87, 110106. [Google Scholar] [CrossRef]
- Naegeli, H.; Sugasawa, K. The xeroderma pigmentosum pathway: Decision tree analysis of DNA quality. DNA Repair 2011, 10, 673–683. [Google Scholar] [CrossRef]
- Park, J.M.; Choi, J.Y.; Yi, J.M.; Chung, J.W.; Leem, S.H.; Koh, S.S.; Kang, T.H. NDR1 modulates the UV-induced DNA-damage checkpoint and nucleotide excision repair. Biochem. Biophys. Res. Commun. 2015, 5, 543–548. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, M.; Draetta, G.F. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003, 4, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Karlsson-Rosenthal, C.; Millar, J.B. Cdc25: Mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006, 16, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, T.; Enomoto, A.; Miyagawa, K. Serine-Threonine Kinase 38 Regulates CDC25A Stability and the DNA Damage-Induced G2/M Checkpoint. Cell. Signal. 2015, 27, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Tong, X.; Ye, X. Cyclin D1 promotes cell cycle progression through enhancing NDR1/2 kinase activity independent of cyclin-dependent kinase 4. J. Biol. Chem. 2013, 288, 26678–26687. [Google Scholar] [CrossRef] [PubMed]
- Cornils, H.; Kohler, R.S.; Hergovich, A.; Hemmings, B.A. Human NDR kinases control G(1)/S cell cycle transition by directly regulating p21 stability. Mol. Cell Biol. 2011, 31, 1382–1395. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Bisikirska, B.C.; Adam, S.J.; Alvarez, M.J.; Rajbhandari, P.; Cox, R.; Lefebvre, C.; Wang, K.; Rieckhof, G.E.; Felsher, D.W.; Califano, A. STK38 Is a Critical Upstream Regulator of MYC’s Oncogenic Activity in Human B-Cell Lymphoma. Oncogene 2013, 32, 5283–5291. [Google Scholar] [CrossRef]
- Shi, D.D.; Shi, H.; Lu, D.; Li, R.; Zhang, Y.; Zhang, J. NDR1/STK38 potentiates NF-κB activation by its kinase activity. Cell Biochem. Funct. 2012, 30, 664–670. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Sig. Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Kido, N.; Ito, M.; Morita, A.; Matsumoto, Y.; Takamatsu, N.; Hosoi, Y.; Miyagawa, K. Negative Regulation of MEKK1/2 Signaling by Serine-Threonine Kinase 38 (STK38). Oncogene 2008, 27, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA damage response. Int. J. Mol. Sci. 2015, 16, 2641–2662. [Google Scholar] [CrossRef]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef]
- Joffre, C.; Dupont, N.; Hoa, L.; Gomez, V.; Pardo, R.; Gonçalves-Pimentel, C.; Achard, P.; Bettoun, A.; Meunier, B.; Bauvy, C.; et al. The Pro-apoptotic STK38 Kinase Is a New Beclin1 Partner Positively Regulating Autophagy. Curr. Biol. 2015, 25, 2479–2492. [Google Scholar] [CrossRef]
- Roşianu, F.; Mihaylov, S.R.; Eder, N.; Martiniuc, A.; Claxton, S.; Flynn, H.R.; Jalal, S.; Domart, M.C.; Collinson, L.; Skehel, M.; et al. Loss of NDR1/2 kinases impairs endomembrane trafficking and autophagy leading to neurodegeneration. Life Sci. Alliance 2022, 6, e202201712. [Google Scholar] [CrossRef]
- Adeyinka, A.; Emberley, E.; Niu, Y.; Snell, L.; Murphy, L.C.; Sowter, H.; Wykoff, C.C.; Harris, A.L.; Watson, P.H. Analysis of gene expression in ductal carcinoma in situ of the breast. Clin. Cancer Res. 2002, 8, 3788–3795. [Google Scholar]
- Welsh, J.B.; Zarrinkar, P.P.; Sapinoso, L.M.; Kern, S.G.; Behling, C.A.; Monk, B.J.; Lockhart, D.J.; Burger, R.A.; Hampton, G.M. Analysis of gene expression profiles in normal and neoplastic ovarian tissue samples identifies candidate molecular markers of epithelial ovarian cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 1176–1181. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Richards, W.G.; Staunton, J.; Li, C.; Monti, S.; Vasa, P.; Ladd, C.; Beheshti, J.; Bueno, R.; Gillette, M.; et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. USA 2001, 98, 13790–13795. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Wan, X.Y.; Liu, C.Q.; Zheng, F.M. NDR1 Increases NOTCH1 Signaling Activity by Impairing Fbw7 Mediated NICD Degradation to Enhance Breast Cancer Stem Cell Properties. Mol. Med. 2022, 28, 49. [Google Scholar] [CrossRef] [PubMed]
- Bettoun, A.; Joffre, C.; Zago, G.; Surdez, D.; Vallerand, D.; Gundogdu, R.; Sharif, A.A.D.; Gomez, M.; Cascone, I.; Meunier, B.; et al. Correction: Mitochondrial clearance by the STK38 kinase supports oncogenic Ras-induced cell transformation. Oncotarget 2018, 9, 22870. [Google Scholar] [CrossRef]
- Ji, C.D.; Wang, Y.X.; Xiang, D.F.; Liu, Q.; Zhou, Z.H.; Qian, F.; Yang, L.; Ren, Y.; Cui, W.; Xu, S.L.; et al. Kir2.1 Interaction with Stk38 Promotes Invasion and Metastasis of Human Gastric Cancer by Enhancing MEKK2-MEK1/2-ERK1/2 Signaling. Cancer Res. 2018, 78, 3041–3053. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, Z.; Zheng, Z.; Li, J.; Yang, K.; Xu, C.; Liu, Q.; Gong, Z.; Yang, Y.; Zhao, Y.; et al. Prognostic and Immunological Role of STK38 across Cancers: Friend or Foe? Int. J. Mol. Sci. 2022, 23, 11590. [Google Scholar] [CrossRef]
- Chen, B.; Liu, B.; Yu, T.; Han, Y.F.; Wu, C.; Wang, Z.Y. Nuclear Dbf2-related Kinase 1 functions as tumor suppressor in glioblastoma by phosphorylation of Yes-associated protein. Chin. Med. J. 2021, 134, 2054–2065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tang, F.; Terracciano, L.; Hynx, D.; Kohler, R.; Bichet, S.; Hess, D.; Cron, P.; Hemmings, B.A.; Hergovich, A.; et al. NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr. Biol. 2015, 25, 296–305. [Google Scholar] [CrossRef]
- Grant, T.J.; Mehta, A.K.; Gupta, A.; Sharif, A.A.D.; Arora, K.S.; Deshpande, V.; Ting, D.T.; Bardeesy, N.; Ganem, N.J.; Hergovich, A.; et al. STK38L kinase ablation promotes loss of cell viability in a subset of KRAS-dependent pancreatic cancer cell lines. Oncotarget 2017, 8, 78556–78572. [Google Scholar] [CrossRef]
- Keller, M.; Dubois, F.; Teulier, S.; Martin, A.P.J.; Levallet, J.; Maille, E.; Brosseau, S.; Elie, N.; Hergovich, A.; Bergot, E.; et al. NDR2 kinase contributes to cell invasion and cytokinesis defects induced by the inactivation of RASSF1A tumor-suppressor gene in lung cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 158. [Google Scholar] [CrossRef]
- Pearl, L.H.; Prodromou, C.; Workman, P. The Hsp90 molecular chaperone: An open and shut case for treatment. Biochem. J. 2008, 410, 439–453. [Google Scholar] [CrossRef]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumor selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Roti Roti, J.L. Cellular responses to hyperthermia (40–46 °C): Cell killing and molecular events. Int. J. Hyperth. 2008, 24, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Zaidi, S.F.; Rehman, R.; Kondo, T. Hyperthermia and protein homeostasis: Cytoprotection and cell death. J. Therm. Biol. 2020, 91, 102615. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Fukasawa, T.; Terunuma, H.; Nakagawa, K.; Yoshizaki, A.; Sato, S.; Miyagawa, K. Decrease in MAP3Ks Expression Enhances the Cell Death Caused by Hyperthermia. Int. J. Hyperth. 2022, 39, 200–208. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukasawa, T.; Enomoto, A.; Yoshizaki-Ogawa, A.; Sato, S.; Miyagawa, K.; Yoshizaki, A. The Role of Mammalian STK38 in DNA Damage Response and Targeting for Radio-Sensitization. Cancers 2023, 15, 2054. https://doi.org/10.3390/cancers15072054
Fukasawa T, Enomoto A, Yoshizaki-Ogawa A, Sato S, Miyagawa K, Yoshizaki A. The Role of Mammalian STK38 in DNA Damage Response and Targeting for Radio-Sensitization. Cancers. 2023; 15(7):2054. https://doi.org/10.3390/cancers15072054
Chicago/Turabian StyleFukasawa, Takemichi, Atsushi Enomoto, Asako Yoshizaki-Ogawa, Shinichi Sato, Kiyoshi Miyagawa, and Ayumi Yoshizaki. 2023. "The Role of Mammalian STK38 in DNA Damage Response and Targeting for Radio-Sensitization" Cancers 15, no. 7: 2054. https://doi.org/10.3390/cancers15072054
APA StyleFukasawa, T., Enomoto, A., Yoshizaki-Ogawa, A., Sato, S., Miyagawa, K., & Yoshizaki, A. (2023). The Role of Mammalian STK38 in DNA Damage Response and Targeting for Radio-Sensitization. Cancers, 15(7), 2054. https://doi.org/10.3390/cancers15072054