
REV7 in Cancer Biology and Management
Abstract
Simple Summary
Abstract
1. Introduction
2. Protein Structure of REV7
3. Role of REV7 in Translesion DNA Synthesis and DNA Damage-Induced Mutagenesis
4. REV7 in Other DNA Repair Pathways
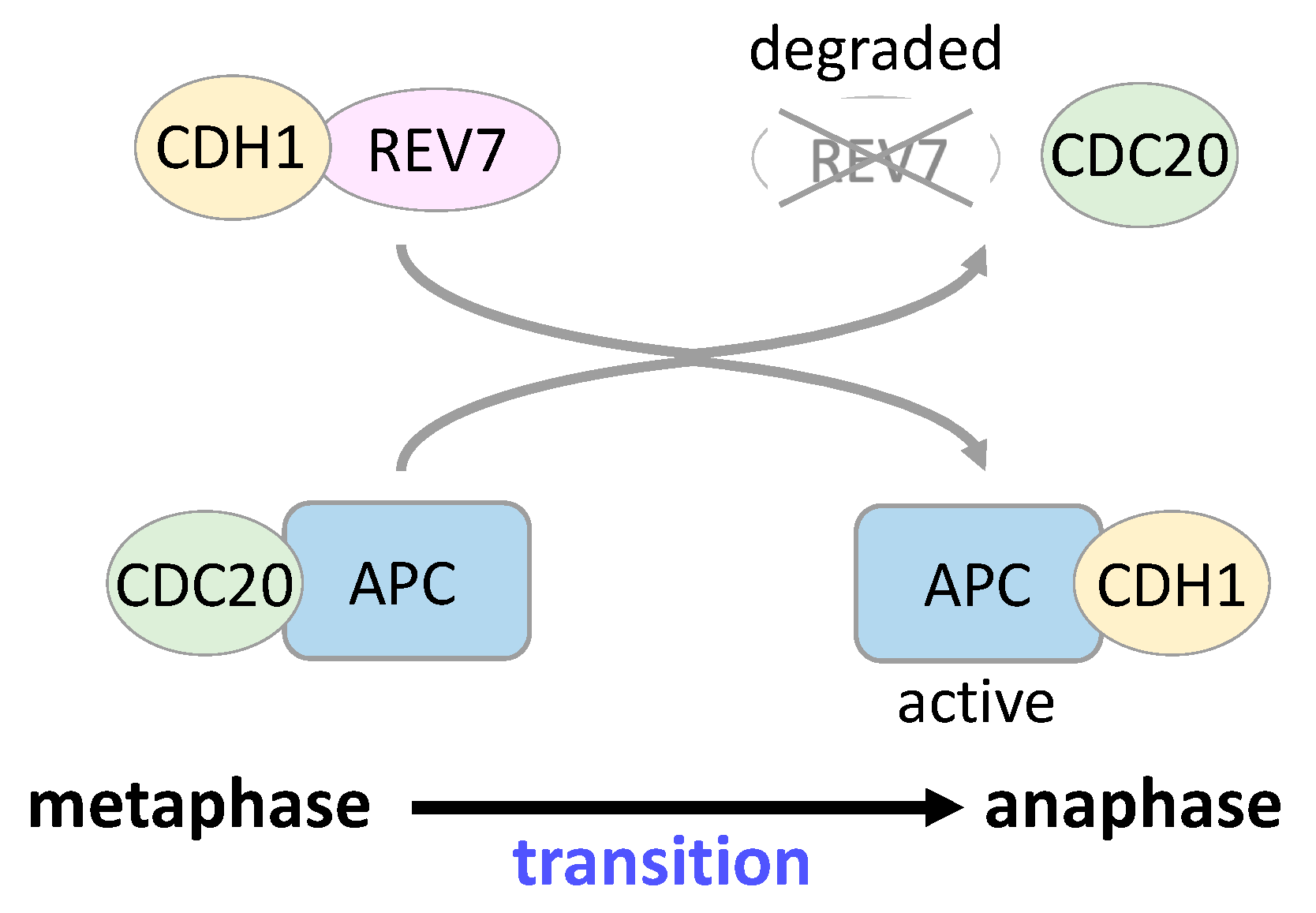
5. REV7 in Cell Cycle Regulation and Gene Transcription
6. REV7 in Cancer
6.1. Possible Role of REV7 in Cancer Development
6.2. Clinical Significance of REV7 in Cancer
6.3. REV7 in Cancer Cell Biology
6.4. Development of Inhibitors Targeting TLS Associated with REV7
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lawrence, C.W.; Das, G.; Christensen, R.B. REV7, a new gene concerned with UV mutagenesis in yeast. Mol. Gen. Genet. 1985, 200, 80–85. [Google Scholar] [CrossRef]
- Lawrence, C.W.; Nisson, P.E.; Christensen, R.B. UV and chemical mutagenesis in rev7 mutants of yeast. Mol. Gen. Genet. 1985, 200, 86–91. [Google Scholar] [CrossRef]
- Torpey, L.E.; Gibbs, P.E.; Nelson, J.; Lawrence, C.W. Cloning and sequence of REV7, a gene whose function is required for DNA damage-induced mutagenesis in Saccharomyces cerevisiae. Yeast 1994, 10, 1503–1509. [Google Scholar] [CrossRef]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Thymine-thymine dimer bypass by yeast DNA polymerase ζ. Science 1996, 272, 1646–1649. [Google Scholar] [CrossRef]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef]
- Shilkin, E.S.; Boldinova, E.O.; Stolyarenko, A.D.; Goncharova, R.I.; Chuprov-Netochin, R.N.; Khairullin, R.F.; Smal, M.P.; Makarova, A.V. Translesion DNA Synthesis and Carcinogenesis. Biochemistry 2020, 85, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; da Costa, L.T.; Carson-Walter, E.B.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Characterization of MAD2B and Other Mitotic Spindle Checkpoint Genes. Genomics 1999, 58, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.K.; Schlondorff, J.; Blobel, C.P. Evidence for an interaction of the metalloprotease—Disintegrin tumour necrosis factor a convertase (TACE) with mitotic arrest deficient 2 (MAD2), and of the metalloprotease—Disintegrin MDC9 with a novel MAD2-related protein, MAD2β. Biochem. J. 1999, 343, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Murakumo, Y.; Roth, T.; Ishii, H.; Rasio, D.; Numata, S.; Croce, C.M.; Fishel, R. A Human REV7 Homolog That Interacts with the Polymerase ζ Catalytic Subunit hREV3 and the Spindle Assembly Checkpoint Protein hMAD2. J. Biol. Chem. 2000, 275, 4391–4397. [Google Scholar] [CrossRef]
- Pfleger, C.M.; Salic, A.; Lee, E.; Kirschner, M.W. Inhibition of Cdh1-APC by the MAD2-related protein MAD2L2: A novel mechanism for regulating Cdh1. Genes Dev. 2001, 15, 1759–1764. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fang, G. MAD2B is an inhibitor of the anaphase-promoting complex. Genes Dev. 2001, 15, 1765–1770. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Wada, Y.; Naoi, Y.; Ishii, C.; Inoue, H. Isolation and genetic characterization of the Neurospora crassa REV1 and REV7 homologs: Evidence for involvement in damage-induced mutagenesis. DNA Repair 2003, 2, 337–346. [Google Scholar] [CrossRef]
- Guo, C.; Fischhaber, P.L.; Luk-Paszyc, M.J.; Masuda, Y.; Zhou, J.; Kamiya, K.; Kisker, C.; Friedberg, E.C. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003, 22, 6621–6630. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, R.; Oshige, M.; Uchida, M.; Ishikawa, G.; Takata, K.; Shimanouchi, K.; Kanai, Y.; Ruike, T.; Morioka, H.; Sakaguchi, K. Purification of Drosophila DNA polymerase ζ by REV1 protein-affinity chromatography. Biochem. J. 2004, 382, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Sakamoto, A.; Sato, S.; Kato, T.; Tabata, S.; Tanaka, A. Roles of Arabidopsis AtREV1 and AtREV7 in translesion synthesis. Plant Physiol. 2005, 138, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Sonoda, E.; Yoshimura, M.; Kawano, Y.; Saya, H.; Kohzaki, M.; Takeda, S. Multiple roles of vertebrate REV genes in DNA repair and recombination. Mol. Cell. Biol. 2005, 25, 6103–6111. [Google Scholar] [CrossRef]
- Clairmont, C.S.; D’Andrea, A.D. REV7 directs DNA repair pathway choice. Trends Cell Biol. 2021, 31, 965–978. [Google Scholar] [CrossRef] [PubMed]
- de Krijger, I.; Boersma, V.; Jacobs, J.J.L. REV7: Jack of many trades. Trends Cell Biol. 2021, 31, 686–701. [Google Scholar] [CrossRef] [PubMed]
- Murakumo, Y. The property of DNA polymerase ζ: REV7 is a putative protein involved in translesion DNA synthesis and cell cycle control. Mutat. Res. 2002, 510, 37–44. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. The HORMA domain: A common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem. Sci. 1998, 23, 284–286. [Google Scholar] [CrossRef]
- Li, Y.; Benezra, R. Identification of a human mitotic checkpoint gene: hsMAD2. Science 1996, 274, 246–248. [Google Scholar] [CrossRef]
- Xia, G.; Luo, X.; Habu, T.; Rizo, J.; Matsumoto, T.; Yu, H. Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. EMBO J. 2004, 23, 3133–3143. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Rosenberg, S.C.; Kugel, C.L.; Kostow, N.; Rog, O.; Davydov, V.; Su, T.Y.; Dernburg, A.F.; Corbett, K.D. The chromosome axis controls meiotic events through a hierarchical assembly of HORMA domain proteins. Dev. Cell. 2014, 31, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kaizuka, T.; Mizushima, N.; Noda, N.N. Structure of the Atg101-Atg13 complex reveals essential roles of Atg101 in autophagy initiation. Nat. Struct. Mol. Biol. 2015, 22, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.C.; Corbett, K.D. The multifaceted roles of the HORMA domain in cellular signaling. J. Cell Biol. 2015, 211, 745–755. [Google Scholar] [CrossRef]
- Prince, J.P.; Martinez-Perez, E. Functions and Regulation of Meiotic HORMA-Domain Proteins. Genes 2022, 13, 777. [Google Scholar] [CrossRef]
- Hara, K.; Hashimoto, H.; Murakumo, Y.; Kobayashi, S.; Kogame, T.; Unzai, S.; Akashi, S.; Takeda, S.; Shimizu, T.; Sato, M. Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase ζ and REV1. J. Biol. Chem. 2010, 285, 12299–12307. [Google Scholar] [CrossRef]
- Hara, K.; Taharazako, S.; Ikeda, M.; Fujita, H.; Mikami, Y.; Kikuchi, S.; Hishiki, A.; Yokoyama, H.; Ishikawa, Y.; Kanno, S.; et al. Dynamic feature of mitotic arrest deficient 2-like protein 2 (MAD2L2) and structural basis for its interaction with chromosome alignment-maintaining phosphoprotein (CAMP). J. Biol. Chem. 2017, 292, 17658–17667. [Google Scholar] [CrossRef] [PubMed]
- Clairmont, C.S.; Sarangi, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’Andrea, A.D. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 2020, 22, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, P.; Clairmont, C.S.; Galli, L.D.; Moreau, L.A.; D’Andrea, A.D. p31comet promotes homologous recombination by inactivating REV7 through the TRIP13 ATPase. Proc. Natl. Acad. Sci. USA 2020, 117, 26795–26803. [Google Scholar] [CrossRef] [PubMed]
- Hanafusa, T.; Habu, T.; Tomida, J.; Ohashi, E.; Murakumo, Y.; Ohmori, H. Overlapping in short motif sequences for binding to human REV7 and MAD2 proteins. Genes Cells 2010, 15, 281–296. [Google Scholar] [CrossRef]
- Tomida, J.; Takata, K.; Lange, S.S.; Schibler, A.C.; Yousefzadeh, M.J.; Bhetawal, S.; Dent, S.Y.; Wood, R.D. REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase ζ. Nucleic Acids Res. 2015, 43, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.C.; Hadden, K. Protein-Protein Interactions in Translesion Synthesis. Molecules 2021, 26, 5544. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Prakash, L.; Prakash, S. Pol31 and Pol32 subunits of yeast DNA polymerase δ are also essential subunits of DNA polymerase ζ. Proc. Natl. Acad. Sci. USA 2012, 109, 12455–12460. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Lada, A.G.; Siebler, H.M.; Zhang, Y.; Pavlov, Y.I.; Tahirov, T.H. DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ. J. Biol. Chem. 2012, 287, 17281–17287. [Google Scholar] [CrossRef]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase ζ complex containing Pol δ accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Gregory, M.T.; Yang, W. Human Pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. Proc. Natl. Acad. Sci. USA 2014, 111, 2954–2959. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Wood, R.D. DNA polymerase ζ in DNA replication and repair. Nucleic Acids Res. 2019, 47, 8348–8361. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, P.E.; McGregor, W.G.; Maher, V.M.; Nisson, P.; Lawrence, C.W. A human homolog of the Saccharomyces cerevisiae REV3 gene, which encodes the catalytic subunit of DNA polymerase ζ. Proc. Natl. Acad. Sci. USA 1998, 95, 6876–6880. [Google Scholar] [CrossRef]
- Xiao, W.; Lechler, T.; Chow, B.L.; Fontanie, T.; Agustus, M.; Carter, K.C.; Wei, Y.F. Identification, chromosomal mapping and tissue-specific expression of hREV3 encoding a putative human DNA polymerase ζ. Carcinogenesis 1998, 19, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Morelli, C.; Mungall, A.J.; Negrini, M.; Barbanti-Brodano, G.; Croce, C.M. Alternative splicing, genomic structure, and fine chromosome localization of REV3L. Cytogenet. Cell Genet. 1998, 83, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wu, X.; Wang, Z. A full-length cDNA of hREV3 is predicted to encode DNA polymerase ζ for damage-induced mutagenesis in humans. Mutat. Res. 1999, 433, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Zhang, Y.; Rajpal, D.K.; Wu, X.; Guo, D.; Wang, M.; Taylor, J.S.; Wang, Z. Specificity of DNA lesion bypass by the yeast DNA polymerase η. J. Biol. Chem. 2000, 275, 8233–8239. [Google Scholar] [CrossRef]
- Johnson, R.E.; Washington, M.T.; Haracska, L.; Prakash, S.; Prakash, L. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 2000, 406, 1015–1019. [Google Scholar] [CrossRef]
- Guo, D.; Wu, X.; Rajpal, D.K.; Taylor, J.S.; Wang, Z. Translesion synthesis by yeast DNA polymerase ζ from templates containing lesions of ultraviolet radiation and acetylaminofluorene. Nucleic Acids Res. 2001, 29, 2875–2883. [Google Scholar] [CrossRef]
- Zhao, B.; Xie, Z.; Shen, H.; Wang, Z. Role of DNA polymerase η in the bypass of abasic sites in yeast cells. Nucleic Acids Res. 2004, 32, 3984–3994. [Google Scholar] [CrossRef]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Deoxycytidyl transferase activity of yeast REV1 protein. Nature 1996, 382, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Murakumo, Y.; Ogura, Y.; Ishii, H.; Numata, S.; Ichihara, M.; Croce, C.M.; Fishel, R.; Takahashi, M. Interantions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7. J. Biol. Chem. 2001, 276, 35644–35651. [Google Scholar] [CrossRef]
- Masuda, Y.; Ohmae, M.; Masuda, K.; Kamiya, K. Structure and enzymatic properties of a stable complex of the human REV1 and REV7 proteins. J. Biol. Chem. 2003, 278, 12356–12360. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Haracska, L.; Johnson, R.E.; Unk, I.; Prakash, S.; Prakash, L. Complex formation of yeast Rev1 and Rev7 proteins: A novel role for the polymerase-associated domain. Mol. Cell. Biol. 2005, 25, 9734–9740. [Google Scholar] [CrossRef] [PubMed]
- Pustovalova, Y.; Bezsonova, I.; Korzhnev, D.M. The C-terminal domain of human Rev1 contains independent binding sites for DNA polymerase η and Rev7 subunit of polymerase ζ. FEBS Lett. 2012, 586, 3051–3056. [Google Scholar] [CrossRef]
- Pustovalova, Y.; Magalhães, M.T.; D’Souza, S.; Rizzo, A.A.; Korza, G.; Walker, G.C.; Korzhnev, D.M. Interaction between the Rev1 C-Terminal Domain and the PolD3 Subunit of Polζ Suggests a Mechanism of Polymerase Exchange upon Rev1/Polζ-Dependent Translesion Synthesis. Biochemistry 2016, 55, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Stith, C.M.; Majka, J.; Burgers, P.M. Proliferating cell nuclear antigen promotes translesion synthesis by DNA polymerase ζ. J. Biol. Chem. 2005, 280, 23446–23450. [Google Scholar] [CrossRef]
- Guo, C.; Sonoda, E.; Tang, T.S.; Parker, J.L.; Bielen, A.B.; Takeda, S.; Ulrich, H.D.; Friedberg, E.C. REV1 protein interacts with PCNA: Significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell. 2006, 23, 265–271. [Google Scholar] [CrossRef]
- Guo, C.; Tang, T.S.; Bienko, M.; Parker, J.L.; Bielen, A.B.; Sonoda, E.; Takeda, S.; Ulrich, H.D.; Dikic, I.; Friedberg, E.C. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol. Cell. Biol. 2006, 26, 8892–8900. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Waters, L.S.; Walker, G.C. Novel conserved motifs in Rev1 C-terminus are required for mutagenic DNA damage tolerance. DNA Repair 2008, 7, 1455–1470. [Google Scholar] [CrossRef]
- Fattah, F.J.; Hara, K.; Fattah, K.R.; Yang, C.; Wu, N.; Warrington, R.; Chen, D.J.; Zhou, P.; Boothman, D.A.; Yu, H. The transcription factor TFII-I promotes DNA translesion synthesis and genomic stability. PLoS Genet. 2014, 10, e1004419. [Google Scholar] [CrossRef]
- Rajpal, D.K.; Wu, X.; Wang, Z. Alteration of ultraviolet-induced mutagenesis in yeast through molecular modulation of the REV3 and REV7 gene expression. Mutat. Res. 2000, 461, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, C.W.; Maher, V.M. Mutagenesis in eukaryotes dependent on DNA polymerase zeta and Rev1p. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 41–46. [Google Scholar] [CrossRef]
- McNally, K.; Neal, J.A.; McManus, T.P.; McCormick, J.J.; Maher, V.M. hRev7, putative subunit of hPolζ, plays a critical role in survival, induction of mutations, and progression through S-phase, of UV((254nm))-irradiated human fibroblasts. DNA Repair 2008, 7, 597–604. [Google Scholar] [CrossRef]
- Cheung, H.W.; Chun, A.C.; Wang, Q.; Deng, W.; Hu, L.; Guan, X.-Y.; Nicholls, J.M.; Ling, M.-T.; Wong, Y.C.; Tsao, S.W.; et al. Inactivation of human MAD2B in nasopharyngeal carcinoma cells leads to chemosensitization to DNA-damaging agents. Cancer Res. 2006, 66, 4357–4367. [Google Scholar] [CrossRef]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Boersma, V.; Moatti, N.; Segura-Bayona, S.; Peuscher, M.H.; van der Torre, J.; Wevers, B.A.; Orthwein, A.; Durocher, D.; Jacobs, J.J.L. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 2015, 521, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e23. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef]
- Findlay, S.; Heath, J.; Luo, V.M.; Malina, A.; Morin, T.; Coulombe, Y.; Djerir, B.; Li, Z.; Samiei, A.; Simo-Cheyou, E.; et al. SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 2018, 37, e100158. [Google Scholar] [CrossRef]
- Setiaputra, D.; Durocher, D. Shieldin—The protector of DNA ends. EMBO Rep. 2019, 20, e47560. [Google Scholar] [CrossRef]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef]
- Mirman, Z.; Sasi, N.K.; King, A.; Chapman, J.R.; de Lange, T. 53BP1-shieldin-dependent DSB processing in BRCA1-deficient cells requires CST-Polα-primase fill-in synthesis. Nat. Cell. Biol. 2022, 24, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Ikeda, M.; Ui, A.; Ouchi, Y.; Mikami, Y.; Kanno, S.I.; Yasui, A.; Tanaka, K. CHAMP1-POGZ counteracts the inhibitory effect of 53BP1 on homologous recombination and affects PARP inhibitor resistance. Oncogene 2022, 41, 2706–2718. [Google Scholar] [CrossRef]
- Li, F.; Sarangi, P.; Iyer, D.R.; Feng, H.; Moreau, L.; Nguyen, H.; Clairmont, C.; D’Andrea, A.D. CHAMP1 binds to REV7/FANCV and promotes homologous recombination repair. Cell Rep. 2022, 40, 111297. [Google Scholar] [CrossRef]
- Sharma, S.; Hicks, J.K.; Chute, C.L.; Brennan, J.R.; Ahn, J.Y.; Glover, T.W.; Canman, C.E. REV1 and polymerase ζ facilitate homologous recombination repair. Nucleic Acids Res. 2012, 40, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Bluteau, D.; Masliah-Planchon, J.; Clairmont, C.; Rousseau, A.; Ceccaldi, R.; d’Enghien, C.D.; Bluteau, O.; Cuccuini, W.; Gachet, S.; de Latour, R.P.; et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J. Clin. Investig. 2016, 126, 3580–3584. [Google Scholar] [CrossRef] [PubMed]
- da Costa, A.A.B.A.; Chowdhury, D.; Shapiro, G.I.; D’Andrea, A.D.; Konstantinopoulos, P.A. Targeting replication stress in cancer therapy. Nat. Rev. Drug. Discov. 2023, 22, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Listovsky, T.; Sale, J.E. Sequestration of CDH1 by MAD2L2 prevents premature APC/C activation prior to anaphase onset. J. Cell Biol. 2013, 203, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.; Wu, Z.; Maher, V.M.; McCormick, J.J.; Xiao, W. Rev7/Mad2B plays a critical role in the assembly of a functional mitotic spindle. Cell Cycle 2015, 14, 3929–3938. [Google Scholar] [CrossRef] [PubMed]
- Medendorp, K.; van Groningen, J.J.M.; Vreede, L.; Hetterschijt, L.; van den Hurk, W.H.; de Bruijn, D.R.H.; Brugmans, L.; van Kessel, A.G. The Mitotic Arrest Deficient Protein MAD2B Interacts with the Small GTPase RAN throughout the Cell Cycle. PLoS ONE 2009, 4, e7020. [Google Scholar] [CrossRef]
- Medendorp, K.; Vreede, L.; van Groningen, J.J.M.; Hetterschijt, L.; Brugmans, L.; Jansen, P.A.M.; van den Hurk, W.H.; de Bruijn, D.R.H.; van Kessel, A.G. The Mitotic Arrest Deficient Protein MAD2B Interacts with the Clathrin Light Chain A during Mitosis. PLoS ONE 2010, 5, e15128. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, S.H.; Sharrocks, A.D. Rev7/MAD2B links c-Jun N-terminal protein kinase pathway signaling to activation of the transcription factor Elk-1. Mol. Cell. Biol. 2007, 27, 2861–2869. [Google Scholar] [CrossRef]
- Hong, C.F.; Chou, Y.T.; Lin, Y.S.; Wu, C.W. MAD2B, a novel TCF4-binding protein, modulates TCF4-mediated epithelial-mesenchymal transdifferentiation. J. Biol. Chem. 2009, 284, 19613–19622. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Wang, X.D.; Li, Z.; McManus, T.P.; McGregor, W.G.; Lawrence, C.W.; Maher, V.M. The function of the human homolog of Saccharomyces cerevisiae REV1 is required for mutagenesis induced by UV light. Proc. Natl. Acad. Sci. USA 2000, 97, 4186–4191. [Google Scholar] [CrossRef]
- Khalaj, M.; Abbasi, A.; Yamanishi, H.; Akiyama, K.; Wakitani, S.; Kikuchi, S.; Hirose, M.; Yuzuriha, M.; Magari, M.; Degheidy, H.A.; et al. A missense mutation in Rev7 disrupts formation of Polζ, impairing mouse development and repair of genotoxic agent-induced DNA lesions. J. Biol. Chem. 2014, 289, 3811–3824. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.; Khalaj, M.; Akiyama, K.; Mukai, Y.; Matsumoto, H.; Acosta, T.J.; Said, N.; Yoshida, M.; Kunieda, T. Lack of Rev7 function results in development of tubulostromal adenomas in mouse ovary. Mol. Cell. Endocrinol. 2015, 412, 19–25. [Google Scholar] [CrossRef]
- Rimkus, C.; Friederichs, J.; Rosenberg, R.; Holzmann, B.; Siewert, J.-R.; Janssen, K.-P. Expression of the mitotic checkpoint gene MAD2L2 has prognostic significance in colon cancer. Int. J. Cancer 2006, 120, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, S.; Wang, H.; Zhang, X.; Kang, T.; Li, Z.; Deng, H.; Yue, W.; Cao, S. Mitotic arrest deficient protein MAD2B is overexpressed in human glioma, with depletion enhancing sensitivity to ionizing radiation. J. Clin. Neurosci. 2011, 18, 827–833. [Google Scholar] [CrossRef]
- Niimi, K.; Murakumo, Y.; Watanabe, N.; Kato, T.; Mii, S.; Enomoto, A.; Asai, M.; Asai, N.; Yamamoto, E.; Kajiyama, H.; et al. Suppression of REV7 enhances cisplatin sensitivity in ovarian clear cell carcinoma cells. Cancer Sci. 2014, 105, 545–552. [Google Scholar] [CrossRef]
- Okina, S.; Yanagisawa, N.; Yokoyama, M.; Sakurai, Y.; Numata, Y.; Umezawa, A.; Higashihara, M.; Murakumo, Y. High expression of REV7 is an independent prognostic indicator in patients with diffuse large B-cell lymphoma treated with rituximab. Int. J. Hematol. 2015, 102, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wei, W.; Heng, Z.; Yantao, H.; Chunbo, W. Knockdown of REV7 Inhibits Breast Cancer Cell Migration and Invasion. Oncol. Res. 2016, 24, 315–325. [Google Scholar] [CrossRef]
- Gu, C.; Luo, J.; Lu, X.; Tang, Y.; Ma, Y.; Yun, Y.; Cao, J.; Cao, J.; Huang, Z.; Zhou, X.; et al. REV7 confers radioresistance of esophagus squamous cell carcinoma by recruiting PRDX2. Cancer Sci. 2019, 110, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Ichinoe, M.; Yoshida, K.; Nakazato, Y.; Saito, S.; Satoh, M.; Nakada, N.; Sanoyama, I.; Umezawa, A.; Numata, Y.; et al. Inactivation of REV7 enhances chemosensitivity and overcomes acquired chemoresistance in testicular germ cell tumors. Cancer Lett. 2020, 489, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Sanoyama, I.; Sakurai, Y.; Ichinoe, M.; Hoshino, A.; Kesen, Y.; Kato, T.; Numata, Y.; Umezawa, A.; Jiang, S.X.; Murakumo, Y. Increased expression of REV7 in small cell lung carcinomas and its association with tumor cell survival and proliferation. Pathol. Int. 2021, 71, 15–23. [Google Scholar] [CrossRef]
- Hoshino, A.; Nakayama, C.; Jiang, S.X.; Sakurai, Y.; Kato, T.; Numata, Y.; Umezawa, A.; Ichinoe, M.; Murakumo, Y. Upregulation of REV7 correlates with progression of malignant melanoma. Pathol. Int. 2022, 72, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; He, X.; Yu, W.; Yue, B.; Yu, Z.; Qin, Y. Mitotic Arrest-Deficient Protein 2B Overexpressed in Lung Cancer Promotes Proliferation, EMT, and Metastasis. Oncol. Res. 2019, 27, 859–869. [Google Scholar] [CrossRef]
- Yang, J.; Ding, W.; Wang, X.; Xiang, Y. Knockdown of DNA polymerase ζ relieved the chemoresistance of glioma via inhibiting the PI3K/AKT signaling pathway. Bioengineered 2021, 12, 3924–3933. [Google Scholar] [CrossRef]
- Pernicone, N.; Peretz, L.; Grinshpon, S.; Listovsky, T. MDA-MB-157 Cell Line Presents High Levels of MAD2L2 and Dysregulated Mitosis. Anticancer Res. 2020, 40, 5471–5480. [Google Scholar] [CrossRef]
- Merlini, A.; Centomo, M.L.; Ferrero, G.; Chiabotto, G.; Miglio, U.; Berrino, E.; Giordano, G.; Brusco, S.; Pisacane, A.; Maldi, E.; et al. DNA damage response and repair genes in advanced bone and soft tissue sarcomas: An 8-gene signature as a candidate predictive biomarker of response to trabectedin and olaparib combination. Front. Oncol. 2022, 12, 844250. [Google Scholar] [CrossRef]
- Cho, Y.E.; Kim, J.H.; Che, Y.H.; Kim, Y.J.; Sung, J.Y.; Kim, Y.W.; Choe, B.G.; Lee, S.; Park, J.H. Role of the WNT/β-catenin/ZKSCAN3 Pathway in Regulating Chromosomal Instability in Colon Cancer Cell lines and Tissues. Int. J. Mol. Sci. 2022, 23, 9302. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Y.; Zou, T.; Yin, J.-Y.; Wang, Z.; Liu, C.; Huang, H.-X.; Ding, F.-X.; Lei, M.-R.; Wang, Y.; Liu, M.; et al. Genetic Variants in Double-Strand Break Repair Pathway Genes to Predict Platinum-Based Chemotherapy Prognosis in Patients With Lung Cancer. Front. Pharmacol. 2022, 13, 915822. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Chen, M.; Yu, X.; Gu, Z.; Qiu, H.; Qin, G.; Long, Q.; Fu, X.; Liu, T.; et al. MAD2L2 inhibits colorectal cancer growth by promoting NCOA3 ubiquitination and degradation. Mol. Oncol. 2018, 12, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, X.; Zhao, X.; Sun, H.; Kong, F.; Li, Y.; Sui, Y.; Xu, F. Oxaliplatin promotes siMAD2L2-induced apoptosis in colon cancer cells. Mol. Med. Rep. 2021, 24, 629. [Google Scholar] [CrossRef]
- Sun, X.; Hou, W.; Liu, X.; Chai, J.; Guo, H.; Yu, J. Targeting REV7 effectively reverses 5-FU and oxaliplatin resistance in colorectal cancer. Cancer Cell Int. 2020, 20, 580. [Google Scholar] [CrossRef] [PubMed]
- Weterman, M.A.; van Groningen, J.J.; Tertoolen, L.; van Kessel, A.G. Impairment of MAD2B-PRCC interaction in mitotic checkpoint defective t(X;1)-positive renal cell carcinomas. Proc. Natl. Acad. Sci. USA 2001, 98, 13808–13813. [Google Scholar] [CrossRef]
- Vassel, F.M.; Bian, K.; Walker, G.C.; Hemann, M.T. Rev7 loss alters cisplatin response and increases drug efficacy in chemotherapy-resistant lung cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 28922–28924. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszek, J.L.; Chatterjee, N.; Najeeb, J.; Ramos, A.; Lee, M.; Bian, K.; Xue, J.Y.; Fenton, B.A.; Park, H.; Li, D.; et al. A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 2019, 178, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Whitman, M.A.; Harris, C.A.; Min, S.M.; Jonas, O.; Lien, E.C.; Luengo, A.; Vander Heiden, M.G.; Hong, J.; Zhou, P.; et al. REV1 inhibitor JH-RE-06 enhances tumor cell response to chemotherapy by triggering senescence hallmarks. Proc. Natl. Acad. Sci. USA 2020, 117, 28918–28921. [Google Scholar] [CrossRef] [PubMed]
- Actis, M.L.; Ambaye, N.D.; Evison, B.J.; Shao, Y.; Vanarotti, M.; Inoue, A.; McDonald, E.T.; Kikuchi, S.; Heath, R.; Hara, K.; et al. Identification of the first small-molecule inhibitor of the REV7 DNA repair protein interaction. Bioorg. Med. Chem. 2016, 24, 4339–4346. [Google Scholar] [CrossRef]
- Pernicone, N.; Elias, M.; Onn, I.; Tobi, D.; Listovsky, T. Disrupting the MAD2L2-Rev1 Complex Enhances Cell Death upon DNA Damage. Molecules 2022, 27, 636. [Google Scholar] [CrossRef]
- Sail, V.; Rizzo, A.A.; Chatterjee, N.; Dash, R.C.; Ozen, Z.; Walker, G.C.; Korzhnev, D.M.; Hadden, M.K. Identification of Small Molecule Translesion Synthesis Inhibitors That Target the Rev1-CT/RIR Protein-Protein Interaction. ACS Chem. Biol. 2017, 12, 1903–1912. [Google Scholar] [CrossRef]
- Ozen, Z.; Dash, R.C.; McCarthy, K.R.; Chow, S.A.; Rizzo, A.A.; Korzhnev, D.M.; Hadden, M.K. Small molecule scaffolds that disrupt the Rev1-CT/RIR protein-protein interaction. Bioorg. Med. Chem. 2018, 26, 4301–4309. [Google Scholar] [CrossRef]
- Dash, R.C.; Ozen, Z.; Rizzo, A.A.; Lim, S.; Korzhnev, D.M.; Hadden, M.K. Structural Approach To Identify a Lead Scaffold That Targets the Translesion Synthesis Polymerase Rev1. J. Chem. Inf. Model. 2018, 58, 2266–2277. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.C.; Ozen, Z.; McCarthy, K.R.; Chatterjee, N.; Harris, C.A.; Rizzo, A.A.; Walker, G.C.; Korzhnev, D.M.; Hadden, M.K. Virtual Pharmacophore Screening Identifies Small-Molecule Inhibitors of the Rev1-CT/RIR Protein-Protein Interaction. ChemMedChem 2019, 14, 1610–1617. [Google Scholar] [CrossRef]
- McPherson, K.S.; Zaino, A.M.; Dash, R.C.; Rizzo, A.A.; Li, Y.; Hao, B.; Bezsonova, I.; Hadden, M.K.; Korzhnev, D.M. Structure-Based Drug Design of Phenazopyridine Derivatives as Inhibitors of Rev1 Interactions in Translesion Synthesis. ChemMedChem 2021, 16, 1126–1132. [Google Scholar] [CrossRef]
- Watanabe, N.; Mii, S.; Asai, N.; Asai, M.; Niimi, K.; Ushida, K.; Kato, T.; Enomoto, A.; Ishii, H.; Takahashi, M.; et al. The Rev7 Subunit of DNA Polymerase ζ Is Essential for Primordial Germ Cell Maintenance in the Mouse. J. Biol. Chem. 2013, 288, 10459–10471. [Google Scholar] [CrossRef] [PubMed]
- Pirouz, M.; Pilarski, S.; Kessel, M. A critical function of Mad2l2 in primordial germ cell development of mice. PLoS Genet. 2013, 9, e1003712. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Screening Target | Mechanism | Effect | References |
|---|---|---|---|---|
| JH-RE-06 | REV1 CTD-REV7 PPI | Promote REV1 dimerization Disrupt REV1-REV7 PPI | Inhibition of cisplatin-induced mutagenic TLS Enhancement of cisplatin, BPDE, 4-NQO, and MMS cytotoxicity in vitro Enhancement of cisplatin chemosensitivity in vivo | [105,106] |
| Compound 7 | REV3-REV7 PPI | Disrupt REV3-REV7 PPI | Inhibition of interstrand cross-link repair Enhancement of cisplatin cytotoxicity in vitro | [107] |
| ZINC97017995, ZINC25496030 | REV7 homodimer | Disrupt REV1-REV7 PPI | Enhancement of cisplatin and doxorubicin cytotoxicity in vitro | [108] |
| Thiophene, piperazine, piperidine, and aryl piperazine compounds | REV1 CTD-Polκ RIR PPI | Disrupt REV1 CTD-RIR PPI | Enhancement of cisplatin and UV cytotoxicity in vitro Inhibition of cisplatin-induced mutagenesis | [109,110] |
| Phenazopyridine compounds | REV1 CTD | Disrupt REV1 CTD-RIR PPI | Enhancement of cisplatin cytotoxicity in vitro | [111,112,113] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murakumo, Y.; Sakurai, Y.; Kato, T.; Hashimoto, H.; Ichinoe, M. REV7 in Cancer Biology and Management. Cancers 2023, 15, 1721. https://doi.org/10.3390/cancers15061721
Murakumo Y, Sakurai Y, Kato T, Hashimoto H, Ichinoe M. REV7 in Cancer Biology and Management. Cancers. 2023; 15(6):1721. https://doi.org/10.3390/cancers15061721
Chicago/Turabian StyleMurakumo, Yoshiki, Yasutaka Sakurai, Takuya Kato, Hiroshi Hashimoto, and Masaaki Ichinoe. 2023. "REV7 in Cancer Biology and Management" Cancers 15, no. 6: 1721. https://doi.org/10.3390/cancers15061721
APA StyleMurakumo, Y., Sakurai, Y., Kato, T., Hashimoto, H., & Ichinoe, M. (2023). REV7 in Cancer Biology and Management. Cancers, 15(6), 1721. https://doi.org/10.3390/cancers15061721