

Phenotypic Plasticity in Circulating Tumor Cells Is Associated with Poor Response to Therapy in Metastatic Breast Cancer Patients

 ,
,  , , ,
, , ,  and
and
Abstract
Simple Summary
Abstract
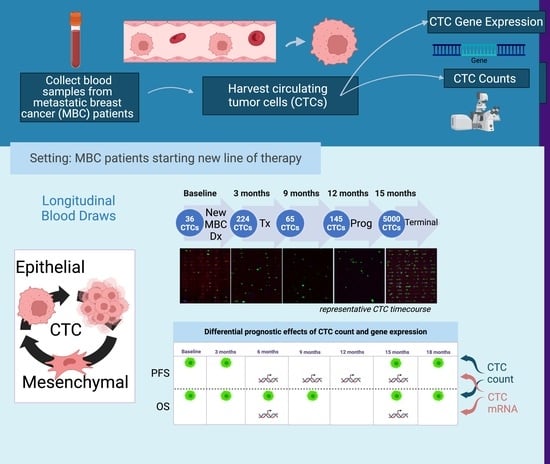
1. Introduction
2. Materials and Methods
2.1. Patients and Healthy Volunteers
2.2. Workflow of Sample Processing
2.2.1. Sample Processing for Enumeration of Captured CTCs
2.2.2. Sample Processing Workflow for Gene Expression Analysis of Captured CTCs
2.2.3. EM Score
2.3. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. Enumeration of CTCs
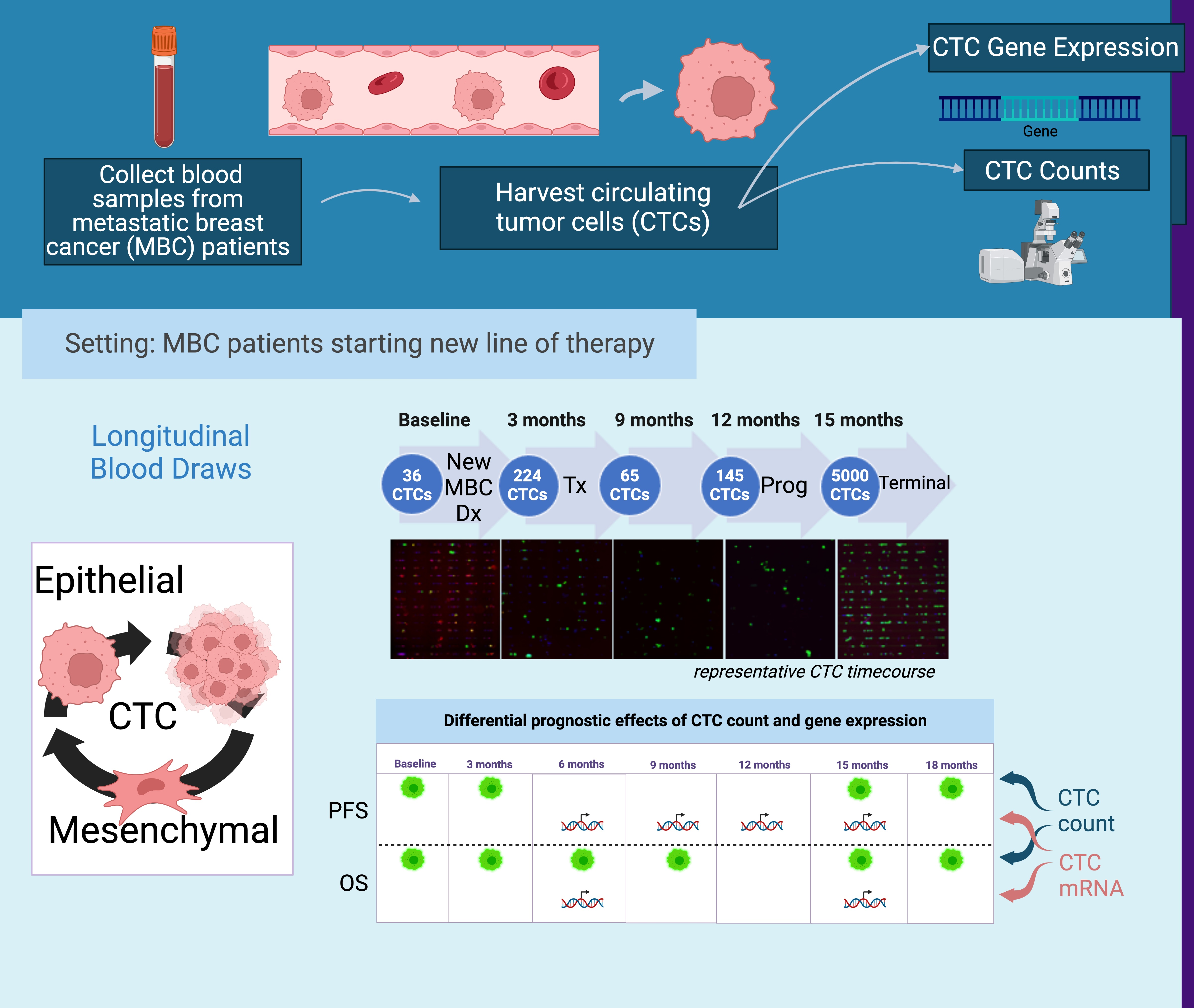
3.2.1. Longitudinal Distribution of CTC Counts during Therapy
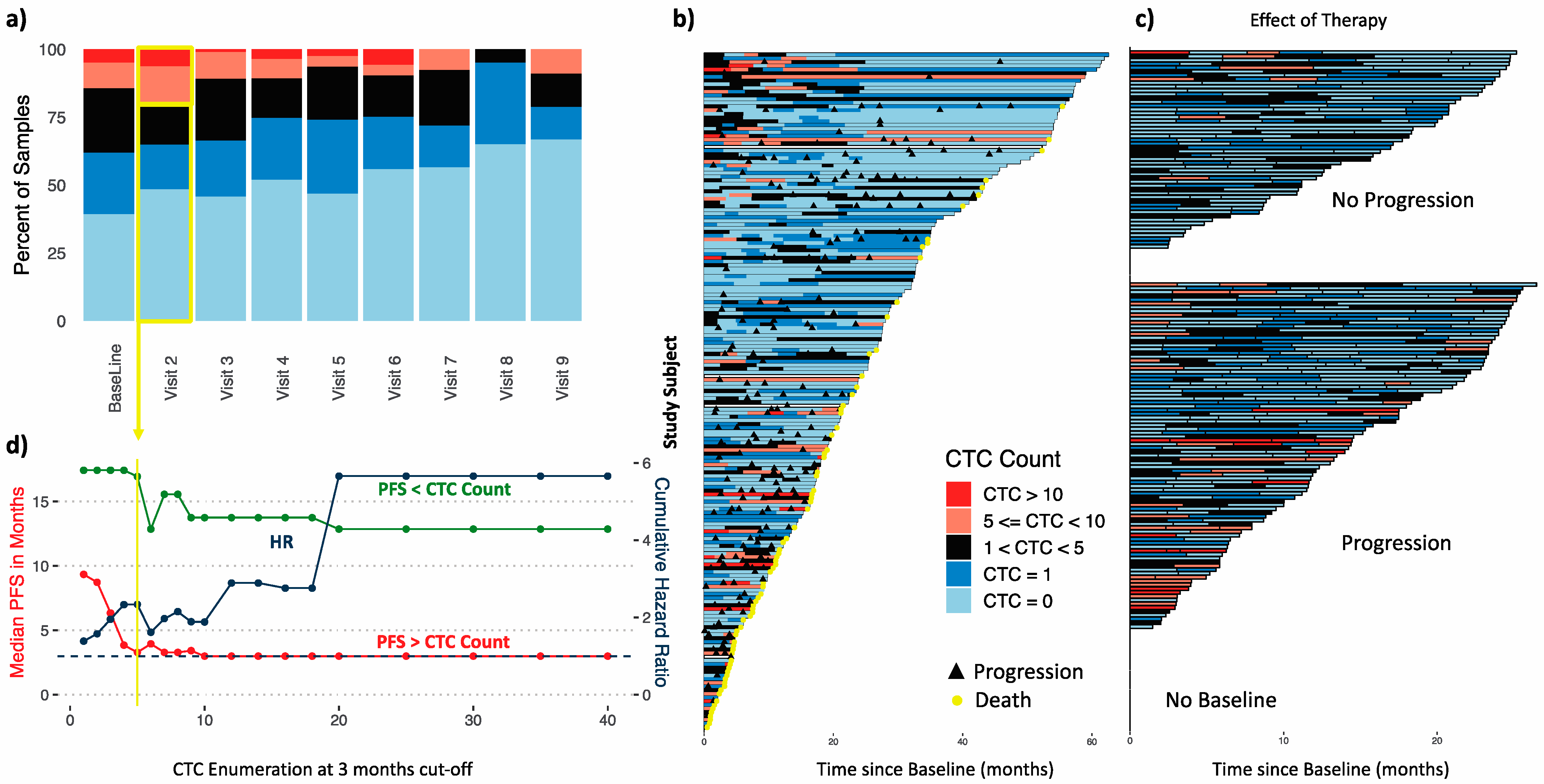
3.2.2. Established CTC Threshold
3.2.3. CTC Enumeration Predicts Survival
3.2.4. Dynamics of CTC Counts during Therapy
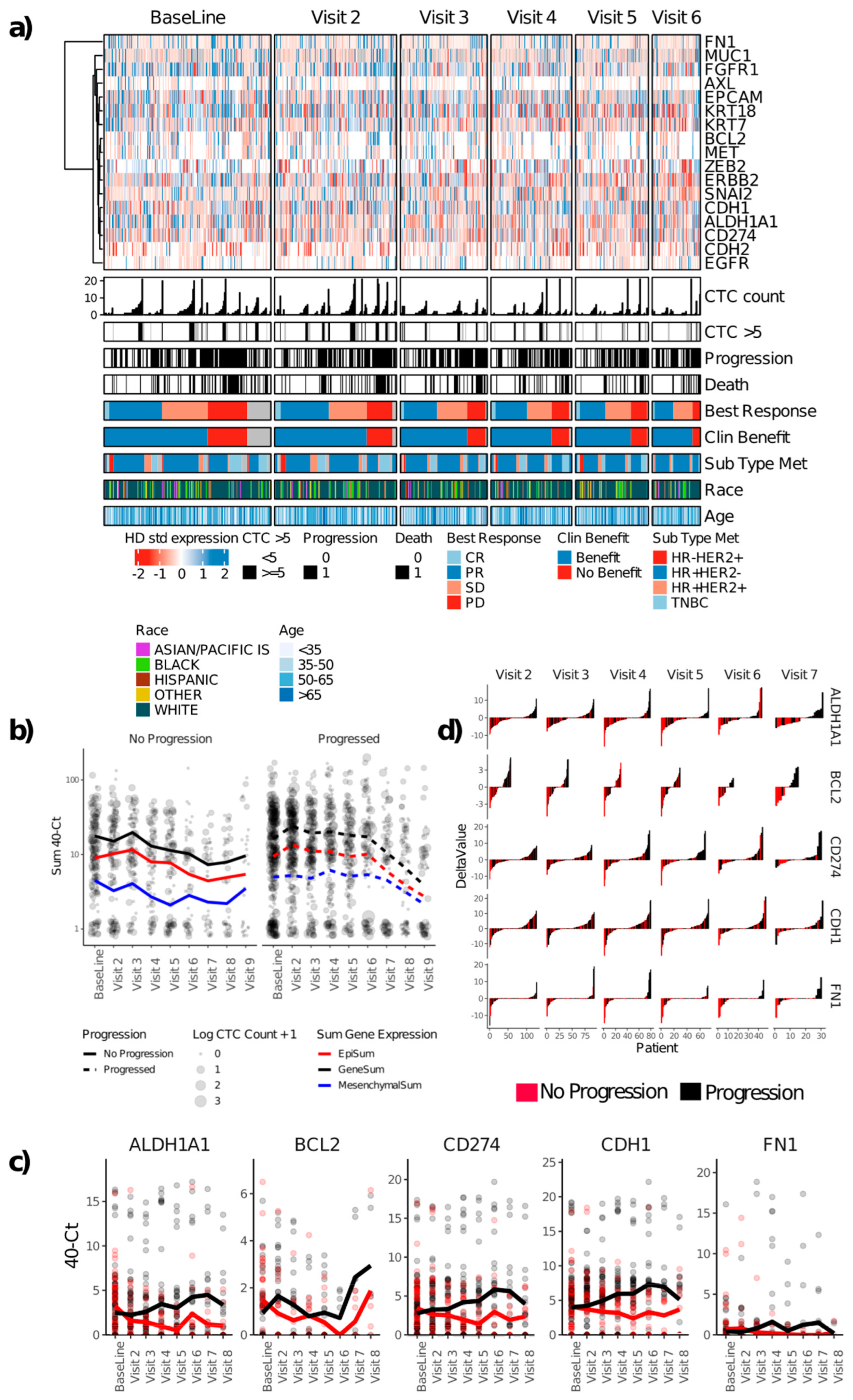
3.3. Gene Expression in Enriched CTCs
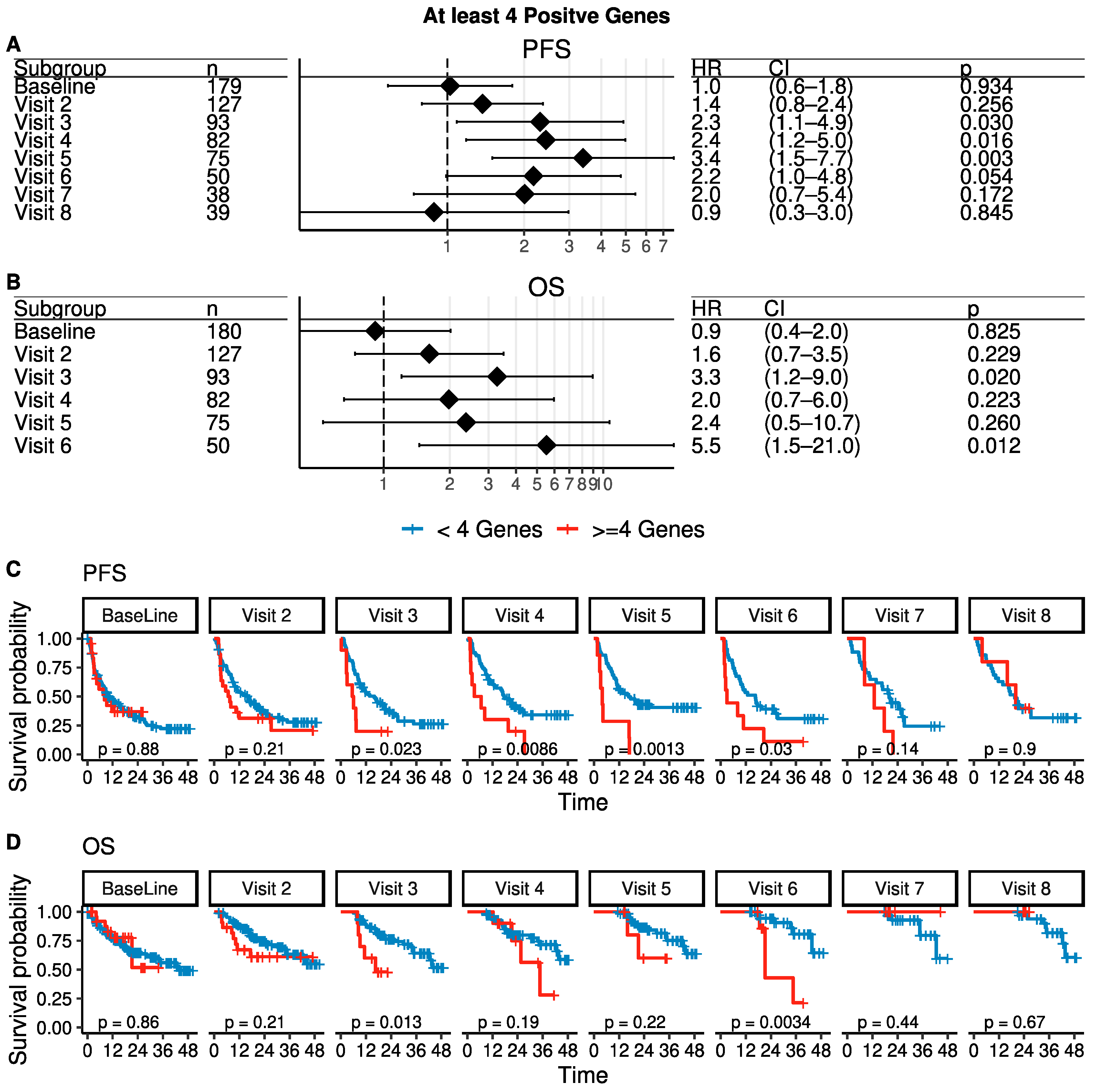
3.4. Time-Dependent Survival Analysis
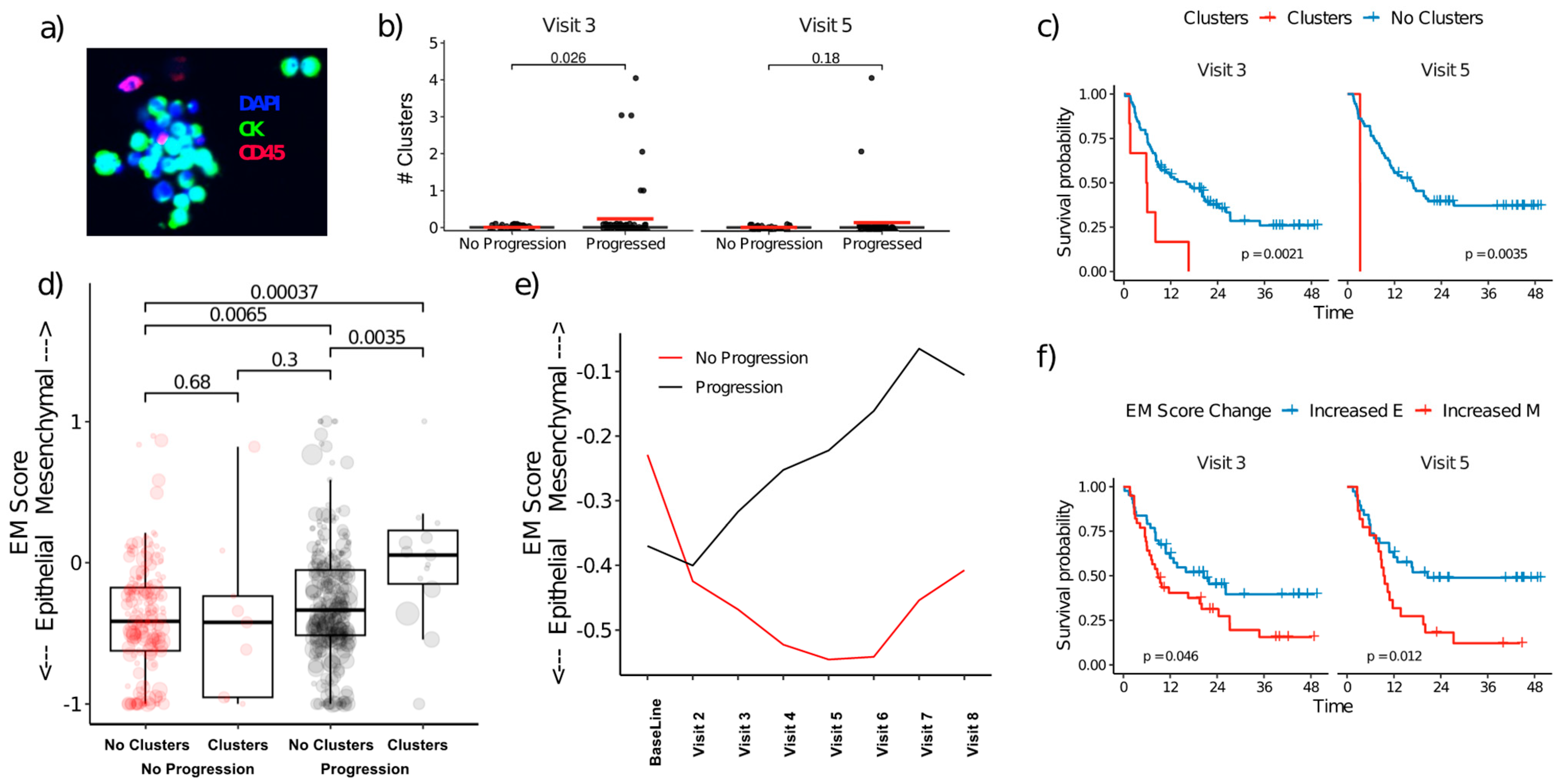
3.5. CTC Clusters and EMT Plasticity
3.6. Selected Biomarker Examples and Case Studies
- (a)
- HER2:
- (b)
- EGFR:
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ozimski, L.L.; Gremmelspacher, D.; Aceto, N. A fatal affair: Circulating tumor cell relationships that shape metastasis. iScience 2021, 24, 103073. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef]
- Plaks, V.; Koopman, C.D.; Werb, Z. Circulating Tumor Cells. Science 2013, 341, 1186–1188. [Google Scholar] [CrossRef]
- Sznurkowska, M.K.; Aceto, N. The gate to metastasis: Key players in cancer cell intravasation. FEBS J. 2022, 289, 4336–4354. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Zhang, Q.; Kong, D.; Yang, Z.; Li, G.; Cheng, S.; Feng, L.; Zhang, K.; Zhang, W. Prognostic value of stem-like circulating tumor cells in patients with cancer: A systematic review and meta-analysis. Clin. Exp. Med. 2023. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Mego, M.; Mani, S.A.; Lee, B.N.; Li, C.; Evans, K.W.; Cohen, E.N.; Gao, H.; Jackson, S.A.; Giordano, A.; Hortobagyi, G.N.; et al. Expression of epithelial-mesenchymal transition-inducing transcription factors in primary breast cancer: The effect of neoadjuvant therapy. Int. J. Cancer 2012, 130, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Banko, P.; Lee, S.Y.; Nagygyorgy, V.; Zrinyi, M.; Chae, C.H.; Cho, D.H.; Telekes, A. Technologies for circulating tumor cell separation from whole blood. J. Hematol. Oncol. 2019, 12, 48. [Google Scholar] [CrossRef]
- Wang, L.; Balasubramanian, P.; Chen, A.P.; Kummar, S.; Evrard, Y.A.; Kinders, R.J. Promise and limits of the CellSearch platform for evaluating pharmacodynamics in circulating tumor cells. Semin. Oncol. 2016, 43, 464–475. [Google Scholar] [CrossRef]
- Hosokawa, M.; Yoshikawa, T.; Negishi, R.; Yoshino, T.; Koh, Y.; Kenmotsu, H.; Naito, T.; Takahashi, T.; Yamamoto, N.; Kikuhara, Y.; et al. Microcavity array system for size-based enrichment of circulating tumor cells from the blood of patients with small-cell lung cancer. Anal. Chem. 2013, 85, 5692–5698. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.N.; Jayachandran, G.; Gao, H.; Qiao, W.; Liu, S.; He, J.; Qiao, Y.; Yao, L.; Lin, S.H.; Reuben, J.M. Enumeration and molecular characterization of circulating tumor cells enriched by microcavity array from stage III non-small cell lung cancer patients. Transl. Lung Cancer Res. 2020, 9, 1974–1985. [Google Scholar] [CrossRef]
- Cohen, E.N.; Jayachandran, G.; Gao, H.; Peabody, P.; McBride, H.B.; Alvarez, F.D.; Lopez Bravo, P.; Qiao, W.; Liu, S.; Yao, L.; et al. Gene expression profiling of circulating tumor cells captured by MicroCavity Array is superior to enumeration in demonstrating therapy response in patients with newly diagnosed advanced and locally advanced non-small cell lung cancer. Transl. Lung Cancer Res. 2023, 12, 109–126. [Google Scholar] [CrossRef]
- Zeune, L.L.; de Wit, S.; Berghuis, A.M.S.; MJ, I.J.; Terstappen, L.; Brune, C. How to Agree on a CTC: Evaluating the Consensus in Circulating Tumor Cell Scoring. Cytom. Part A 2018, 93, 1202–1206. [Google Scholar] [CrossRef]
- Bustin, S.A.; Beaulieu, J.F.; Huggett, J.; Jaggi, R.; Kibenge, F.S.; Olsvik, P.A.; Penning, L.C.; Toegel, S. MIQE precis: Practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time PCR experiments. BMC Mol. Biol. 2010, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Prentice, R.L.; Williams, B.J.; Peterson, A.V. On the Regression-Analysis of Multivariate Failure Time Data. Biometrika 1981, 68, 373–379. [Google Scholar] [CrossRef]
- Nelson, W. Recurrent Events Data Analysis for Product Repairs, Disease Recurrences, and Other Applications; Society for Industrial and Applied Mathematics: Philadelphia, PA, USA, 2003. [Google Scholar]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef]
- Hayes, D.F.; Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Miller, M.C.; Matera, J.; Allard, W.J.; Doyle, G.V.; Terstappen, L.W. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin. Cancer Res. 2006, 12, 4218–4224. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Pierga, J.Y.; Reuben, J.; Rademaker, A.; Davis, A.A.; Peeters, D.J.; Fehm, T.; Nole, F.; Gisbert-Criado, R.; Mavroudis, D.; et al. The clinical use of circulating tumor cells (CTCs) enumeration for staging of metastatic breast cancer (MBC): International expert consensus paper. Crit. Rev. Oncol. Hematol. 2019, 134, 39–45. [Google Scholar] [CrossRef]
- Smerage, J.B.; Barlow, W.E.; Hortobagyi, G.N.; Winer, E.P.; Leyland-Jones, B.; Srkalovic, G.; Tejwani, S.; Schott, A.F.; O’Rourke, M.A.; Lew, D.L.; et al. Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J. Clin. Oncol. 2014, 32, 3483–3489. [Google Scholar] [CrossRef]
- Bidard, F.C.; Jacot, W.; Kiavue, N.; Dureau, S.; Kadi, A.; Brain, E.; Bachelot, T.; Bourgeois, H.; Goncalves, A.; Ladoire, S.; et al. Efficacy of Circulating Tumor Cell Count-Driven vs Clinician-Driven First-line Therapy Choice in Hormone Receptor-Positive, ERBB2-Negative Metastatic Breast Cancer: The STIC CTC Randomized Clinical Trial. JAMA Oncol. 2021, 7, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.N.; Jayachandran, G.; Hardy, M.R.; Venkata Subramanian, A.M.; Meng, X.; Reuben, J.M. Antigen-agnostic microfluidics-based circulating tumor cell enrichment and downstream molecular characterization. PLoS ONE 2020, 15, e0241123. [Google Scholar] [CrossRef] [PubMed]
- Pore, A.A.; Bithi, S.S.; Zeinali, M.; Navaid, H.B.; Nagrath, S.; Layeequr Rahman, R.; Vanapalli, S.A. Phenotyping of rare circulating cells in the blood of non-metastatic breast cancer patients using microfluidic Labyrinth technology. Biomicrofluidics 2022, 16, 064107. [Google Scholar] [CrossRef] [PubMed]
- Mego, M.; Karaba, M.; Minarik, G.; Benca, J.; Silvia, J.; Sedlackova, T.; Manasova, D.; Kalavska, K.; Pindak, D.; Cristofanilli, M.; et al. Circulating Tumor Cells with Epithelial-to-mesenchymal Transition Phenotypes Associated With Inferior Outcomes in Primary Breast Cancer. Anticancer Res. 2019, 39, 1829–1837. [Google Scholar] [CrossRef]
- Topa, J.; Gresner, P.; Zaczek, A.J.; Markiewicz, A. Breast cancer circulating tumor cells with mesenchymal features-an unreachable target? Cell. Mol. Life Sci. 2022, 79, 81. [Google Scholar] [CrossRef]
- Zhao, L.; Zheng, Z.; Liu, Y.; Liu, F.; Li, X.; Wu, Z. The mesenchymal circulating tumor cells as biomarker for prognosis prediction and supervision in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2023. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Chen, X.; Liao, R.; Li, D.; Sun, J. Induced cancer stem cells generated by radiochemotherapy and their therapeutic implications. Oncotarget 2017, 8, 17301–17312. [Google Scholar] [CrossRef]
- Debeb, B.G.; Lacerda, L.; Xu, W.; Larson, R.; Solley, T.; Atkinson, R.; Sulman, E.P.; Ueno, N.T.; Krishnamurthy, S.; Reuben, J.M.; et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/beta-catenin signaling. Stem Cells 2012, 30, 2366–2377. [Google Scholar] [CrossRef] [PubMed]
- Eslami, S.Z.; Cortes-Hernandez, L.E.; Thomas, F.; Pantel, K.; Alix-Panabieres, C. Functional analysis of circulating tumour cells: The KEY to understand the biology of the metastatic cascade. Br. J. Cancer 2022, 127, 800–810. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Cohen, E.N.; Gao, H.; Anfossi, S.; Mego, M.; Reddy, N.G.; Debeb, B.; Giordano, A.; Tin, S.; Wu, Q.; Garza, R.J.; et al. Inflammation Mediated Metastasis: Immune Induced Epithelial-To-Mesenchymal Transition in Inflammatory Breast Cancer Cells. PLoS ONE 2015, 10, e0132710. [Google Scholar] [CrossRef] [PubMed]
- Miklikova, S.; Minarik, G.; Sedlackova, T.; Plava, J.; Cihova, M.; Jurisova, S.; Kalavska, K.; Karaba, M.; Benca, J.; Smolkova, B.; et al. Inflammation-Based Scores Increase the Prognostic Value of Circulating Tumor Cells in Primary Breast Cancer. Cancers 2020, 12, 1134. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.A.; Mala, A.; Merodoulaki, A.C.; Vassilakopoulou, M.; Mavroudis, D.; Agelaki, S. Investigating the Role of CTCs with Stem/EMT-like Features in Metastatic Breast Cancer Patients Treated with Eribulin Mesylate. Cancers 2022, 14, 3903. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Wilde, A.; Kuestner, R.E.; Skelton, S.M.; MacIntosh, E.; d’Herouel, A.F.; Ertaylan, G.; Del Sol, A.; Skupin, A.; Huang, S. Loss of inter-cellular cooperation by complete epithelial-mesenchymal transition supports favorable outcomes in basal breast cancer patients. Oncotarget 2018, 9, 20018–20033. [Google Scholar] [CrossRef]
- Lopresti, A.; Acquaviva, C.; Boudin, L.; Finetti, P.; Garnier, S.; Aulas, A.; Liberatoscioli, M.L.; Cabaud, O.; Guille, A.; de Nonneville, A.; et al. Identification of Atypical Circulating Tumor Cells with Prognostic Value in Metastatic Breast Cancer Patients. Cancers 2022, 14, 932. [Google Scholar] [CrossRef]
- Ivanova, E.; Ward, A.; Wiegmans, A.P.; Richard, D.J. Circulating Tumor Cells in Metastatic Breast Cancer: From Genome Instability to Metastasis. Front. Mol. Biosci. 2020, 7, 134. [Google Scholar] [CrossRef]
- Ring, A.; Nguyen-Strauli, B.D.; Wicki, A.; Aceto, N. Biology, vulnerabilities and clinical applications of circulating tumour cells. Nat. Rev. Cancer 2023, 23, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Toner, M.; Maheswaran, S.; Haber, D.A. En Route to Metastasis: Circulating Tumor Cell Clusters and Epithelial-to-Mesenchymal Transition. Trends Cancer 2015, 1, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Sawabata, N.; Kawaguchi, T.; Watanabe, T.; Yohikawa, D.; Ouji-Sageshima, N.; Ito, T. Pure Solid Pattern of Non-Small Cell Lung Cancer and Clustered Circulating Tumor Cells. Cancers 2022, 14, 4514. [Google Scholar] [CrossRef] [PubMed]
- Murlidhar, V.; Reddy, R.M.; Fouladdel, S.; Zhao, L.; Ishikawa, M.K.; Grabauskiene, S.; Zhang, Z.; Lin, J.; Chang, A.C.; Carrott, P.; et al. Poor Prognosis Indicated by Venous Circulating Tumor Cell Clusters in Early-Stage Lung Cancers. Cancer Res. 2017, 77, 5194–5206. [Google Scholar] [CrossRef]
- Lu, L.; Hu, W.; Liu, B.; Yang, T. Insights into Circulating Tumor Cell Clusters: A Barometer for Treatment Effects and Prognosis for Prostate Cancer Patients. Cancers 2022, 14, 3985. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Muinelo-Romay, L.; Cebey-Lopez, V.; Pereira-Veiga, T.; Martinez-Pena, I.; Abreu, M.; Abalo, A.; Lago-Leston, R.M.; Abuin, C.; Palacios, P.; et al. Analysis of a Real-World Cohort of Metastatic Breast Cancer Patients Shows Circulating Tumor Cell Clusters (CTC-clusters) as Predictors of Patient Outcomes. Cancers 2020, 12, 1111. [Google Scholar] [CrossRef]
- Larsson, A.M.; Jansson, S.; Bendahl, P.O.; Levin Tykjaer Jorgensen, C.; Loman, N.; Graffman, C.; Lundgren, L.; Aaltonen, K.; Ryden, L. Longitudinal enumeration and cluster evaluation of circulating tumor cells improve prognostication for patients with newly diagnosed metastatic breast cancer in a prospective observational trial. Breast Cancer Res. 2018, 20, 48. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Zhang, Q.; Gerratana, L.; Shah, A.N.; Zhan, Y.; Qiang, W.; Finkelman, B.S.; Flaum, L.; Behdad, A.; Gradishar, W.J.; et al. Association of a novel circulating tumor DNA next-generating sequencing platform with circulating tumor cells (CTCs) and CTC clusters in metastatic breast cancer. Breast Cancer Res. 2019, 21, 137. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mu, Z.; Chervoneva, I.; Austin, L.; Ye, Z.; Rossi, G.; Palazzo, J.P.; Sun, C.; Abu-Khalaf, M.; Myers, R.E.; et al. Longitudinally collected CTCs and CTC-clusters and clinical outcomes of metastatic breast cancer. Breast Cancer Res. Treat. 2017, 161, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Krol, I.; Schwab, F.D.; Carbone, R.; Ritter, M.; Picocci, S.; De Marni, M.L.; Stepien, G.; Franchi, G.M.; Zanardi, A.; Rissoglio, M.D.; et al. Detection of clustered circulating tumour cells in early breast cancer. Br. J. Cancer 2021, 125, 23–27. [Google Scholar] [CrossRef]
- Reduzzi, C.; Di Cosimo, S.; Gerratana, L.; Motta, R.; Martinetti, A.; Vingiani, A.; D’Amico, P.; Zhang, Y.; Vismara, M.; Depretto, C.; et al. Circulating Tumor Cell Clusters Are Frequently Detected in Women with Early-Stage Breast Cancer. Cancers 2021, 13, 2356. [Google Scholar] [CrossRef] [PubMed]
- Edd, J.F.; Mishra, A.; Dubash, T.D.; Herrera, S.; Mohammad, R.; Williams, E.K.; Hong, X.; Mutlu, B.R.; Walsh, J.R.; Machado de Carvalho, F.; et al. Microfluidic concentration and separation of circulating tumor cell clusters from large blood volumes. Lab A Chip 2020, 20, 558–567. [Google Scholar] [CrossRef]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, J.; Munoz-Couselo, E.; Soberino, J.; Racca, F.; Cortes, J. Targeting FGFR pathway in breast cancer. Breast 2018, 37, 126–133. [Google Scholar] [CrossRef]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Akhand, S.S.; Chen, H.; Purdy, S.C.; Liu, Z.; Anderson, J.C.; Willey, C.D.; Wendt, M.K. Fibroblast growth factor receptor facilitates recurrence of minimal residual disease following trastuzumab emtansine therapy. NPJ Breast Cancer 2021, 7, 5. [Google Scholar] [CrossRef]
- Santolla, M.F.; Maggiolini, M. The FGF/FGFR System in Breast Cancer: Oncogenic Features and Therapeutic Perspectives. Cancers 2020, 12, 3029. [Google Scholar] [CrossRef]
- Reuben, J.M.; Lee, B.N.; Li, C.; Gao, H.; Broglio, K.R.; Valero, V.; Jackson, S.A.; Ueno, N.T.; Krishnamurthy, S.; Hortobagyi, G.N.; et al. Circulating tumor cells and biomarkers: Implications for personalized targeted treatments for metastatic breast cancer. Breast J. 2010, 16, 327–330. [Google Scholar] [CrossRef]
- Hayashi, N.; Nakamura, S.; Tokuda, Y.; Shimoda, Y.; Yagata, H.; Yoshida, A.; Ota, H.; Hortobagyi, G.N.; Cristofanilli, M.; Ueno, N.T. Prognostic value of HER2-positive circulating tumor cells in patients with metastatic breast cancer. Int. J. Clin. Oncol. 2012, 17, 96–104. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Wang, T.; Bian, L.; Hu, H.; Xu, C.; Liu, B.; Liu, Y.; Cristofanilli, M.; Jiang, Z. Real-time HER2 status detected on circulating tumor cells predicts different outcomes of anti-HER2 therapy in histologically HER2-positive metastatic breast cancer patients. BMC Cancer 2016, 16, 526. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mu, Z.; Ye, Z.; Zhang, Z.; Abu-Khalaf, M.M.; Silver, D.P.; Palazzo, J.P.; Jagannathan, G.; Fellin, F.M.; Bhattacharya, S.; et al. Prognostic value of HER2 status on circulating tumor cells in advanced-stage breast cancer patients with HER2-negative tumors. Breast Cancer Res. Treat. 2020, 181, 679–689. [Google Scholar] [CrossRef]
- Zeune, L.; van Dalum, G.; Decraene, C.; Proudhon, C.; Fehm, T.; Neubauer, H.; Rack, B.; Alunni-Fabbroni, M.; Terstappen, L.; van Gils, S.A.; et al. Quantifying HER-2 expression on circulating tumor cells by ACCEPT. PLoS ONE 2017, 12, e0186562. [Google Scholar] [CrossRef]
- Jordan, N.V.; Bardia, A.; Wittner, B.S.; Benes, C.; Ligorio, M.; Zheng, Y.; Yu, M.; Sundaresan, T.K.; Licausi, J.A.; Desai, R.; et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016, 537, 102–106. [Google Scholar] [CrossRef]
- Jacot, W.; Cottu, P.; Berger, F.; Dubot, C.; Venat-Bouvet, L.; Lortholary, A.; Bourgeois, H.; Bollet, M.; Servent, V.; Luporsi, E.; et al. Actionability of HER2-amplified circulating tumor cells in HER2-negative metastatic breast cancer: The CirCe T-DM1 trial. Breast Cancer Res. 2019, 21, 121. [Google Scholar] [CrossRef]
- Vidlarova, M.; Rehulkova, A.; Stejskal, P.; Prokopova, A.; Slavik, H.; Hajduch, M.; Srovnal, J. Recent Advances in Methods for Circulating Tumor Cell Detection. Int. J. Mol. Sci. 2023, 24, 3902. [Google Scholar] [CrossRef]
- Danila, D.C.; Samoila, A.; Patel, C.; Schreiber, N.; Herkal, A.; Anand, A.; Bastos, D.; Heller, G.; Fleisher, M.; Scher, H.I. Clinical Validity of Detecting Circulating Tumor Cells by AdnaTest Assay Compared with Direct Detection of Tumor mRNA in Stabilized Whole Blood, as a Biomarker Predicting Overall Survival for Metastatic Castration-Resistant Prostate Cancer Patients. Cancer J. 2016, 22, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Andreopoulou, E.; Yang, L.Y.; Rangel, K.M.; Reuben, J.M.; Hsu, L.; Krishnamurthy, S.; Valero, V.; Fritsche, H.A.; Cristofanilli, M. Comparison of assay methods for detection of circulating tumor cells in metastatic breast cancer: AdnaGen AdnaTest BreastCancer Select/Detect versus Veridex CellSearch system. Int. J. Cancer 2012, 130, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Schneck, H.; Gierke, B.; Uppenkamp, F.; Behrens, B.; Niederacher, D.; Stoecklein, N.H.; Templin, M.F.; Pawlak, M.; Fehm, T.; Neubauer, H.; et al. EpCAM-Independent Enrichment of Circulating Tumor Cells in Metastatic Breast Cancer. PLoS ONE 2015, 10, e0144535. [Google Scholar] [CrossRef]
- Gribko, A.; Stiefel, J.; Liebetanz, L.; Nagel, S.M.; Kunzel, J.; Wandrey, M.; Hagemann, J.; Stauber, R.H.; Freese, C.; Gul, D. IsoMAG-An Automated System for the Immunomagnetic Isolation of Squamous Cell Carcinoma-Derived Circulating Tumor Cells. Diagnostics 2021, 11, 2040. [Google Scholar] [CrossRef]
- Dao, L.; Ragoonanan, D.; Batth, I.; Satelli, A.; Foglesong, J.; Wang, J.; Zaky, W.; Gill, J.B.; Liu, D.; Albert, A.; et al. Prognostic Value of Cell-Surface Vimentin-Positive CTCs in Pediatric Sarcomas. Front. Oncol. 2021, 11, 760267. [Google Scholar] [CrossRef] [PubMed]
- Batth, I.S.; Dao, L.; Satelli, A.; Mitra, A.; Yi, S.; Noh, H.; Li, H.; Brownlee, Z.; Zhou, S.; Bond, J.; et al. Cell surface vimentin-positive circulating tumor cell-based relapse prediction in a long-term longitudinal study of postremission neuroblastoma patients. Int. J. Cancer 2020, 147, 3550–3559. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Batth, I.; Brownlee, Z.; Mitra, A.; Zhou, S.; Noh, H.; Rojas, C.R.; Li, H.; Meng, Q.H.; Li, S. EMT circulating tumor cells detected by cell-surface vimentin are associated with prostate cancer progression. Oncotarget 2017, 8, 49329–49337. [Google Scholar] [CrossRef]
- Liu, M.; Wang, R.; Sun, X.; Liu, Y.; Wang, Z.; Yan, J.; Kong, X.; Liang, S.; Liu, Q.; Zhao, T.; et al. Prognostic significance of PD-L1 expression on cell-surface vimentin-positive circulating tumor cells in gastric cancer patients. Mol. Oncol. 2020, 14, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Zhang, X.; Zhang, Q.; Yang, J.; Chen, Q.; Wang, J.; Li, X.; Chen, J.; Ma, T.; Li, G.; et al. Vimentin-positive circulating tumor cells as a biomarker for diagnosis and treatment monitoring in patients with pancreatic cancer. Cancer Lett. 2019, 452, 237–243. [Google Scholar] [CrossRef]
- Xie, X.; Wang, L.; Wang, X.; Fan, W.H.; Qin, Y.; Lin, X.; Xie, Z.; Liu, M.; Ouyang, M.; Li, S.; et al. Evaluation of Cell Surface Vimentin Positive Circulating Tumor Cells as a Diagnostic Biomarker for Lung Cancer. Front. Oncol. 2021, 11, 672687. [Google Scholar] [CrossRef]
- Satelli, A.; Brownlee, Z.; Mitra, A.; Meng, Q.H.; Li, S. Circulating tumor cell enumeration with a combination of epithelial cell adhesion molecule- and cell-surface vimentin-based methods for monitoring breast cancer therapeutic response. Clin. Chem. 2015, 61, 259–266. [Google Scholar] [CrossRef]
- Ffrench, B.; Kashdan, E.; Huang, Y.; Spillane, C.D.; Cocchiglia, S.; Charmsaz, S.; Vareslija, D.; O’Brien, C.; Scholz, D.; Martin, C.; et al. CTC-5: A novel digital pathology approach to characterise circulating tumour cell biodiversity. Heliyon 2023, 9, e13044. [Google Scholar] [CrossRef]
- Werner, S.L.; Graf, R.P.; Landers, M.; Valenta, D.T.; Schroeder, M.; Greene, S.B.; Bales, N.; Dittamore, R.; Marrinucci, D. Analytical Validation and Capabilities of the Epic CTC Platform: Enrichment-Free Circulating Tumour Cell Detection and Characterization. J. Circ. Biomark. 2015, 4, 3. [Google Scholar] [CrossRef]
- Fujii, T.; Reuben, J.M.; Huo, L.; Espinosa Fernandez, J.R.; Gong, Y.; Krupa, R.; Suraneni, M.V.; Graf, R.P.; Lee, J.; Greene, S.; et al. Androgen receptor expression on circulating tumor cells in metastatic breast cancer. PLoS ONE 2017, 12, e0185231. [Google Scholar] [CrossRef]
- Lang, J.E.; Ring, A.; Porras, T.; Kaur, P.; Forte, V.A.; Mineyev, N.; Tripathy, D.; Press, M.F.; Campo, D. RNA-Seq of Circulating Tumor Cells in Stage II-III Breast Cancer. Ann. Surg. Oncol. 2018, 25, 2261–2270. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.; Campo, D.; Porras, T.B.; Kaur, P.; Forte, V.A.; Tripathy, D.; Lu, J.; Kang, I.; Press, M.F.; Jeong, Y.J.; et al. Circulating Tumor Cell Transcriptomics as Biopsy Surrogates in Metastatic Breast Cancer. Ann. Surg. Oncol. 2022, 29, 2882–2894. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.F.; Wu, L.; Liu, S.P.; Jiang, M.M.; Hu, B.; Zhou, K.Q.; Guo, W.; Xu, Y.; Zhong, Y.; Zhou, X.R.; et al. Dissecting spatial heterogeneity and the immune-evasion mechanism of CTCs by single-cell RNA-seq in hepatocellular carcinoma. Nat. Commun. 2021, 12, 4091. [Google Scholar] [CrossRef] [PubMed]
- Diamantopoulou, Z.; Castro-Giner, F.; Schwab, F.D.; Foerster, C.; Saini, M.; Budinjas, S.; Strittmatter, K.; Krol, I.; Seifert, B.; Heinzelmann-Schwarz, V.; et al. The metastatic spread of breast cancer accelerates during sleep. Nature 2022, 607, 156–162. [Google Scholar] [CrossRef]
- Ruan, H.; Zhou, Y.; Shen, J.; Zhai, Y.; Xu, Y.; Pi, L.; Huang, R.; Chen, K.; Li, X.; Ma, W.; et al. Circulating tumor cell characterization of lung cancer brain metastases in the cerebrospinal fluid through single-cell transcriptome analysis. Clin. Transl. Med. 2020, 10, e246. [Google Scholar] [CrossRef]
- Davis, R.T.; Blake, K.; Ma, D.; Gabra, M.B.I.; Hernandez, G.A.; Phung, A.T.; Yang, Y.; Maurer, D.; Lefebvre, A.; Alshetaiwi, H.; et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat. Cell Biol. 2020, 22, 310–320. [Google Scholar] [CrossRef]
- Franken, A.; Kraemer, A.; Sicking, A.; Watolla, M.; Rivandi, M.; Yang, L.; Warfsmann, J.; Polzer, B.M.; Friedl, T.W.P.; Meier-Stiegen, F.; et al. Comparative analysis of EpCAM high-expressing and low-expressing circulating tumour cells with regard to their clonal relationship and clinical value. Br. J. Cancer 2023. [Google Scholar] [CrossRef]
- Descamps, L.; Le Roy, D.; Deman, A.L. Microfluidic-Based Technologies for CTC Isolation: A Review of 10 Years of Intense Efforts towards Liquid Biopsy. Int. J. Mol. Sci. 2022, 23, 1981. [Google Scholar] [CrossRef]
- Au, S.H.; Edd, J.; Stoddard, A.E.; Wong, K.H.K.; Fachin, F.; Maheswaran, S.; Haber, D.A.; Stott, S.L.; Kapur, R.; Toner, M. Microfluidic Isolation of Circulating Tumor Cell Clusters by Size and Asymmetry. Sci. Rep. 2017, 7, 2433. [Google Scholar] [CrossRef]
- Hosokawa, M.; Kenmotsu, H.; Koh, Y.; Yoshino, T.; Yoshikawa, T.; Naito, T.; Takahashi, T.; Murakami, H.; Nakamura, Y.; Tsuya, A.; et al. Size-based isolation of circulating tumor cells in lung cancer patients using a microcavity array system. PLoS ONE 2013, 8, e67466. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | Number of Patients | 184 |
| Number of samples | 755 | |
| Number of healthy volunteers | 57 | |
| Number of healthy volunteer samples | 118 | |
| Patient Age | <35 | 15 (8.2%) |
| 35–49 | 57 (31.0%) | |
| 50–65 | 75 (40.8%) | |
| >65 | 37 (20.1%) | |
| Met Histologic Subtype | HR+ HER2− | 106 (57.6%) |
| HR− HER2+ | 10 (5.4%) | |
| HR+ HER2+ | 14 (7.6%) | |
| TNBC | 37 (20.1%) | |
| NA * | 17 (9.2%) | |
| IBC | IBC | 23 (12.5%) |
| Non-IBC | 161 (87.5%) | |
| Lines of Treatment at Enrollment | 1 | 140 (76.1%) |
| >1 | 44 (23.9%) | |
| De novo Stage IV | De novo Stage IV | 45 (24.5%) |
| Previous DX | 139 (75.5%) | |
| Status | Alive | 104 (56.5%) |
| NED | 14 (7.6%) | |
| Progressed | 105 (57.1%) | |
| Deceased | 80 (43.5%) | |
| Race | White | 137 (74.5%) |
| Hispanic | 10 (5.4%) | |
| Black | 21 (11.4%) | |
| Asian | 7 (3.8%) | |
| Other | 9 (4.9%) | |
| Clinical Benefit of Met therapy Best Response | Yes (CR + PR + SD) | 115 (62.5%) |
| No (PD) | 43 (23.4%) | |
| NA | 26 (14.1%) |
| PFS | OS | ||||
|---|---|---|---|---|---|
| Covariate | Level | HR (95% CI) | p-Value | HR (95% CI) | p-Value |
| CTC count | <5 | 1.000 | 1 | ||
| ≥5 | 2.251 (1.628–3.113) | <0.0001 | 4.894 (2.854–8.393) | <0.0001 | |
| HR/HER2 | HR+/HER2+ | 1.000 | 1 | ||
| (metastatic site) | HR+/HER2− | 0.717 (0.409–1.258) | 0.1968 | 0.482 (0.115–2.024) | 0.3191 |
| HR−/HER2+ | 1.057 (0.571–1.959) | 0.859 | 0.893 (0.267–2.993) | 0.8549 | |
| HR−/HER2− | 1.725 (1.267–2.348) | 0.0005 | 3.389 (1.973–5.820) | <0.0001 | |
| FGFR1 | Low | 1 | |||
| High | 1.470 (1.074–2.012) | 0.0138 | - | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohen, E.N.; Jayachandran, G.; Gao, H.; Peabody, P.; McBride, H.B.; Alvarez, F.D.; Kai, M.; Song, J.; Shen, Y.; Willey, J.S.; et al. Phenotypic Plasticity in Circulating Tumor Cells Is Associated with Poor Response to Therapy in Metastatic Breast Cancer Patients. Cancers 2023, 15, 1616. https://doi.org/10.3390/cancers15051616
Cohen EN, Jayachandran G, Gao H, Peabody P, McBride HB, Alvarez FD, Kai M, Song J, Shen Y, Willey JS, et al. Phenotypic Plasticity in Circulating Tumor Cells Is Associated with Poor Response to Therapy in Metastatic Breast Cancer Patients. Cancers. 2023; 15(5):1616. https://doi.org/10.3390/cancers15051616
Chicago/Turabian StyleCohen, Evan N., Gitanjali Jayachandran, Hui Gao, Phillip Peabody, Heather B. McBride, Franklin D. Alvarez, Megumi Kai, Juhee Song, Yu Shen, Jie S. Willey, and et al. 2023. "Phenotypic Plasticity in Circulating Tumor Cells Is Associated with Poor Response to Therapy in Metastatic Breast Cancer Patients" Cancers 15, no. 5: 1616. https://doi.org/10.3390/cancers15051616
APA StyleCohen, E. N., Jayachandran, G., Gao, H., Peabody, P., McBride, H. B., Alvarez, F. D., Kai, M., Song, J., Shen, Y., Willey, J. S., Lim, B., Valero, V., Ueno, N. T., & Reuben, J. M. (2023). Phenotypic Plasticity in Circulating Tumor Cells Is Associated with Poor Response to Therapy in Metastatic Breast Cancer Patients. Cancers, 15(5), 1616. https://doi.org/10.3390/cancers15051616