
Circulating Tumor DNA: The Dawn of a New Era in the Optimization of Chemotherapeutic Strategies for Metastatic Colo-Rectal Cancer Focusing on RAS Mutation


Abstract
Simple Summary
Abstract
1. Introduction
2. Liquid Biopsy
3. ctDNA
3.1. Approaches for ctDNA Detection
3.2. Assessment of Prognosis
3.3. Detection of Recurrence: Minimal Residual Disease
3.4. Predicting Response to Treatment and Monitoring Acquired Resistance
4. Anti-EGFR Monoclonal Antibody Rechallenge
4.1. Trials of anti-EGFR Monoclonal Antibody Rechallenge
4.2. Ongoing Trials of anti-EGFR Monoclonal Antibody Rechallenge Based on Liquid Biopsy
5. NeoRAS
Ongoing Trials for the Treatment of NeoRAS Wild-Type Metastatic Colorectal Cancer
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Lièvre, A.; Bachet, J.B.; Boige, V.; Cayre, A.; Le Corre, D.; Buc, E.; Ychou, M.; Bouché, O.; Landi, B.; Louvet, C.; et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
- Taieb, J.; Le Malicot, K.; Shi, Q.; Penault-Llorca, F.; Bouché, O.; Tabernero, J.; Mini, E.; Goldberg, R.M.; Folprecht, G.; Luc Van Laethem, J.; et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS Stage III colon cancer. J. Natl. Cancer Inst. 2017, 109, djw272. [Google Scholar] [CrossRef]
- Zhu, G.; Pei, L.; Xia, H.; Tang, Q.; Bi, F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol. Cancer. 2021, 20, 143. [Google Scholar] [CrossRef]
- Mauri, G.; Bonazzina, E.; Amatu, A.; Tosi, F.; Bencardino, K.; Gori, V.; Massihnia, D.; Cipani, T.; Spina, F.; Ghezzi, S.; et al. The Evolutionary Landscape of Treatment for BRAFV600E Mutant Metastatic Colorectal Cancer. Cancers 2021, 13, 137. [Google Scholar] [CrossRef]
- Maughan, T.S.; Adams, R.A.; Smith, C.G.; Meade, A.M.; Seymour, M.T.; Wilson, R.H.; Idziaszczyk, S.; Harris, R.; Fisher, D.; Kenny, S.L.; et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: Results of the randomised phase 3 MRC COIN trial. Lancet 2011, 377, 2103–2114. [Google Scholar] [CrossRef]
- Tie, J.; Gibbs, P.; Lipton, L.; Christie, M.; Jorissen, R.N.; Burgess, A.W.; Croxford, M.; Jones, I.; Langland, R.; Kosmider, S.; et al. Optimizing targeted therapeutic development: Analysis of a colorectal cancer patient population with the BRAF(V600E) mutation. Int. J. Cancer 2011, 128, 2075–2084. [Google Scholar] [CrossRef]
- Bando, H.; Kagawa, Y.; Kato, T.; Akagi, K.; Denda, T.; Nishina, T.; Komatsu, Y.; Oki, E.; Kudo, T.; Kumamoto, H.; et al. A multicentre, prospective study of plasma circulating tumour DNA test for detecting RAS mutation in patients with metastatic colorectal cancer. Br. J. Cancer 2019, 120, 982–986. [Google Scholar] [CrossRef]
- Woodhouse, R.; Li, M.; Hughes, J.; Delfosse, D.; Skoletsky, J.; Ma, P.; Meng, W.; Dewal, N.; Milbury, C.; Clark, T.; et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS ONE 2020, 15, e0237802. [Google Scholar] [CrossRef]
- Overman, M.J.; Modak, J.; Kopetz, S.; Murthy, R.; Yao, J.C.; Hicks, M.E.; Abbruzzese, J.L.; Tam, A.L. Use of research biopsies in clinical trials: Are risks and benefits adequately discussed? J. Clin. Oncol. 2013, 31, 17–22. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Venesio, T.; Siravegna, G.; Bardelli, A.; Sapino, A. Liquid biopsies for monitoring temporal genomic heterogeneity in breast and colon cancers. Pathobiology 2018, 85, 146–154. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Nuclear acids in human blood plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Analysis of the size distributions of fetal and maternal cell-free DNA by paired-end sequencing. Clin. Chem. 2010, 56, 1279–1286. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Birkenkamp-Demtröder, K.; Nordentoft, I.; Christensen, E.; Høyer, S.; Reinert, T.; Vang, S.; Borre, M.; Agerbæk, M.; Jensen, J.B.; Ørntoft, T.F.; et al. Genomic alterations in liquid biopsies from patients with bladder cancer. Eur. Urol. 2016, 70, 75–82. [Google Scholar] [CrossRef]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.B.; Relan, V.; Clarke, B.E.; Duhig, E.E.; Windsor, M.N.; Matar, K.S.; Naidoo, R.; Passmore, L.; McCaul, E.; Courtney, D.; et al. Pleural fluid cell-free DNA integrity index to identify cytologically negative malignant pleural effusions including mesotheliomas. BMC Cancer 2012, 12, 428. [Google Scholar] [CrossRef] [PubMed]
- Villatoro, S.; Mayo-de-Las-Casas, C.; Jordana-Ariza, N.; Viteri-Ramírez, S.; Garzón-Ibañez, M.; Moya-Horno, I.; García-Peláez, B.; González-Cao, M.; Malapelle, U.; Balada-Bel, A.; et al. Prospective detection of mutations in cerebrospinal fluid, pleural effusion, and ascites of advanced cancer patients to guide treatment decisions. Mol. Oncol. 2019, 13, 2633–2645. [Google Scholar] [CrossRef] [PubMed]
- Mithani, S.K.; Smith, I.M.; Zhou, S.; Gray, A.; Koch, W.M.; Maitra, A.; Califano, J.A. Mitochondrial resequencing arrays detect tumor-specific mutations in salivary rinses of patients with head and neck cancer. Clin. Cancer Res. 2007, 13, 7335–7340. [Google Scholar] [CrossRef] [PubMed]
- Avanzini, S.; Kurtz, D.M.; Chabon, J.J.; Moding, E.J.; Hori, S.S.; Gambhir, S.S.; Alizadeh, A.A.; Diehn, M.; Reiter, J.G. A mathematical model of ctDNA shedding predicts tumor detection size. Sci. Adv. 2020, 6, eabc4308. [Google Scholar] [CrossRef]
- Ito, K.; Hibi, K.; Ando, H.; Hidemura, K.; Yamazaki, T.; Akiyama, S.; Nakao, A. Usefulness of analytical CEA doubling time and half-life time for overlooked synchronous metastases in colorectal carcinoma. Jpn. J. Clin. Oncol. 2002, 32, 54–58. [Google Scholar] [CrossRef]
- Duffy, M.J.; Crown, J. Circulating tumor DNA as a biomarker for monitoring patients with solid cancers: Comparison with standard protein biomarkers. Clin. Chem. 2022, 68, 1381–1390. [Google Scholar] [CrossRef]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical utility of comprehensive cell-free DNA analysis to identify genomic biomarkers in patients with newly diagnosed metastatic non-small cell lung cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N.; et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med. 2020, 26, 1859–1864. [Google Scholar] [CrossRef]
- Benavides, M.; Alcaide-Garcia, J.; Torres, E.; Gil-Calle, S.; Sevilla, I.; Wolman, R.; Durán, G.; Álvarez, M.; Reyna-Fortes, C.; Ales, I.; et al. Clinical utility of comprehensive circulating tumor DNA genotyping compared with standard of care tissue testing in patients with newly diagnosed metastatic colorectal cancer. ESMO Open 2022, 7, 100481. [Google Scholar] [CrossRef]
- Gray, J.E.; Okamoto, I.; Sriuranpong, V.; Vansteenkiste, J.; Imamura, F.; Lee, J.S.; Pang, Y.K.; Cobo, M.; Kasahara, K.; Cheng, Y.; et al. Tissue and plasma EGFR Mutation Analysis in the FLAURA Trial: Osimertinib versus Comparator EGFR tyrosine kinase Inhibitor as First-Line Treatment in Patients with EGFR-Mutated Advanced non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6644–6652. [Google Scholar] [CrossRef]
- Rothwell, D.G.; Ayub, M.; Cook, N.; Thistlethwaite, F.; Carter, L.; Dean, E.; Smith, N.; Villa, S.; Dransfield, J.; Clipson, A.; et al. Utility of ctDNA to support patient selection for early phase clinical trials: The TARGET study. Nat. Med. 2019, 25, 738–743. [Google Scholar] [CrossRef]
- Rose Brannon, A.; Jayakumaran, G.; Diosdado, M.; Patel, J.; Razumova, A.; Hu, Y.; Meng, F.; Haque, M.; Sadowska, J.; Murphy, B.J.; et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat. Commun. 2021, 12, 3770. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Kagawa, Y.; Elez, E.; García-Foncillas, J.; Bando, H.; Taniguchi, H.; Vivancos, A.; Akagi, K.; García, A.; Denda, T.; Ros, J.; et al. Combined analysis of concordance between liquid and tumor tissue biopsies for RAS mutations in colorectal cancer with a single metastasis site: The METABEAM study. Clin. Cancer Res. 2021, 27, 2515–2522. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Ooki, A.; Shimozaki, K.; Kamiimabeppu, D.; Nakayama, I.; Wakatsuki, T.; Ogura, M.; Takahari, D.; Chin, K.; et al. Correlation between circulating tumor DNA and carcinoembryonic antigen levels in patients with metastatic colorectal cancer. Cancer Med. 2021, 10, 8820–8828. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Pantel, K.; Kemper, B.; Beeger, C.; Otterbach, F.; Kimmig, R.; Kasimir-Bauer, S. Comparative evaluation of cell-free tumor DNA in blood and disseminated tumor cells in bone marrow of patients with primary breast cancer. Breast Cancer Res. 2009, 11, R71. [Google Scholar] [CrossRef]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 2017, 21, 374–382.e4. [Google Scholar] [CrossRef]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef]
- Ptashkin, R.N.; Mandelker, D.L.; Coombs, C.C.; Bolton, K.; Yelskaya, Z.; Hyman, D.M.; Solit, D.B.; Baselga, J.; Arcila, M.E.; Ladanyi, M.; et al. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA Oncol. 2018, 4, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.T.; Nagayama, S.; Chin, Y.M.; Otaki, M.; Hayashi, R.; Kiyotani, K.; Fukunaga, Y.; Ueno, M.; Nakamura, Y.; Low, S.K. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol. Oncol. 2020, 14, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hu, S.; Zhang, L.; Xin, J.; Sun, C.; Wang, L.; Ding, K.; Wang, B. Tumor circulome in the liquid biopsies for cancer diagnosis and prognosis. Theranostics 2020, 10, 4544–4556. [Google Scholar] [CrossRef] [PubMed]
- Osumi, H.; Shinozaki, E.; Yamaguchi, K.; Zembutsu, H. Clinical utility of circulating tumor DNA for colorectal cancer. Cancer Sci. 2019, 110, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jing, C.; Wu, J.; Ni, J.; Sha, H.; Xu, X.; Du, Y.; Lou, R.; Dong, S.; Feng, J. Circulating tumor DNA detection: A potential tool for colorectal cancer management. Oncol. Lett. 2019, 17, 1409–1416. [Google Scholar] [CrossRef]
- Nikanjam, M.; Kato, S.; Kurzrock, R. Liquid biopsy: Current technology and clinical applications. J. Hematol. Oncol. 2022, 15, 131. [Google Scholar] [CrossRef]
- Vessies, D.C.L.; Greuter, M.J.E.; van Rooijen, K.L.; Linders, T.C.; Lanfermeijer, M.; Ramkisoensing, K.L.; Meijer, G.A.; Koopman, M.; Coupé, V.M.H.; Vink, G.R.; et al. Performance of four platforms for KRAS mutation detection in plasma cell-free DNA: ddPCR, Idylla, COBAS z480 and BEAMing. Sci. Rep. 2020, 10, 8122. [Google Scholar] [CrossRef]
- Garcia, J.; Forestier, J.; Dusserre, E.; Wozny, A.S.; Geiguer, F.; Merle, P.; Tissot, C.; Ferraro-Peyret, C.; Jones, F.S.; Edelstein, D.L.; et al. Cross-platform comparison for the detection of RAS mutations in cfDNA (ddPCR BioRad detection assay, BEAMing assay, and NGS strategy). Oncotarget 2018, 9, 21122–21131. [Google Scholar] [CrossRef]
- Vivancos, A.; Aranda, E.; Benavides, M.; Élez, E.; Gómez-España, M.A.; Toledano, M.; Alvarez, M.; Parrado, M.R.C.; García-Barberán, V.; Diaz-Rubio, E. Comparison of the clinical sensitivity of the idylla platform and the OncoBEAM RAS CRC assay for KRAS mutation detection in liquid biopsy samples. Sci. Rep. 2019, 9, 8976. [Google Scholar] [CrossRef]
- Schmiegel, W.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- García-Foncillas, J.; Alba, E.; Aranda, E.; Díaz-Rubio, E.; López-López, R.; Tabernero, J.; Vivancos, A. Incorporating BEAMing technology as a liquid biopsy into clinical practice for the management of colorectal cancer patients: An expert taskforce review. Ann. Oncol. 2017, 28, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.B.; Siu, L.L.; et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 2019, 30, 1580–1590. [Google Scholar] [CrossRef]
- Lin, M.T.; Mosier, S.L.; Thiess, M.; Beierl, K.F.; Debeljak, M.; Tseng, L.H.; Chen, G.; Yegnasubramanian, S.; Ho, H.; Cope, L.; et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 856–866. [Google Scholar] [CrossRef]
- Kamps, R.; Brandão, R.D.; Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-generation sequencing in oncology: Genetic diagnosis, risk prediction and cancer classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef]
- Revolutionizing Cancer Care|Caris Life Sciences. Available online: https://www.carislifesciences.com/ (accessed on 17 December 2022).
- Puccini, A.; Martelli, V.; Pastorino, A.; Sciallero, S.; Sobrero, A. ctDNA to guide treatment of colorectal cancer: Ready for standard of care? Curr. Treat. Options Oncol. 2023, 24, 76–92. [Google Scholar] [CrossRef]
- Signatera. Circulating Tumor DNA Blood Test | Natera. Available online: https://www.natera.com/oncology/signatera-advanced-cancer-detection/ (accessed on 17 December 2022).
- Tabernero, J.; Lenz, H.J.; Siena, S.; Sobrero, A.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: A retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol. 2015, 16, 937–948. [Google Scholar] [CrossRef]
- Manca, P.; Corallo, S.; Lonardi, S.; Fucà, G.; Busico, A.; Leone, A.G.; Corti, F.; Antoniotti, C.; Procaccio, L.; Smiroldo, V.; et al. Variant allele frequency in baseline circulating tumour DNA to measure tumour burden and to stratify outcomes in patients with RAS wild-type metastatic colorectal cancer: A translational objective of the Valentino study. Br. J. Cancer. 2022, 126, 449–455. [Google Scholar] [CrossRef]
- Janku, F.; Zhang, S.; Waters, J.; Liu, L.; Huang, H.J.; Subbiah, V.; Hong, D.S.; Karp, D.D.; Fu, S.; Cai, X.; et al. Development and validation of an ultradeep next-generation sequencing assay for testing of plasma cell-free DNA from patients with advanced cancer. Clin. Cancer Res. 2017, 23, 5648–5656. [Google Scholar] [CrossRef]
- El Messaoudi, S.; Mouliere, F.; Du Manoir, S.; Bascoul-Mollevi, C.; Gillet, B.; Nouaille, M.; Fiess, C.; Crapez, E.; Bibeau, F.; Theillet, C.; et al. Circulating DNA as a strong multimarker prognostic tool for metastatic colorectal cancer patient management care. Clin. Cancer Res. 2016, 22, 3067–3077. [Google Scholar] [CrossRef] [PubMed]
- Pairawan, S.; Hess, K.R.; Janku, F.; Sanchez, N.S.; Mills Shaw, K.R.; Eng, C.; Damodaran, S.; Javle, M.; Kaseb, A.O.; Hong, D.S.; et al. Cell-free circulating tumor DNA variant allele frequency associates with survival in metastatic cancer. Clin. Cancer Res. 2020, 26, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Garlan, F.; Laurent-Puig, P.; Sefrioui, D.; Siauve, N.; Didelot, A.; Sarafan-Vasseur, N.; Michel, P.; Perkins, G.; Mulot, C.; Blons, H.; et al. Early evaluation of circulating tumor DNA as marker of therapeutic efficacy in metastatic colorectal cancer patients (PLACOL study). Clin. Cancer Res. 2017, 23, 5416–5425. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Price, T.; Boedigheimer, M.; Kim, T.W.; Ruff, P.; Gibbs, P.; Thomas, A.; Demonty, G.; Hool, K.; Ang, A. Evaluation of emergent mutations in circulating cell-free DNA and clinical outcomes in patients with metastatic colorectal cancer treated with panitumumab in the ASPECCT study. Clin. Cancer Res. 2019, 25, 1216–1225. [Google Scholar] [CrossRef]
- Luo, H.; Zhao, Q.; Wei, W.; Zheng, L.; Yi, S.; Li, G.; Wang, W.; Sheng, H.; Pu, H.; Mo, H.; et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci. Transl. Med. 2020, 12, eaax7533. [Google Scholar] [CrossRef]
- Manca, P.; Corallo, S.; Busico, A.; Lonardi, S.; Corti, F.; Antoniotti, C.; Procaccio, L.; Clavarezza, M.; Smiroldo, V.; Tomasello, G.; et al. The added value of baseline circulating tumor DNA profiling in patients with molecularly hyperselected, left-sided metastatic colorectal cancer. Clin. Cancer Res. 2021, 27, 2505–2514. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef]
- Tarazona, N.; Gimeno-Valiente, F.; Gambardella, V.; Zuñiga, S.; Rentero-Garrido, P.; Huerta, M.; Roselló, S.; Martinez-Ciarpaglini, C.; Carbonell-Asins, J.A.; Carrasco, F.; et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann. Oncol. 2019, 30, 1804–1812. [Google Scholar] [CrossRef]
- Reinert, T.; Schøler, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2016, 65, 625–634. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating tumor DNA analyses as markers of recurrence risk and benefit of adjuvant therapy for Stage III colon cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating tumor DNA analysis guiding adjuvant therapy in Stage II colon cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Coombes, R.C.; Page, K.; Salari, R.; Hastings, R.K.; Armstrong, A.; Ahmed, S.; Ali, S.; Cleator, S.; Kenny, L.; Stebbing, J.; et al. Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin. Cancer Res. 2019, 25, 4255–4263. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Christensen, E.; Birkenkamp-Demtröder, K.; Sethi, H.; Shchegrova, S.; Salari, R.; Nordentoft, I.; Wu, H.T.; Knudsen, M.; Lamy, P.; Lindskrog, S.V.; et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. J. Clin. Oncol. 2019, 37, 1547–1557. [Google Scholar] [CrossRef]
- Taniguchi, H.; Nakamura, Y.; Kotani, D.; Yukami, H.; Mishima, S.; Sawada, K.; Shirasu, H.; Ebi, H.; Yamanaka, T.; Aleshin, A.; et al. CIRCULATE-Japan: Circulating tumor DNA-guided adaptive platform trials to refine adjuvant therapy for colorectal cancer. Cancer Sci. 2021, 112, 2915–2920. [Google Scholar] [CrossRef]
- Spindler, K.L.; Pallisgaard, N.; Vogelius, I.; Jakobsen, A. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin. Cancer Res. 2012, 18, 1177–1185. [Google Scholar] [CrossRef]
- Spindler, K.L.; Sorensen, M.M.; Pallisgaard, N.; Andersen, R.F.; Havelund, B.M.; Ploen, J.; Lassen, U.; Jakobsen, A.K. Phase II trial of temsirolimus alone and in combination with irinotecan for KRAS mutant metastatic colorectal cancer: Outcome and results of KRAS mutational analysis in plasma. Acta Oncol. 2013, 52, 963–970. [Google Scholar] [CrossRef]
- Ueda, K.; Yamada, T.; Ohta, R.; Matsuda, A.; Sonoda, H.; Kuriyama, S.; Takahashi, G.; Iwai, T.; Takeda, K.; Miyasaka, T.; et al. BRAF V600E mutations in right-side colon cancer: Heterogeneity detected by liquid biopsy. Eur. J. Surg. Oncol. 2022, 48, 1375–1383. [Google Scholar] [CrossRef]
- Gilson, P.; Merlin, J.L.; Harlé, A. Detection of microsatellite instability: State of the art and future applications in circulating tumour DNA (ctDNA). Cancers 2021, 13, 1491. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Marsavela, G.; Lee, J.; Calapre, L.; Wong, S.Q.; Pereira, M.R.; McEvoy, A.C.; Reid, A.L.; Robinson, C.; Warburton, L.; Abed, A.; et al. Circulating tumor DNA predicts outcome from first-, but not second-line treatment and identifies melanoma patients who may benefit from combination immunotherapy. Clin. Cancer Res. 2020, 26, 5926–5933. [Google Scholar] [CrossRef] [PubMed]
- Seremet, T.; Jansen, Y.; Planken, S.; Njimi, H.; Delaunoy, M.; El Housni, H.; Awada, G.; Schwarze, J.K.; Keyaerts, M.; Everaert, H.; et al. Undetectable circulating tumor DNA (ctDNA) levels correlate with favorable outcome in metastatic melanoma patients treated with anti-PD1 therapy. J. Transl. Med. 2019, 17, 303. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Okamoto, W.; Kato, T.; Esaki, T.; Kato, K.; Komatsu, Y.; Yuki, S.; Masuishi, T.; Nishina, T.; Ebi, H.; et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: A phase 2 trial. Nat. Med. 2021, 27, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Lazzari, L.; Crisafulli, G.; Sartore-Bianchi, A.; Mussolin, B.; Cassingena, A.; Martino, C.; Lanman, R.B.; Nagy, R.J.; Fairclough, S.; et al. Radiologic and genomic evolution of individual metastases during HER2 blockade in colorectal cancer. Cancer Cell 2018, 34, 148–162.e7. [Google Scholar] [CrossRef]
- Siravegna, G.; Sartore-Bianchi, A.; Nagy, R.J.; Raghav, K.; Odegaard, J.I.; Lanman, R.B.; Trusolino, L.; Marsoni, S.; Siena, S.; Bardelli, A. Plasma HER2 (ERBB2) Copy Number Predicts Response to HER2-targeted Therapy in Metastatic Colorectal Cancer. Clin. Cancer Res. 2019, 25, 3046–3053. [Google Scholar] [CrossRef]
- Rolfo, C.; Drilon, A.; Hong, D.; McCoach, C.; Dowlati, A.; Lin, J.J.; Russo, A.; Schram, A.M.; Liu, S.V.; Nieva, J.J.; et al. NTRK1 Fusions identified by non-invasive plasma next-generation sequencing (NGS) across 9 cancer types. Br. J. Cancer 2022, 126, 514–520. [Google Scholar] [CrossRef]
- Callesen, L.B.; Hamfjord, J.; Boysen, A.K.; Pallisgaard, N.; Guren, T.K.; Kure, E.H.; Spindler, K.G. Circulating tumour DNA and its clinical utility in predicting treatment response or survival in patients with metastatic colorectal cancer: A systematic review and meta-analysis. Br. J. Cancer 2022, 127, 500–513. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef]
- Morelli, M.P.; Overman, M.J.; Dasari, A.; Kazmi, S.M.A.; Mazard, T.; Vilar, E.; Morris, V.K.; Lee, M.S.; Herron, D.; Eng, C.; et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann. Oncol. 2015, 26, 731–736. [Google Scholar] [CrossRef]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef]
- Strickler, J.H.; Loree, J.M.; Ahronian, L.G.; Parikh, A.R.; Niedzwiecki, D.; Pereira, A.A.L.; McKinney, M.; Korn, W.M.; Atreya, C.E.; Banks, K.C.; et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov. 2018, 8, 164–173. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting oncogenic BRAF: Past, present, and future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef]
- Santini, D.; Vincenzi, B.; Addeo, R.; Garufi, C.; Masi, G.; Scartozzi, M.; Mancuso, A.; Frezza, A.M.; Venditti, O.; Imperatori, M.; et al. Cetuximab rechallenge in metastatic colorectal cancer patients: How to come away from acquired resistance? Ann. Oncol. 2017, 28, 2906. [Google Scholar] [CrossRef]
- Avallone, A.; Giuliani, F.; Nasti, G.; Montesarchio, V.; Santabarbara, G.; Leo, S.; De Stefano, A.; Rosati, G.; Lolli, I.; Tamburini, E.; et al. Randomized intermittent or continuous panitumumab plus FOLFIRI (FOLFIRI/PANI) for first-line treatment of patients (pts) with RAS/BRAF wild-type (wt) metastatic colorectal cancer (mCRC): The IMPROVE study. J. Clin. Oncol. 2022, 40 (Suppl. S16), 3503. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef]
- Parseghian, C.M.; Loree, J.M.; Morris, V.K.; Liu, X.; Clifton, K.K.; Napolitano, S.; Henry, J.T.; Pereira, A.A.; Vilar, E.; Johnson, B.; et al. Anti-EGFR-resistant clones decay exponentially after progression: Implications for anti-EGFR re-challenge. Ann. Oncol. 2019, 30, 243–249. [Google Scholar] [CrossRef]
- Mayer, R.J.; Van Cutsem, E.; Falcone, A.; Yoshino, T.; Garcia-Carbonero, R.; Mizunuma, N.; Yamazaki, K.; Shimada, Y.; Tabernero, J.; Komatsu, Y.; et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N. Engl. J. Med. 2015, 372, 1909–1919. [Google Scholar] [CrossRef]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Kuboki, Y.; Nishina, T.; Shinozaki, E.; Yamazaki, K.; Shitara, K.; Okamoto, W.; Kajiwara, T.; Matsumoto, T.; Tsushima, T.; Mochizuki, N.; et al. TAS-102 plus bevacizumab for patients with metastatic colorectal cancer refractory to standard therapies (C-TASK FORCE): An investigator-initiated, open-label, single-arm, multicentre, phase 1/2 study. Lancet Oncol. 2017, 18, 1172–1181. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Yilmaz, M.; Möller, S.; Zitnjak, D.; Krogh, M.; Petersen, L.N.; Poulsen, L.Ø.; Winther, S.B.; Thomsen, K.G.; Qvortrup, C. TAS-102 with or without bevacizumab in patients with chemorefractory metastatic colorectal cancer: An investigator-initiated, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 412–420. [Google Scholar] [CrossRef]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for patients with RAS and BRAF wild-type metastatic colorectal cancer with acquired resistance to first-line cetuximab and irinotecan: A phase 2 single-arm clinical trial. JAMA Oncol. 2019, 5, 343–350. [Google Scholar] [CrossRef]
- Osawa, H.; Shinozaki, E.; Nakamura, M.; Ohhara, Y.; Shindo, Y.; Shiozawa, M.; Uetake, H.; Matsumoto, H.; Ureshino, N.; Satake, H.; et al. Phase II study of cetuximab rechallenge in patients with ras wild-type metastatic colorectal cancer: E-rechallenge. Ann. Oncol. 2018, 29 (Suppl. S8), viii161. [Google Scholar] [CrossRef]
- Sunakawa, Y.; Nakamura, M.; Ishizaki, M.; Kataoka, M.; Satake, H.; Kitazono, M.; Yanagisawa, H.; Kawamoto, Y.; Kuramochi, H.; Ohori, H.; et al. RAS mutations in circulating tumor DNA and clinical outcomes of rechallenge treatment with anti-EGFR antibodies in patients with metastatic colorectal cancer. JCO Precis. Oncol. 2020, 4, 898–911. [Google Scholar] [CrossRef]
- Martinelli, E.; Martini, G.; Famiglietti, V.; Troiani, T.; Napolitano, S.; Pietrantonio, F.; Ciardiello, D.; Terminiello, M.; Borrelli, C.; Vitiello, P.P.; et al. Cetuximab rechallenge plus avelumab in pretreated patients with RAS wild-type metastatic colorectal cancer: The Phase 2 single-arm clinical CAVE trial. JAMA Oncol. 2021, 7, 1529–1535. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Crisafulli, G.; Bartolini, A.; Fenocchio, E.; Amatu, A.; Manca, P.; et al. Circulating tumor DNA to guide rechallenge with panitumumab in metastatic colorectal cancer: The phase 2 CHRONOS trial. Nat. Med. 2022, 28, 1612–1618. [Google Scholar] [CrossRef]
- Kagawa, Y.; Kotani, D.; Bando, H.; Takahashi, N.; Hamaguchi, T.; Kanazawa, A.; Kato, T.; Ando, K.; Satake, H.; Shinozaki, E.; et al. Plasma RAS dynamics and anti-EGFR rechallenge efficacy in patients with RAS/BRAF wild-type metastatic colorectal cancer: REMARRY and PURSUIT trials. J. Clin. Oncol. 2022, 40 (Suppl. 16), 3518. [Google Scholar] [CrossRef]
- Ciardiello, D.; Martini, G.; Famiglietti, V.; Napolitano, S.; De Falco, V.; Troiani, T.; Latiano, T.P.; Ros, J.; Elez Fernandez, E.; Vitiello, P.P.; et al. Biomarker-guided anti-egfr rechallenge therapy in metastatic colorectal cancer. Cancers 2021, 13, 1941. [Google Scholar] [CrossRef]
- Sorah, J.D.; Moore, D.T.; Reilley, M.J.; Salem, M.E.; Triglianos, T.; Sanoff, H.K.; McRee, A.J.; Lee, M.S. Phase II single-arm study of palbociclib and cetuximab rechallenge in patients with KRAS/NRAS/BRAF wild-type colorectal cancer. Oncologist 2022, 27, 1006-e930. [Google Scholar] [CrossRef]
- García-Foncillas, J.; Tabernero, J.; Élez, E.; Aranda, E.; Benavides, M.; Camps, C.; Jantus-Lewintre, E.; López, R.; Muinelo-Romay, L.; Montagut, C.; et al. Prospective multicenter real-world RAS mutation comparison between OncoBEAM-based liquid biopsy and tissue analysis in metastatic colorectal cancer. Br. J. Cancer 2018, 119, 1464–1470. [Google Scholar] [CrossRef]
- Sugimachi, K.; Sakimura, S.; Kuramitsu, S.; Hirata, H.; Niida, A.; Iguchi, T.; Eguchi, H.; Masuda, T.; Morita, M.; Toh, Y.; et al. Serial mutational tracking in surgically resected locally advanced colorectal cancer with neoadjuvant chemotherapy. Br. J. Cancer 2018, 119, 419–423. [Google Scholar] [CrossRef]
- Klein-Scory, S.; Wahner, I.; Maslova, M.; Al-Sewaidi, Y.; Pohl, M.; Mika, T.; Ladigan, S.; Schroers, R.; Baraniskin, A. Evolution of RAS mutational status in liquid biopsies during first-line chemotherapy for metastatic colorectal cancer. Front. Oncol. 2020, 10, 1115. [Google Scholar] [CrossRef]
- Osumi, H.; Vecchione, L.; Keilholz, U.; Vollbrecht, C.; Alig, A.H.S.; von Einem, J.C.; Stahler, A.; Striefler, J.K.; Kurreck, A.; Kind, A.; et al. NeoRAS wild-type in metastatic colorectal cancer: Myth or truth?-Case series and review of the literature. Eur. J. Cancer. 2021, 153, 86–95. [Google Scholar] [CrossRef]
- Raimondi, C.; Nicolazzo, C.; Belardinilli, F.; Loreni, F.; Gradilone, A.; Mahdavian, Y.; Gelibter, A.; Giannini, G.; Cortesi, E.; Gazzaniga, P. Transient disappearance of RAS mutant clones in plasma: A counterintuitive clinical use of EGFR inhibitors in RAS mutant metastatic colorectal cancer. Cancers 2019, 11, 42. [Google Scholar] [CrossRef]
- Nicolazzo, C.; Barault, L.; Caponnetto, S.; Macagno, M.; De Renzi, G.; Gradilone, A.; Belardinilli, F.; Cortesi, E.; Di Nicolantonio, F.; Gazzaniga, P. Circulating methylated DNA to monitor the dynamics of RAS mutation clearance in plasma from metastatic colorectal cancer patients. Cancers 2020, 12, 3633. [Google Scholar] [CrossRef]
- Moati, E.; Blons, H.; Taly, V.; Garlan, F.; Wang-Renault, S.F.; Pietrasz, D.; Didelot, A.; Garrigou, S.; Saint, A.; Pernot, S.; et al. Plasma clearance of RAS mutation under therapeutic pressure is a rare event in metastatic colorectal cancer. Int. J. Cancer 2020, 147, 1185–1189. [Google Scholar] [CrossRef]
- Henry, J.; Willis, J.; Parseghian, C.M.; Raghav, K.P.S.; Johnson, B.; Dasari, A.; Stone, D.; Jeyakumar, N.; Coker, O.; Raymond, V.M.; et al. NeoRAS: Incidence of RAS reversion from RAS mutated to RAS wild type. J. Clin. Oncol. 2020, 38 (Suppl. S4), 180. [Google Scholar] [CrossRef]
- Fernández Montes, A.; Martínez Lago, N.; de la Cámara Gómez, J.; Covela Rúa, M.; Cousillas Castiñeiras, A.; González Villarroel, P.; Méndez Méndez, J. FOLFIRI plus panitumumab as second-line treatment in mutated RAS metastatic colorectal cancer patients who converted to wild type RAS after receiving first-line FOLFOX/CAPOX plus bevacizumab-based treatment: Phase II CONVERTIX trial. Ann. Oncol. 2019, 30 (Suppl. S4), iv23–iv24. [Google Scholar] [CrossRef]
- Sunakawa, Y.; Usher, J.; Satake, H.; Jaimes, Y.; Miyamoto, Y.; Nakamura, M.; Kataoka, M.; Shiozawa, M.; Takagane, A.; Terazawa, T.; et al. Gene mutation status in circulating tumor DNA (ctDNA) and first-line FOLFOXIRI plus bevacizumab (bev) in metastatic colorectal cancer (mCRC) harboring RAS mutation. Ann. Oncol. 2018, 29 (Suppl. S8), viii181–viii182. [Google Scholar] [CrossRef]
- Nicolazzo, C.; Barault, L.; Caponnetto, S.; De Renzi, G.; Belardinilli, F.; Bottillo, I.; Bargiacchi, S.; Macagno, M.; Grammatico, P.; Giannini, G.; et al. True conversions from RAS mutant to RAS wild-type in circulating tumor DNA from metastatic colorectal cancer patients as assessed by methylation and mutational signature. Cancer Lett. 2021, 507, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Bouchahda, M.; Saffroy, R.; Karaboué, A.; Hamelin, J.; Innominato, P.; Saliba, F.; Lévi, F.; Bosselut, N.; Lemoine, A. Undetectable RAS-mutant clones in plasma: Possible implication for anti-EGFR therapy and prognosis in patients with RAS-mutant metastatic colorectal cancer. JCO Precis. Oncol. 2020, 4, 1070–1079. [Google Scholar] [CrossRef]
- Osumi, H.; Ishizuka, N.; Takashima, A.; Kumekawa, Y.; Nakano, D.; Shiozawa, M.; Denda, T.; Sawada, R.; Ouchi, K.; Wakatsuki, T.; et al. Multicentre single-arm phase II trial evaluating the safety and effiCacy of panitumumab and iRinOtecan in NeoRAS Wild-type mEtaStatic colorectal cancer patientS (C-PROWESS trial): Study protocol. BMJ Open 2022, 12, e063071. [Google Scholar] [CrossRef]



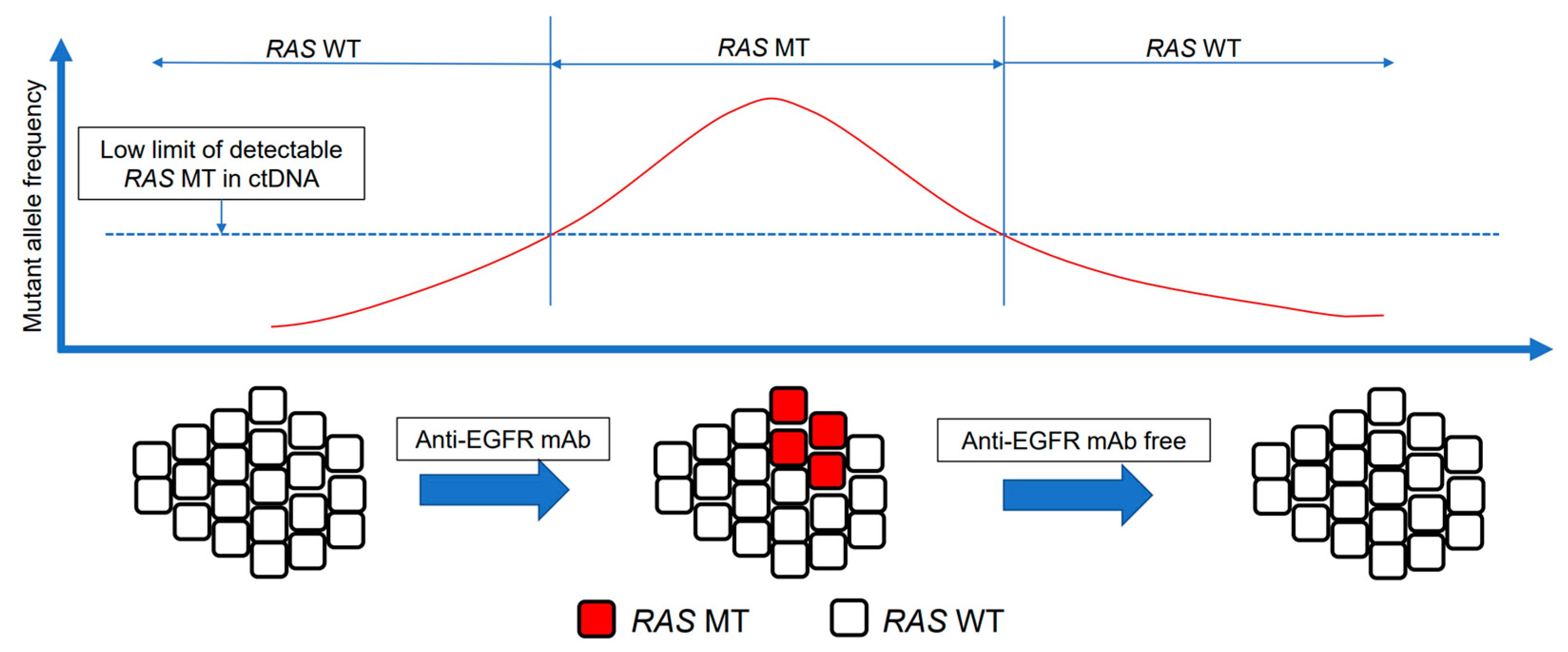{kind=link}
{kind=link}
| Study | Phase | Assay | Number | Primary Endpoint | Treatment | ORR (%) | mPFS (Months) | mOS (Months) |
|---|---|---|---|---|---|---|---|---|
| CRIKET [105] | Single-arm phase II | Droplet digital PCR | 28 (ctDNA: 25) | ORR | CET + IRI | 21 | 3.4 | 9.8 |
| E-Rechallenge [106] | Single-arm phase II | Droplet digital PCR | 33 (ctDNA: 24) | ORR | PANI + IRI | 15.2 | 2.9 | 8.7 |
| JACCRO CC-08/09AR [107] | Single-arm phase II | BEAMing (OncoBEAMTM RAS CRC KIT) | 16 | 3-month PFS | CET + IRI (08) PANI + IRI (09) | 0 | 2.4 | 8.2 |
| CAVE [108] | Single-arm phase II | Quantitative PCR | 77 (ctDNA: 67) | OS | Avelumab + CET | 7.8 | 3.6 | 11.6 |
| CHRONOS [109] | Single-arm phase II | Droplet digital PCR | 27 | ORR | PANI | 30 | N/A | 12.8 |
| PURSUIT [110] | Single-arm phase II | BEAMing (OncoBEAMTM RAS CRC KIT) | 50 | ORR | PANI + IRI | 14 | 3.6 | N/A |
| VELO [111] | Randomized phase II | Real-time PCR (IdyllaTM) | 62 | PFS | PANI + TAS102 vs. TAS102 | 9.7 | 4.0 vs. 2.5 | N/A |
| Study | Phase | Number | Assay | Estimated Enrollment | Treatment | Primary Endpoint | Study Completion Date |
|---|---|---|---|---|---|---|---|
| PULSE | Randomized phase II | NCT03992456 | NGS (Guardant360® assay) | 120 | PANI vs. TAS102 or regorafenib | OS | 7 October 2023 |
| PARERE | Randomized phase II | NCT04787341 | NGS (OncomineTM Colon cfDNA Assay) | 214 | PANI → regorafenib vs. regorafenib → PANI | OS | 15 June 2024 |
| CAPRI II GOIM | Single-arm phase II | NCT05312398 | NGS | 200 | The therapeutic algorithm by liquid biopsy assessment of RAS/BRAF status | ORR | 15 June 2026 |
| CAVE II GOIM | Randomized phase II | NCT05291156 | NGS (FoundationOne® Liquid) | 173 | CET + avelumab vs. CET | OS | 1 July 2025 |
| Study | Phase | Number | Assay | Estimated Enrollment | Treatment | Primary Endpoint | Study Completion Date |
|---|---|---|---|---|---|---|---|
| CETIDYL | Single-arm phase II | NCT 04189055 | Real-time PCR (IdyllaTM) | 72 | CET (Cohort1) or CET + IRI (Cohort2) | ORR | 7/1/2023 |
| KAIROS | Single-arm phase II | EudraCT 2019-001328-36 | Real-time PCR (Idylla TM) | 112 | CET + chemotherapy | ORR | N/A |
| MoLiMoR | Randomized phase II | NCT 04554836 | Droplet digital PCR | 144 | FOLFIRI + intermittent CET vs. FOLFIRI | PFS | 10/1/2024 |
| C-PROWESS [118] | Single-arm phase II | jRCTs 031210565 | BEAMing (the OncoBEAM TM RAS CRC KIT) | 30 | PANI + IRI | ORR | 1/1/2025 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udagawa, S.; Ooki, A.; Shinozaki, E.; Fukuda, K.; Yamaguchi, K.; Osumi, H. Circulating Tumor DNA: The Dawn of a New Era in the Optimization of Chemotherapeutic Strategies for Metastatic Colo-Rectal Cancer Focusing on RAS Mutation. Cancers 2023, 15, 1473. https://doi.org/10.3390/cancers15051473
Udagawa S, Ooki A, Shinozaki E, Fukuda K, Yamaguchi K, Osumi H. Circulating Tumor DNA: The Dawn of a New Era in the Optimization of Chemotherapeutic Strategies for Metastatic Colo-Rectal Cancer Focusing on RAS Mutation. Cancers. 2023; 15(5):1473. https://doi.org/10.3390/cancers15051473
Chicago/Turabian StyleUdagawa, Shohei, Akira Ooki, Eiji Shinozaki, Koshiro Fukuda, Kensei Yamaguchi, and Hiroki Osumi. 2023. "Circulating Tumor DNA: The Dawn of a New Era in the Optimization of Chemotherapeutic Strategies for Metastatic Colo-Rectal Cancer Focusing on RAS Mutation" Cancers 15, no. 5: 1473. https://doi.org/10.3390/cancers15051473
APA StyleUdagawa, S., Ooki, A., Shinozaki, E., Fukuda, K., Yamaguchi, K., & Osumi, H. (2023). Circulating Tumor DNA: The Dawn of a New Era in the Optimization of Chemotherapeutic Strategies for Metastatic Colo-Rectal Cancer Focusing on RAS Mutation. Cancers, 15(5), 1473. https://doi.org/10.3390/cancers15051473