
Propagated Circulating Tumor Cells Uncover the Potential Role of NFκB, EMT, and TGFβ Signaling Pathways and COP1 in Metastasis

 , , , , ,
, , , , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. CTCs Can Be Established from Multiple Cancer Types
2.2. CDXs Can Be Established from CTCs Expanded In Vitro
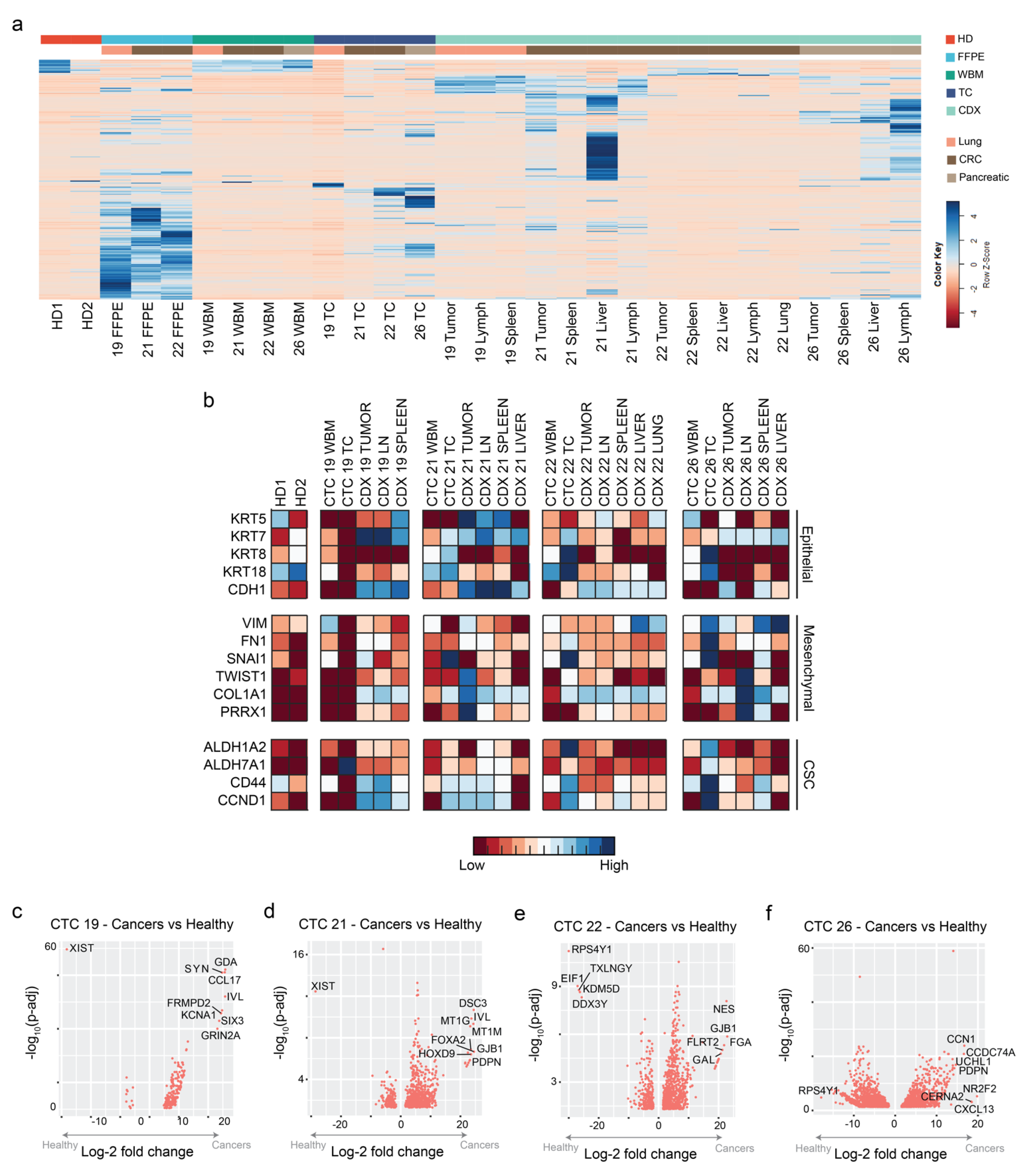
2.3. CTCs and CDXs Express Transcriptomic Heterogeneity
2.4. Five Genes Are Associated with High-Risk CTCs
2.5. NF-κB, EMT and TGFβ as Putative Pathways for CTCs
2.6. CDXs Can Be Queried for Metastasis-Driving Mutations
3. Discussion
4. Methods
4.1. Patient Enrollment
4.2. CTC Isolation from Whole Blood Using FiColl-Paque
4.3. CTC Cultures
4.4. CTC-Derived Xenograft Models
4.5. RNA/DNA Extraction
4.6. Immunohistochemistry and Immunofluorescence
4.7. Quantitative Real-Time PCR
4.8. Next-Generation RNA and Whole-Exome Sequencing Library Preparation and Sequencing
4.9. Bulk RNA-Sequencing Bioinformatics Analysis
4.10. Whole-Exome-Sequencing Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CTC | Circulating Tumor Cell |
| CDX | Circulating Tumor Cell-derived Xenograft |
| EMT | Epithelial Mesenchymal Transition |
| TCGA | The Cancer Genome Atlas |
| FFPE | Formalin-fixed Paraffin-embedded |
| qRT-PCR | Quantitative Real-Time Polymerase Chain Reaction |
| IF | Immunofluorescent |
| H&E | Hematoxylin & Eosin |
| HD | Healthy donor |
| WBM | Whole blood matched |
| TC | Tissue Culture |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GSEA | Gene Set Enrichment Analysis |
| GO | Gene Ontology |
| SNP | Single Nucleotide Polymorphism |
| LN | Lymph Node |
| WES | Whole exome sequencing |
| BWA | Burrows-Wheeler Aligner |
| IRB | Institutional Review Board |
| SRBSR | Survey, Recruitment, and Biospecimen Collection Shared Resource |
| HBSS | Hank’s Balanced Salt Solution |
| PBS | Phosphate Buffered Solution |
| IACUC | Institutional Animal Care and Use Committee |
References
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Dillekas, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer. Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, C.; Soto, A.M. Cancer Metastases: So Close and So Far. J. Natl. Cancer. Inst. 2015, 107, djv236. [Google Scholar] [CrossRef]
- Ashworth, T.R. A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Australas. Med. J. 1869, 14, 146–147. [Google Scholar]
- Anderson, R.L.; Balasa, T.; Callaghan, J.; Coombes, R.; Evans, J.; Hall, J.A.; Kinrade, S.; Jones, D.; Jones, P.S.; Jones, R.; et al. A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 2019, 16, 185–204. [Google Scholar] [CrossRef]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kunda, R.; et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 2022, 185, 563–575.e11. [Google Scholar] [CrossRef]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Lang, L.; Shay, C.; Xiong, Y.; Thakkar, P.; Chemmalakuzhy, R.; Wang, X.; Teng, Y. Combating head and neck cancer metastases by targeting Src using multifunctional nanoparticle-based saracatinib. J. Hematol. Oncol. 2018, 11, 85. [Google Scholar] [CrossRef]
- Posadas, E.M.; Ahmad, R.S.; Karrison, T.; Szmulewitz, R.Z.; O’Connell, P.H.; Wade, J.L.; Shen, J.; Gururajan, M.; Sievert, M.; Stadler, W.M. Saracatinib as a metastasis inhibitor in metastatic castration-resistant prostate cancer: A University of Chicago Phase 2 Consortium and DOD/PCF Prostate Cancer Clinical Trials Consortium Study. Prostate 2016, 76, 286–293. [Google Scholar] [CrossRef]
- Xiao, J.; Pohlmann, P.R.; Isaacs, C.; Weinberg, B.A.; He, A.R.; Schlegel, R.; Agarwal, S. Circulating Tumor Cells: Technologies and Their Clinical Potential in Cancer Metastasis. Biomedicines 2021, 9, 1111. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Liu, H.E.; Vuppalapaty, M.; Wilkerson, C.; Renier, C.; Chiu, M.; Lemaire, C.; Che, J.; Matsumoto, M.; Carroll, J. Detection of EGFR Mutations in cfDNA and CTCs, and Comparison to Tumor Tissue in Non-Small-Cell-Lung-Cancer (NSCLC) Patients. Front. Oncol. 2020, 10, 572895. [Google Scholar] [CrossRef]
- De Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V.; Terstappen, L.W.W.M.; Pienta, K.J.; Raghavan, D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef] [PubMed]
- Alvarez Cubero, M.J.; Lorente, J.A.; Robles-Fernandez, I.; Rodriguez-Martinez, A.; Puche, J.L.; Serrano, M.J. Circulating Tumor Cells: Markers and Methodologies for Enrichment and Detection. Methods Mol. Biol. 2017, 1634, 283–303. [Google Scholar]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5, 180ra48. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bauerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef]
- Cayrefourcq, L.; Mazard, T.; Joosse, S.; Solassol, J.; Ramos, J.; Assenat, E.; Schumacher, U.; Costes, V.; Maudelonde, T.; Pantel, K.; et al. Establishment and characterization of a cell line from human circulating colon cancer cells. Cancer Res. 2015, 75, 892–901. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Brungs, D.; Minaei, E.; Piper, A.; Perry, J.; Splitt, A.; Carolan, M.; Ryan, S.; Wu, X.J.; Corde, S.; Tehei, M.; et al. Establishment of novel long-term cultures from EpCAM positive and negative circulating tumour cells from patients with metastatic gastroesophageal cancer. Sci. Rep. 2020, 10, 539. [Google Scholar] [CrossRef]
- Rivera-Baez, L.; Lohse, I.; Lin, E.; Raghavan, S.; Owen, S.; Harouaka, R.; Herman, K.; Mehta, G.; Lawrence, T.S.; Morgan, M.A.; et al. Expansion of Circulating Tumor Cells from Patients with Locally Advanced Pancreatic Cancer Enable Patient Derived Xenografts and Functional Studies for Personalized Medicine. Cancers 2020, 12, 1011. [Google Scholar] [CrossRef]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.; Saturno, G.; Furney, S.J.; et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Drapkin, B.J.; George, J.; Christensen, C.L.; Mino-Kenudson, M.; Dries, R.; Sundaresan, T.; Phat, S.; Myers, D.T.; Zhong, J.; Igo, P.; et al. Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts. Cancer Discov. 2018, 8, 600–615. [Google Scholar] [CrossRef] [PubMed]
- Faugeroux, V.; Pailler, E.; Oulhen, M.; Deas, O.; Brulle-Soumare, L.; Hervieu, C.; Marty, V.; Alexandrova, K.; Andree, K.C.; Stoecklein, N.H.; et al. Genetic characterization of a unique neuroendocrine transdifferentiation prostate circulating tumor cell-derived eXplant model. Nat. Commun. 2020, 11, 1884. [Google Scholar] [CrossRef]
- Xiao, J.; McGill, J.R.; Stanton, K.; Kassner, J.D.; Choudhury, S.; Schlegel, R.; Sauna, Z.E.; Pohlmann, P.R.; Agarwal, S. Efficient Propagation of Circulating Tumor Cells: A First Step for Probing Tumor Metastasis. Cancers 2020, 12, 2784. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.; Kuske, A.; Joosse, S.A.; Yigit, G.; Sflomos, G.; Thaler, S.; Smit, D.J.; Werner, S.; Borgmann, K.; Gärtner, S.; et al. Characterization of circulating breast cancer cells with tumorigenic and metastatic capacity. EMBO Mol. Med. 2020, 12, e11908. [Google Scholar] [CrossRef]
- Lallo, A.; Schenk, M.W.; Frese, K.K.; Blackhall, F.; Dive, C. Circulating tumor cells and CDX models as a tool for preclinical drug development. Transl. Lung Cancer Res. 2017, 6, 397–408. [Google Scholar] [CrossRef]
- Hodgkinson, C.L.; Morrow, C.J.; Li, Y.; Metcalf, R.L.; Rothwell, D.G.; Trapani, F.; Polanski, R.; Burt, D.J.; Simpson, K.L.; Morris, K.; et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat. Med. 2014, 20, 897–903. [Google Scholar] [CrossRef]
- Carmona-Ule, N.; González-Conde, M.; Abuín, C.; Cueva, J.F.; Palacios, P.; López-López, R.; Costa, C.; Dávila-Ibáñez, A.B. Short-Term Ex Vivo Culture of CTCs from Advance Breast Cancer Patients: Clinical Implications. Cancers 2021, 13, 2668. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Rossi, E.; Rugge, M.; Facchinetti, A.; Pizzi, M.; Nardo, G.; Barbieri, V.; Manicone, M.; Faveri, S.D.; Scaini, M.C.; Basso, U.; et al. Retaining the long-survive capacity of Circulating Tumor Cells (CTCs) followed by xeno-transplantation: Not only from metastatic cancer of the breast but also of prostate cancer patients. Oncoscience 2014, 1, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Veiga, T.; Abreu, M.; Robledo, D.; Matias-Guiu, X.; Santacana, M.; Sanchez, L.; Cueva, J.; Palacios, P.; Abdulkader, I.; Lopez-Lopez, R.; et al. CTCs-derived xenograft development in a triple negative breast cancer case. Int. J. Cancer 2019, 144, 2254–2265. [Google Scholar] [CrossRef]
- Morrow, C.J.; Trapani, F.; Metcalf, R.L.; Bertolini, G.; Hodgkinson, C.L.; Khandelwal, G.; Kelly, P.; Galvin, M.; Carter, L.; Simpson, K.L.; et al. Tumourigenic non-small-cell lung cancer mesenchymal circulating tumour cells: A clinical case study. Ann. Oncol. 2016, 27, 1155–1160. [Google Scholar] [CrossRef]
- Tayoun, T.; Faugeroux, V.; Oulhen, M.; Déas, O.; Michels, J.; Brulle-Soumare, L.; Cairo, S.; Scoazec, J.-Y.; Marty, V.; Aberlenc, A.; et al. Targeting genome integrity dysfunctions impedes metastatic potency in non-small cell lung cancer circulating tumor cell-derived explants. JCI Insight 2022, 7, e155804. [Google Scholar] [CrossRef]
- Liu, X. Lymphomagenesis in Circulating Tumor Cell-Derived Xenograft Models. Biomed. J. Sci. Technical. Res. 2019, 18, 13247–13249. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.T.; Bardia, A.; Spring, L.M.; Giobbie-Hurder, A.; Kalinich, M.; Dubash, T.; Sundaresan, T.; Hong, X.; LiCausi, J.A.; Ho, U.; et al. A Digital RNA Signature of Circulating Tumor Cells Predicting Early Therapeutic Response in Localized and Metastatic Breast Cancer. Cancer Discov. 2018, 8, 1286–1299. [Google Scholar] [CrossRef]
- Kalinich, M.; Bhan, I.; Kwan, T.T.; Miyamoto, D.T.; Javaid, S.; LiCausi, J.A.; Milner, J.D.; Hong, X.; Goyal, L.; Sil, S.; et al. An RNA-based signature enables high specificity detection of circulating tumor cells in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 1123–1128. [Google Scholar] [CrossRef]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.-F.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; Vijver, M.J.; van de Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic. Acids. Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Agnoletto, C.; Corrà, F.; Minotti, L.; Baldassari, F.; Crudele, F.; Cook, W.J.J.; Leva, G.D.; d’Adamo, A.P.; Gasparini, P.; Volinia, S. Heterogeneity in Circulating Tumor Cells: The Relevance of the Stem-Cell Subset. Cancers 2019, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhou, J.; Fan, J.; Yang, X. Single-cell RNA sequencing reveals spatial heterogeneity and immune evasion of circulating tumor cells. Cancer Biol. Med. 2021, 18, 934. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef]
- Kulemann, B.; Rösch, S.; Seifert, S.; Timme, S.; Bronsert, P.; Seifert, G.; Martini, V.; Kuvendjiska, J.; Glatz, T.; Hussung, S.; et al. Author Correction: Pancreatic cancer: Circulating Tumor Cells and Primary Tumors show Heterogeneous KRAS Mutations. Sci. Rep. 2017, 7, 14631. [Google Scholar] [CrossRef]
- Deng, G.; Krishnakumar, S.; Powell, A.A.; Zhang, H.; Mindrinos, M.N.; Telli, M.L.; Davis, R.W.; Jeffrey, S.S. Single cell mutational analysis of PIK3CA in circulating tumor cells and metastases in breast cancer reveals heterogeneity, discordance, and mutation persistence in cultured disseminated tumor cells from bone marrow. BMC Cancer 2014, 14, 456. [Google Scholar] [CrossRef] [PubMed]
- Maheswaran, S.; Sequist, L.V.; Nagrath, S.; Ulkus, L.; Brannigan, B.; Collura, C.V.; Inserra, E.; Diederichs, S.; Iafrate, A.J.; Bell, D.W.; et al. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl. J. Med. 2008, 359, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Auer, M.; Gasch, C.; Pichler, M.; Ulz, P.; Hoffmann, E.M.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Lackner, C.; et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013, 73, 2965–2975. [Google Scholar] [CrossRef] [PubMed]
- Lyberopoulou, A.; Aravantinos, G.; Efstathopoulos, E.P.; Nikiteas, N.; Bouziotis, P.; Isaakidou, A.; Papalois, A.; Marinos, E.; Gazouli, M. Mutational analysis of circulating tumor cells from colorectal cancer patients and correlation with primary tumor tissue. PLoS ONE 2015, 10, e0123902. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Stott, S.L.; Hsu, C.-H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef]
- Lowes, L.E.; Goodale, D.; Xia, Y.; Postenka, C.; Piaseczny, M.M.; Paczkowski, F.; Allan, A.L. Epithelial-mesenchymal transitioned circulating tumor cells capture for detecting tumor progression. Clin Cancer Res. 2015, 21, 899–906. [Google Scholar]
- Ting, D.T.; Wittner, B.S.; Ligorio, M.; Jordan, N.V.; Shah, A.M.; Miyamoto, D.T.; Aceto, N.; Bersani, F.; Brannigan, B.W.; Xega, K.; et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014, 8, 1905–1918. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-alpha therapies: The next generation. Nat. Rev. Drug. Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef]
- Li, F.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. NF-kappaB in cancer therapy. Arch. Toxicol. 2015, 89, 711–731. [Google Scholar] [CrossRef]
- Guo, A.K.; Itahana, Y.; Seshachalam, V.P.; Chow, H.Y.; Ghosh, S.; Itahana, K. Mutant TP53 interacts with BCAR1 to contribute to cancer cell invasion. Br. J. Cancer. 2021, 124, 299–312. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Zhang, J.; Zhong, J.; Yang, R. COL1A1 promotes metastasis in colorectal cancer by regulating the WNT/PCP pathway. Mol. Med. Rep. 2018, 17, 5037–5042. [Google Scholar] [CrossRef]
- Ma, H.-P.; Chang, H.-L.; Bamodu, O.A.; Yadav, V.K.; Huang, T.-Y.; Wu, A.T.H.; Yeh, C.-T.; Tsai, S.-H.; Lee, W.-H. Collagen 1A1 (COL1A1) Is a Reliable Biomarker and Putative Therapeutic Target for Hepatocellular Carcinogenesis and Metastasis. Cancers 2019, 11, 786. [Google Scholar] [CrossRef] [PubMed]
- Sheng, P.; Zhu, H.; Zhang, W.; Xu, Y.; Peng, W.; Sun, J.; Gu, M.; Jiang, H. The immunoglobulin superfamily member 3 (IGSF3) promotes hepatocellular carcinoma progression through activation of the NF-kappaB pathway. Ann. Transl. Med. 2020, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Lee, I.; Kim, S.T.; Lee, J.; Kim, K.-M.; Park, J.O.; Kang, W.K. RRAD expression in gastric and colorectal cancer with peritoneal carcinomatosis. Sci. Rep. 2019, 9, 19439. [Google Scholar] [CrossRef]
- Shang, R.; Wang, J.; Sun, W.; Dai, B.; Ruan, B.; Zhang, Z.; Yang, X.; Gao, Y.; Qu, S.; Lv, X.; et al. RRAD inhibits aerobic glycolysis, invasion, and migration and is associated with poor prognosis in hepatocellular carcinoma. Tumour. Biol. 2016, 37, 5097–5105. [Google Scholar] [CrossRef]
- Mo, J.; Zhao, X.; Wang, W.; Zhao, N.; Dong, X.; Zhang, Y.; Cheng, R.; Sun, B. TFPI2 Promotes Perivascular Migration in an Angiotropism Model of Melanoma. Front Oncol. 2021, 11, 662434. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Huang, W.; Li, W.; Chen, S.; Chen, W.; Zhou, Y.; Peng, P.; Gu, W. TFPI-2 suppresses breast cancer cell proliferation and invasion through regulation of ERK signaling and interaction with actinin-4 and myosin-9. Sci. Rep. 2018, 8, 14402. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Kripke, M.L. Metastasis results from preexisting variant cells within a malignant tumor. Science 1977, 197, 893–895. [Google Scholar] [CrossRef]
- Shao, J.; Teng, Y.; Padia, R.; Hong, S.; Noh, H.; Xie, X.; Mumm, J.S.; Dong, Z.; Ding, H.-F.; Cowell, J.; et al. COP1 and GSK3beta cooperate to promote c-Jun degradation and inhibit breast cancer cell tumorigenesis. Neoplasia 2013, 15, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Hinz, N.; Jucker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal 2019, 17, 154. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Chen, M.; Broude, E.V.; Roninson, I.B. Role of transcription-regulating kinase CDK8 in colon cancer metastasis. Oncotarget 2019, 10, 622–623. [Google Scholar] [CrossRef]
- Digumarthy, S.R.; Mendoza, D.P.; Padole, A.; Chen, T.; Peterson, P.G.; Piotrowska, Z.; Sequist, L.V. Diffuse Lung Metastases in EGFR-Mutant Non-Small Cell Lung Cancer. Cancers 2019, 11, 1360. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, S.I.; Rimoldi, D.; Iseli, C.; Valsesia, A.; Robyr, D.; Gehrig, C.; Harshman, K.; Guipponi, M.; Bukach, O.; Zoete, V.; et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat. Genet. 2011, 44, 133–139. [Google Scholar] [CrossRef]
- Sethi, N.; Kang, Y. Notch signalling in cancer progression and bone metastasis. Br. J. Cancer 2011, 105, 1805–1810. [Google Scholar] [CrossRef]
- Lv, S.; Xu, W.; Zhang, Y.; Zhang, J.; Dong, X. NUF2 as an anticancer therapeutic target and prognostic factor in breast cancer. Int. J. Oncol. 2020, 57, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. BioRxiv 2021, 060012. [Google Scholar]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Anagnostou, V.; Lytle, K.; Parpart-Li, S.; Nesselbush, M.; Riley, D.R.; Shukla, M.; Chesnick, B.; Kadan, M.; Papp, E.; et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci. Transl. Med. 2015, 7, 283ra53. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Mutation |
Variant Frequency in
Biopsy | CDX Metastasis | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient FFPE | CTC Culture | CDX Tumor | Spleen | LN | Liver | Lung | |||
| 21 | APC c.1631T > C | 51% | Y * | Y | Y | ||||
| APC c.2521T > G | 8% | N ** | N | N | |||||
| APC c.2805C > A | 34% | Y | N | N | |||||
| APC c.6873A > T | 47% | Y | Y | Y | |||||
| PPP2R1A c.548G > A | 38% | Y | N | N | |||||
| TP53 c.743G > A | 53% | Y | Y | Y | |||||
| 22 | TP53 c.743G > T | 48% | Y | Y | N | N | N | N | N |
| 26 | SF3B1 c.2098A > G | 11% | Y | Y | Y | Y | Y | Y | |
| TP53 c.536A > G | 28% | Y | N | N | N | N | N | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, J.; Sharma, U.; Arab, A.; Miglani, S.; Bhalla, S.; Suguru, S.; Suter, R.; Mukherji, R.; Lippman, M.E.; Pohlmann, P.R.; et al. Propagated Circulating Tumor Cells Uncover the Potential Role of NFκB, EMT, and TGFβ Signaling Pathways and COP1 in Metastasis. Cancers 2023, 15, 1831. https://doi.org/10.3390/cancers15061831
Xiao J, Sharma U, Arab A, Miglani S, Bhalla S, Suguru S, Suter R, Mukherji R, Lippman ME, Pohlmann PR, et al. Propagated Circulating Tumor Cells Uncover the Potential Role of NFκB, EMT, and TGFβ Signaling Pathways and COP1 in Metastasis. Cancers. 2023; 15(6):1831. https://doi.org/10.3390/cancers15061831
Chicago/Turabian StyleXiao, Jerry, Utsav Sharma, Abolfazl Arab, Sohit Miglani, Sonakshi Bhalla, Shravanthy Suguru, Robert Suter, Reetu Mukherji, Marc E. Lippman, Paula R. Pohlmann, and et al. 2023. "Propagated Circulating Tumor Cells Uncover the Potential Role of NFκB, EMT, and TGFβ Signaling Pathways and COP1 in Metastasis" Cancers 15, no. 6: 1831. https://doi.org/10.3390/cancers15061831
APA StyleXiao, J., Sharma, U., Arab, A., Miglani, S., Bhalla, S., Suguru, S., Suter, R., Mukherji, R., Lippman, M. E., Pohlmann, P. R., Zeck, J. C., Marshall, J. L., Weinberg, B. A., He, A. R., Noel, M. S., Schlegel, R., Goodarzi, H., & Agarwal, S. (2023). Propagated Circulating Tumor Cells Uncover the Potential Role of NFκB, EMT, and TGFβ Signaling Pathways and COP1 in Metastasis. Cancers, 15(6), 1831. https://doi.org/10.3390/cancers15061831