Simple Summary
Recently, PRUNE2 mutations were indicated in the pathogenesis of aggressive parathyroid neoplasms. Here, we report novel, rare PRUNE2 mutations located in exons 3, 8, 9, and 12 in patients with parathyroid tumors in the genetically homogenous Finnish population. We identified PRUNE2 mutations in patients with parathyroid carcinoma, atypical parathyroid tumors, and adenomas. While further research is needed, mutations of the PRUNE2 gene could play a role in the pathogenesis of parathyroid tumors.
Abstract
Parathyroid tumors are mostly sporadic but can also occur in familial forms, including different kinds of genetic syndromes with varying phenotypes and penetrance. Recently, somatic mutations of the tumor suppressor gene PRUNE2 were found to be frequent in parathyroid cancer (PC). The germline mutation status of PRUNE2 was investigated in a large cohort of patients with parathyroid tumors from the genetically homogenous Finnish population, 15 of which had PC, 16 atypical parathyroid tumors (APT), and 6 benign parathyroid adenomas (PA). Mutations in previously established hyperparathyroidism-related genes were screened with a targeted gene panel analysis. Nine PRUNE2 germline mutations with a minor allele frequency (MAF) of <0.05 were found in our cohort. Five of these were predicted to be potentially damaging and were identified in two patients with PC, two with APT, and three with PA. The mutational status was not associated with the tumor group nor related to the clinical picture or severity of the disease. Still, the frequent finding of rare germline mutations of PRUNE2 may point to the gene playing a role in the pathogenesis of parathyroid neoplasms.
1. Introduction
Parathyroid carcinoma (PC) is a rare cause of primary hyperparathyroidism (PHPT). In Finland, around 2–3 new cases are diagnosed every year [1]. In contrast, PHPT due to benign adenomas is very common with an incidence of 1–2/1000 people [2,3]. The clinical picture of PC is similar to that of PHPT caused by benign parathyroid adenomas (PA). However, PC tumors are often larger than benign adenomas, and the clinical picture is usually more severe. PHPT due to benign adenomas is most common in postmenopausal women (gender ratio 3:1), while for PC, there is no gender difference, and incidence peaks at 40–50 years [4,5]. The diagnosis of PC is set based on unequivocal invasive characteristics on histopathological examination. Surgery is the only possible curative treatment. Of note, so called en bloc surgery, with the removal of the ipsilateral thyroid lobe and parathyroid gland gives the patient a better prognosis [6,7]. Atypical parathyroid tumors (APT), also called atypical parathyroid adenomas, are tumors which lack invasive histological characteristics but feature properties such as intratumoral fibrosis and nuclear atypia not regularly found in conventional parathyroid adenomas [8,9]. Around 5% of parathyroid adenomas (up to 15%, according to some studies) can be classified as APT [2,9,10].
The vast majority of PC cases are sporadic (80–90%) and associated with somatic mutations, although PC can also be hereditary [8]. Familial PHPT can occur as isolated PHPT or syndromes, presenting with both benign parathyroid adenomas as well as PC. Germline inactivation of the CDC73 gene, coding for the protein parafibromin, is associated with hyperparathyroidism jaw–tumor syndrome (HPT-JT). The parafibromin protein functions as a tumor suppressor, and HPT-JT presents with early-onset PHPT, ossifying jaw fibromas, as well as tumors of the kidney and uterus. However, CDC73 mutation carriers display incomplete penetrance, and germline CDC73 mutations can also cause isolated familial PHPT without the HPT-JT phenotype [4,11]. Up to 30% of PC patients carry inactivating germline CDC73 mutations [9]. Due to incomplete penetrance, these PC patients do not always present with a family history of PHPT. Somatic inactivation of CDC73 in tumor DNA is common in sporadic PC, occurring in up to 75% of cases [12,13].
Parathyroid neoplasms can also be associated with multiple endocrine neoplasia (MEN) syndromes MEN1, MEN2A, and MEN4, caused by mutations in the genes MEN-1, RET, and CDKN1B, respectively [8,14,15,16]. While loss-of-function mutations in calcium-signaling-related genes, such as CASR, GNA11, and AP2S1, are associated with familial hypocalciuric hypercalcemia (FHH) and neonatal severe hyperparathyroidism (NSHPT), these mutations are not associated with parathyroid neoplasms [17,18,19,20].
Non-syndromic hereditary hyperparathyroidism, called familial isolated hyperparathyroidism (FIHP), can be caused by the incomplete manifestation of mutations of syndrome-related genes such as MEN-1, CDC73, GCM2, CDKN1A, CDKN1B, or CDKN2C [17,21]. However, up to 70% of FIHP cases lack mutations in known susceptibility genes, indicating that there are many factors not yet discovered in the pathogenesis of parathyroid tumors [17,21]. Additional germline mutations reported in PC patients include genes PRUNE2, CCD1, ADCK1, and genes of the PI3K/AKT/mTOR pathway [22,23,24,25].
Prune homolog 2 with BCH domain (PRUNE2, also known as BMCC1) (9q21.2, ENSG00000106772) belongs to the B-cell CLL/lymphoma 2 and adenovirus E1B 19kDa interacting family. Members of this gene family are involved in several cellular processes, such as apoptosis, cell transformation, and synaptic function [26,27]. PRUNE2 is a tumor suppressor gene of particular significance shown in prostate cancer, where its expression is regulated by prostate cancer antigen 3 (PCA3), a non-protein coding gene located on the opposite DNA strand in an intron of PRUNE2 [28]. PCA3 has been investigated as a biomarker in prostate cancer [29,30]. Increased PCA3 levels have also been found in other cancers such as choriocarcinoma as well as ovarian and thyroid cancer, indicating involvement of PRUNE2 in the pathogenesis of these cancers [31]. Increased PRUNE2 protein levels are associated with a favorable prognosis in neuroblastoma and leiomyosarcoma and have also been associated with lower tumorigenic activity in colorectal cancer [26,27]. Somatic PRUNE2 mutations have been reported in up to 18% of sporadic PCs. These mutations, in combination with the loss of heterozygosity (LOH) events in the tumors, suggest PRUNE2 involvement in the pathogenesis of PC [24,25]. Moreover, Yu et al. identified a rare germline missense mutation in one PC patient without other known PC-driving mutations. The remaining wild-type allele was silenced in the patient’s tumor by LOH, a feature characteristic of tumor suppressor genes. However, the role of PRUNE2 in the genetic predisposition of PC is yet to be confirmed [25].
As somatic PRUNE2 mutations are considered significant in the pathogenesis of PC, we wanted to study whether germline PRUNE2 mutations might be identified in patients diagnosed with aggressive parathyroid tumors in the genetically homogenous Finnish population, an ideal population for the discovery of rare genetic defects of monogenic diseases [32].
2. Materials and Methods
Our study cohort consists of 37 patients with PHPT, 15 of which have been diagnosed with PC, 16 with APT, and 6 with PA. Three patients are known to have an ethnicity other than Finnish. All patients have been diagnosed and treated for PHPT during the years 2004–2021 at the Endocrine Department of Helsinki University Hospital. Clinical history, surgical and pathological reports as well as laboratory results at diagnosis were collected from the Helsinki University Central Hospital databases. Patients with previously known CDC73 mutations were reported earlier and are not included in this study [11].
PC patient 152164 is the mother of APT patient 152177; otherwise, the patients are not related to each other and do not have known family history of PHPT. All patients were summoned for laboratory tests. Serum ionized calcium (S-Ca-ion) as well as fasting plasma parathyroid hormone (fP-PTH), serum vitamin D (S-D-25-OH), and serum creatinine were also analyzed to assess possible recurrence of primary hyperparathyroidism, as some patients had not been participating in regular follow-ups. For some patients, due to problems with sample delivery, DNA was not extracted from whole blood but from saliva samples sent to the patients’ home addresses, using the Ora-Collect OCR-100 sample collection kit (Ottawa, Ont., Canada)).
All patients gave their written informed consent to study participation. The study protocol was approved by the Ethics Committee of the Helsinki University Hospital (Dnro 1803/2018).
EDTA whole-blood samples were sent to Blueprint Genetics (www.blueprintgenetics.com, accessed on 3 January 2023, Helsinki, Finland)) for DNA extraction and whole exome sequencing (WES). WES was performed using BpG according to in-house methods and sequence alignment to human reference genome GRCh37/hg19 with Illumina’s bcl2fastq2 Software v2.20 (Illumina, Inc. San Diego, CA, USA).
Mutations in previously known PHPT-related genes were analyzed and reported according to a customized extended BpG hyperparathyroidism gene panel (www.blueprintgenetics.com, accessed on 3 January 2023, Helsinki, Finland, including genes AIP, AIRE, AKAP9, AP2S1, APC, BRCA2, CASR, CDC73, CDKN1A, CDKN1B, CDKN2B, CDKN2C, CEP152, CTNNB1, DICER1, EZH2, FLNA, GCM2, GNA11, KDM5C, MEN1, MTOR, NF1, NTRK1, PIK3CA, PTEN, PTH, RB1, RET, SDHA, SETD1B, SMARCA4, STK11, TERT, TNRC6A, TP53, TRPV6, TSC1, TSC2, WT1, and ZEB1.
The gnomAD38 v.2.1.1 database (https://gnomad.broadinstitute.org/(accessed on 3 January 2023)) was used as a resource for population frequencies of the germline mutations. Previously known mutation consequences were assessed through the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/(accessed on 3 January 2023)) [33]. For assessing the pathogenicity of the variants PolyPhen-239 v.2.2.3 (http://genetics.bwh.harvard.edu/pph2/(accessed on 4 January 2023)) and SIFT v5.1.1 (http://sift-dna.org (accessed on 4 January 2023)) mutation consequence predictors were used. PhyloP conservation scores were gathered from the UCSC database (http://genome.ucsc.edu (PhyloP version 3.19, accessed on 4 January 2023)) [34]. Protein sequence and structural information about the PRUNE2 gene was gathered from the Ensembl database (v.108) [35].
Visualization of PRUNE2 single nucleotide variants and small insertions/deletions were performed with the in-house developed analysis and visualization program BasePlayer [36]. Minimum coverage for variant calling was set at four reads, and the mutated allele was required to be present in at least 20% of the reads. Output processing and analysis was performed using RStudio v.1.1.463 (RStudio, Inc., Boston, MA, USA) and IBM SPSS Statistics v.27 (SPSS, Inc., Chicago, IL, USA). p-values of <0.05 (two-tailed) were considered statistically significant. The χ2-test with Fisher’s exact test, as appropriate, was used to investigate differences in categorical variables between groups, while the Kruskal–Wallis test was used for continuous variables. For PRUNE2 mutations, those with a frequency of less than 5% in the global and/or Finnish population are reported. Genome position is reported according to PRUNE2 transcript ENST00000376718.
3. Results
3.1. Overview of the Main Study Results and Patient Characteristics
A flowchart depicting the study design and the main findings is shown in Figure 1.
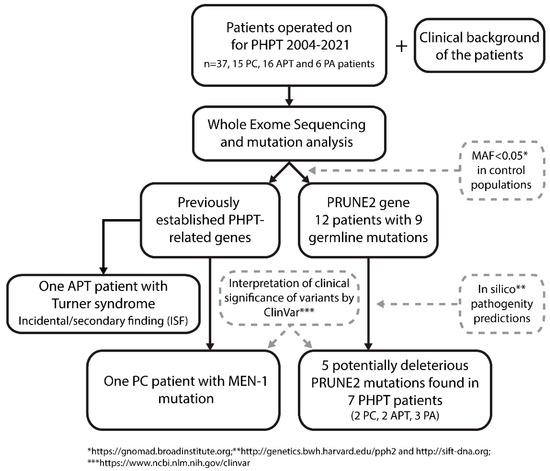
Figure 1.
Flowchart of PRUNE2 germline mutation screening in patients with parathyroid tumors. The whole exome sequencing (WES) was performed from normal DNA. After WES analysis, the variant calls were filtered to have MAF < 0.05. Mutation consequences were assessed using pathogenicity predictor software and the ClinVar database. Five potentially deleterious PRUNE2 mutations were identified, distributed among seven PHPT patients. PC = parathyroid cancer; APT = atypical parathyroid tumor; PA = parathyroid adenoma; MAF = minor allele frequency.
The patient characteristics are shown in Table 1.

Table 1.
Characteristics of the patient cohort.
3.2. Hyperparathyroidism Gene Panel Findings
Gene panel findings in the cohort are listed in Table 2.
Table 2.
Results of the customized Blueprint Genetics (BpG) hyperparathyroidism gene panel analysis.
The BpG custom hyperparathyroidism gene panel (www.blueprintgenetics.com, accessed on 3 January 2023, Helsinki, Finland did not reveal any additional germline CDC73 mutations in our cohort. One PC patient (152164) is the mother of one of the APT patients (152177). Otherwise, the patients are not related to each other and do not have any known cases of hyperparathyroidism in their respective families. Three PC patients are known to have an ethnicity other than Finnish. All patients were alive at follow-up. More than one surgery was performed on nine PC patients; five patients had local or distant PC recurrence. Additional en bloc surgery was performed as a preventative measure on four PC patients, as en bloc was not performed as the primary surgery. Three APT patients had more than one surgery; in all these cases, additional surgery was performed due to persistent hypercalcemia after the initial surgery, or the patient presented with additional parathyroid pathology (adenoma or hyperplasia). None of these patients were carriers of any potentially damaging germline mutations found in this study.
One PC patient (patient ID 152172) was found to have a likely pathogenic heterozygous germline MEN-1 mutation (c.1280G > T, p.Ser427Ile, ENST00000312049.6), although displayed no other clinical features of the MEN1 syndrome (Figure 1, Table 2) [37,38]. One APT patient had a family history of MEN1, but no pathogenic MEN-1 variants were detected in this patient. The custom WES panel revealed monosomy X (Turner syndrome) in one APT patient (patient ID 152142) as an incidental or secondary finding (ISF) with clinical significance (Figure 1). Moreover, a heterozygous germline APC missense mutation (c.2222A > G, p.Asn741Ser, rs150209825) was found in PA patient 152129. The variant is considered of unknown significance (VUS) [39,40,41]. This patient does not have any history of other neoplasia, and the possible family history of cancer is unknown. A 5′UTR variant of the APC gene (c.-128G > A, rs543098847) was found in PC patient 152174. This multiallelic single nucleotide variant is considered likely benign VUS (https://www.ncbi.nlm.nih.gov/clinvar (accessed on 3 January 2023)). A RET mutation (c.604G > A, p.Val202Met, rs751572082) of unknown significance was identified in PC patient 152165. An amino acid deletion of BRCA2 with unknown significance (c.3900_3902del, p.Met1300_Thr1301delinsIle, rs397507697) was found in APT patient 152175, and APT patient 152146 had a VUS missense AIP mutation (c.940C > T, p.Arg314Trp, rs375740557). In PA patient 152125, a multiallelic 5′UTR SDHA variant (c.-11C > T, rs1396057630) was identified.
3.3. PRUNE2 Mutations
Altogether, 25 non-synonymous PRUNE2 germline variants were found in the patient cohort. Out of those, nine variants had a minor allele frequency (MAF) <0.05 in a control population (https://gnomad.broadinstitute.org (accessed on 21 December 2022)) (Figure 1). The details on these nine PRUNE2 variants are listed in Table 3.
Table 3.
Identified rare PRUNE2 germline mutations (MAF < 0.05) according to underlying tumor in 37 patients with primary hyperparathyroidism.
The nine variants were distributed among twelve patients, of which three had PC, six APT and three PA. None of the patients with rare PRUNE2 variants had mutations of previously established PHPT-related genes (Table 2). All discovered mutations were heterozygous missense changes. Altogether, seven patients harbored a PRUNE2 variant in silico predicted to be likely deleterious (Figure 1, Table 3). Interestingly, the germline variant p.Ser595Tyr found in APT patient 152142 was previously also identified in prostate cancer patients [42]. Otherwise, the rare PRUNE2 germline variants found in our cohort have not been associated with any kind of pathology (https://www.ncbi.nlm.nih.gov/clinvar/(accessed on 3 January 2023)). APT patient 152142 with Turner syndrome, who was 59 years old at the time of her diagnosis, had three different germline PRUNE2 variants; however, only (c.1784G > T, p.Ser595Tyr) was predicted to be damaging (Table 3). More than one PRUNE2 mutation was also found in patients 152127 (PA, 38 years old at diagnosis) and 152171 (PC, 66 years at diagnosis). Similarly, in these cases, only one mutation per patient was predicted to be pathogenic. In addition, patients 152155 (PC, 49 years at diagnosis), 152121 (PA, 44 years at diagnosis), 152123 (PA, 54 years at diagnosis), and 152131 (APT, 63 years at diagnosis) each harbored one PRUNE2 germline mutation that was predicted to be damaging (Table 3). The location of the PRUNE2 mutations found in our cohort in relation to the PRUNE2 gene and previously found PRUNE2 mutations in PC (somatic and germline) are visualized in Figure 2.
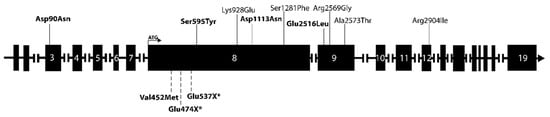
Figure 2.
Schematic depiction of the PRUNE2 gene and its exons. PRUNE2 variants found in our cohort are annotated above the gene, and previously identified PRUNE2 mutations (somatic and germline) in PC patients are annotated below the gene. The respective sizes of the exons are not to scale. Somatic mutations are marked with an asterisk (*). Mutations predicted to be damaging are marked in bold.
Most mutations were located in exon 8, the longest exon of the gene, but mutations were also observed in exons 3, 9, and 12. The cohort was screened for gene variants of ADCK1, CCD1, FAT3, and THRAP3 that have previously been associated with PC as described in the literature but that were not included in the BpG hyperparathyroidism gene panel assessment [4,16,25,27]. No previously described variants of these genes were found in our cohort.
PRUNE2 mutation status did not correlate with clinical parameters such as severity of disease (Ca-ion or PTH levels at diagnosis), tumor size, or parafibromin staining on immunohistochemistry, neither by looking at all rare prune mutations in our cohort nor by separately analyzing the variants considered damaging. Neither were the PRUNE2 mutations associated with a lower age at diagnosis.
4. Discussion
The majority of PC cases are associated with somatic alterations, although hereditary predisposition also plays a role in the genesis of this rare malignancy [14,43]. Somatic recurrent PRUNE2 mutations have been reported in up to 18% of PC cases. A reported germline PRUNE2 mutation and inactivation of the wild-type allele by LOH in the patient’s tumor indicates that PRUNE2 alterations might predispose to PC [24,25]. To clarify the contribution of inherited PRUNE2 mutations in the development of parathyroid tumors, the germline mutation status of PRUNE2 was analyzed in a cohort of 37 mostly Finnish PHPT patients. We identified nine rare PRUNE2 germline variants (MAF < 0.05) in twelve patients, of which five mutations were predicted to be deleterious by disrupting the function of the PRUNE2 protein. These five mutations were distributed among seven patients, including individuals from all tumor groups (PC, APT, and PA). Interestingly, one of the mutations (c.270C > T, p.Asp90ASn) was shared between three unrelated patients: two with PC and one with PA. The MAF of the variant was 0.0148 among the Finnish controls and 0.0092 in the global control population (Table 3). This recurrent mutation might be a Finnish founder mutation, and enrichment of the mutant allele in our patient cohort may imply a causal relationship between the c.270C > T (p.Asp90ASn) mutation and the disease phenotype. The homogenous Finnish population has a unique genetic background, and founder mutations exist at high frequencies [44]. As such, Finnish population-based cohorts are valuable for the discovery of the causative genetic mutations of monogenic diseases.
Most of the rare PRUNE2 variants found in our cohort are localized to PRUNE2 exon 8, which is, by far, the largest exon of the gene, harboring approximately half of all the amino acids in its sequence. The mutations found in our study do not seem to be targeting any specific regions or previously established domains of PRUNE2 [45].
According to the literature, germline mutations of the PRUNE2 gene have been very scarcely investigated in other forms of cancers, despite somatic mutations playing a role in the pathogenesis of prostate cancer, leiomyosarcoma, and colorectal cancer among others [26,27,29,30,31]. Of note, a recent study found PRUNE2 germline mutations in 2.8% of patients with familial prostate cancer, proposing PRUNE2 as a new prostate cancer predisposition gene [42]. The PRUNE2 germline variant c. 1784G > T (p.Ser595Tyr) found in our APT patient was also identified in the familial prostate cancer study. This mutant allele was classified as a variant of unclear association with the prostate cancer risk [42].
The patients with rare PRUNE2 germline mutations did not carry any germline mutations in previously established PHPT-driving genes. One patient was found to have monosomy X/Turner syndrome. This is relevant in the setting of parathyroid tumors due to the X chromosome harboring loci for the FLNA and KDM5C genes. The FLNA protein participates in the regulation of the calcium-sensing receptor and has been associated with increased aggressiveness in a wide range of cancers [46,47,48]. FLNA expression has also been investigated in parathyroid tumors, with unclear conclusions [49,50]. Inactivation of the KDM5C gene encoding for the histone demethylase protein JARID1C is frequent in renal cell carcinoma, and somatic KDM5C mutations have also been found in PC [25,51]. The monosomy X patient is thus susceptible to somatic mutations in these genes. One patient (152172) was discovered to have a likely pathogenic mutation of the MEN-1 gene. Other mutations of the same codon (c.1281T > A, p.Ser427Arg, rs1114167528) were previously reported in several MEN1 patients [37,38]. The family history of the MEN-1 mutation-positive patient identified in this study is not known. The patient has no other medical history of cancer except for PC, and the age at diagnosis (67 years) was not conspicuous. Still, due to the likely pathogenicity of this gene mutation, further clinical follow-up might be needed. Similarly, despite the germline APC mutations discovered in our cohort not being previously reported to be associated with pathogenicity, these patients and their relatives might also require further investigations, as APC mutations are so pronouncedly associated with hereditary colorectal malignancy [41,52].
Patients with previously known CDC73 mutations were excluded from this study, but as germline CDC73 mutations are also quite common in patients with sporadic PC it is perhaps rather surprising that none of the patients in the present cohort were found to carry CDC73 mutations. As CDC73 mutations may underlie both benign and malignant parathyroid tumors with varying penetrance and phenotypes, one can, therefore, speculate that mutations of the PRUNE2 gene could also give rise to similarly varying parathyroid tumor phenotypes with similarly varying penetrance [6,53,54]. The incomplete penetrance manifested by PRUNE2 germline defects might be the reason for the observed lack of family history of the disease.
The shortcoming of the study was the lack of tumor material; hence, the investigation of the biallelic inactivation of PRUNE2 in tumors was not achievable. Our patient cohort is of a reasonable size considering the rarity of PC. Another strength of our material is the detailed clinical, surgical, and histopathological characterization of the patients. However, the patient number is too small for the relevant assessment of the possible relationships between gene variants and clinical or biochemical parameters. All our patients were alive at the time of the study, with a median follow-up time of 7 years. Globally, the 5-year survival of PC is considered around 85%, indicating that our patients have had a rather favorable course of disease [1,55,56]. En bloc surgery was performed in 13 of our 15 PC patients (86%), either as primary or secondary surgery. The excellent prognosis of the patients likely reflects the high awareness of PC as a cause of PHPT in our tertiary centre and the close collaboration with our endocrine surgeons, preventing diagnostic delay, as well as en bloc surgery, ensuring margin-free resection. Recently, margin-free resection was reported to predict excellent long-term outcomes in PC [7].
In this study, we report that Finnish PHPT patients with CDC73 mutation-negative parathyroid tumors frequently display rare germline mutations in the PRUNE2 tumor suppressor gene. However, further work is needed to examine whether PRUNE2 plays a causative role in the genetic predisposition of parathyroid neoplasia. Clarification of this question would require additional sample sets and more extensive molecular workup. The identification of new parathyroid tumor-predisposing genes is important to improve the risk assessment of patients, and it would enable targeted testing of family members at risk.
5. Conclusions
Rare germline PRUNE2 variants are frequent in Finnish patients with parathyroid neoplasms, regardless of tumor type (PC, APT, or PA). Further studies are needed to clarify the role of PRUNE2 in patients with parathyroid tumors.
Author Contributions
Conceptualization, S.S., A.K. and C.S.-J.; Methodology, S.S., E.R., A.K. and C.S.-J.; Formal analysis, S.S., A.K. and C.S-J.; Investigation, S.S., E.R. and C.S.-J.; Resources, C.S.-J.; Writing—original draft, S.S.; Writing—review & editing, S.S., E.R., A.K. and C.S.-J.; Supervision, A.K. and C.S.-J.; Project administration, C.S.-J.; Funding acquisition, C.S.-J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Helsinki University Hospital Research Funds (TYH2019254, to C.S.-J.) and Finska Läkaresällskapet (to C.S.-J.).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of the Helsinki University Hospital (Dnro 1803/2018).
Informed Consent Statement
Written informed consent was obtained from all study subjects.
Data Availability Statement
The data is available on request from the corresponding author.
Acknowledgments
We wish to thank Blueprint Genetics (BpG) for DNA extractions, WeS sequencing, and gene panel analysis. We also want to thank the study nurse Hanna Talosela for her excellent assistance in patient-related matters, including paperwork and practicalities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ryhänen, E.M.; Leijon, H.; Metso, S.; Eloranta, E.; Korsoff, P.; Ahtiainen, P.; Kekäläinen, P.; Tamminen, M.; Ristamäki, R.; Knutar, O.; et al. A nationwide study on parathyroid carcinoma. Acta Oncol. 2017, 56, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.E.; Healy, J.; Lebastchi, A.H.; Brown, T.C.; Stein, J.E.; Prasad, M.L.; Callender, G.G.; Carling, T.; Udelsman, R. Modern experience with aggressive parathyroid tumors in a high-volume New England referral center. J. Am. Coll. Surg. 2015, 220, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.D. Epidemiology of parathyroid disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Cetani, F.; Pardi, E.; Marcocci, C. Parathyroid Carcinoma. Front. Horm. Res. 2018, 51, 63–76. [Google Scholar] [CrossRef]
- Marcocci, C.; Cetani, F.; Rubin, M.R.; Silverberg, S.J.; Pinchera, A.; Bilezikian, J.P. Parathyroid carcinoma. J. Bone Miner. Res. 2008, 23, 1869–1880. [Google Scholar] [CrossRef]
- Duan, K.; Hernandez, K.G.; Mete, O. Clinicopathological correlates of hyperparathyroidism. J. Clin. Pathol. 2015, 68, 771–787. [Google Scholar] [CrossRef]
- Schulte, K.; Talat, N.; Galat, G. Margin Free Resection Achieves Excellent Long Term Outcomes in Parathyroid Cancer. Cancers 2023, 15, 199. [Google Scholar] [CrossRef]
- Duan, K.; Mete, Ö. Parathyroid carcinoma: Diagnosis and clinical implications. Turk. J. Pathol. 2015, 31, 80–97. [Google Scholar] [CrossRef]
- Erickson, L.A.; Mete, O.; Juhlin, C.C.; Perren, A.; Gill, A.J. Overview of the 2022 WHO Classification of Parathyroid Tumors. Endocr. Pathol. 2022, 33, 64–89. [Google Scholar] [CrossRef]
- Cetani, F.; Marcocci, C.; Torregrossa, L.; Pardi, E. Atypical parathyroid adenomas: Challenging lesions in the differential diagnosis of endocrine tumors. Endocr. Relat. Cancer 2019, 26, R441–R464. [Google Scholar] [CrossRef]
- Korpi-Hyövälti, E.; Cranston, T.; Ryhänen, E.; Arola, J.; Aittomäki, K.; Sane, T.; Thakker, R.V.; Schalin-Jäntti, C. CDC73 intragenic deletion in familial primary hyperparathyroidism associated with parathyroid carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, 3044–3048. [Google Scholar] [CrossRef]
- Cetani, F.; Banti, C.; Pardi, E.; Borsari, S.; Viacava, P.; Miccoli, P.; Torregrossa, L.; Basolo, F.; Pelizzo, M.R.; Rugge, M.; et al. CDC73 mutational status and loss of parafibromin in the outcome of parathyroid cancer. Endocr. Connect. 2013, 2, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Segiet, O.A.; Deska, M.; Michalski, M.; Gawrychowski, J.; Wojnicz, R. Molecular profiling in primary hyperparathyroidism. Head Neck 2015, 37, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Juhlin, C.C.; Erickson, L.A. Genomics and Epigenomics in Parathyroid Neoplasia: From Bench to Surgical Pathology Practice. Endocr. Pathol. 2021, 32, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Minnetti, M.; Grossman, A. Somatic and germline mutations in NETs: Implications for their diagnosis and management. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 115–127. [Google Scholar] [CrossRef]
- Clarke, C.N.; Katsonis, P.; Hsu, T.-K.; Koire, A.M.; Silva-Figueroa, A.; Christakis, I.; Williams, M.D.; Kutahyalioglu, M.; Kwatampora, L.; Xi, Y.; et al. Comprehensive Genomic Characterization of Parathyroid Cancer Identifies Novel Candidate Driver Mutations and Core Pathways. J. Endocr. Soc. 2019, 3, 544–559. [Google Scholar] [CrossRef]
- Sharretts, J.M.; Simonds, W.F. Clinical and Molecular Genetics of Parathyroid Neoplasms John. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 24, 491–502. [Google Scholar] [CrossRef]
- Brewer, K.; Costa-Guda, J.; Arnold, A. Molecular genetic insights into sporadic primary hyperparathyroidism. Endocr. Relat. Cancer 2019, 26, R53–R72. [Google Scholar] [CrossRef]
- Cetani, F.; Pinchera, A.; Pardi, E.; Cianferotti, L.; Vignali, E.; Picone, A.; Miccoli, P.; Viacava, P.; Marcocci, C. No evidence for mutations in the calcium-sensing receptor gene in sporadic parathyroid adenomas. J. Bone Miner. Res. 1999, 14, 878–882. [Google Scholar] [CrossRef]
- Witteveen, J.E.; Hamdy, N.A.T.; Dekkers, O.M.; Kievit, J.; Van Wezel, T.; Teh, B.T.; Romijn, J.A.; Morreau, H. Downregulation of CASR expression and global loss of parafibromin staining are strong negative determinants of prognosis in parathyroid carcinoma. Mod. Pathol. 2011, 24, 688–697. [Google Scholar] [CrossRef]
- Cardoso, L.; Stevenson, M.; Thakker, R.V. Molecular genetics of syndromic and non-syndromic forms of parathyroid carcinoma. Hum. Mutat. 2017, 38, 1621–1648. [Google Scholar] [CrossRef] [PubMed]
- Bollerslev, J.; Schalin-Jäntti, C.; Rejnmark, L.; Siggelkow, H.; Morreau, H.; Thakker, R.; Sitges-Serra, A.; Cetani, F.; Marcocci, C.; Guistina, A.; et al. Unmet therapeutic, educational and scientific needs in parathyroid disorders: Consensus statement from the first European Society of Endocrinology Workshop (PARAT). Eur. J. Endocrinol. 2019, 181, P1–P19. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, X.; Wang, O.; Bi, Y.; Xing, X.; Cui, M.; Wang, M.; Tao, W.; Liao, Q.; Zhao, Y. The genomic profile of parathyroid carcinoma based on whole-genome sequencing. Int. J. Cancer 2020, 147, 2446–2457. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; McPherson, J.R.; Stevenson, M.; Van Eijk, R.; Heng, H.L.; Newey, P.; Gan, A.; Ruano, D.; Huang, D.; Poon, S.L.; et al. Whole-exome sequencing studies of parathyroid carcinomas reveal novel PRUNE2 mutations, distinctive mutational spectra related to APOBEC-catalyzed DNA mutagenesis and mutational enrichment in kinases associated with cell migration and invasion. J. Clin. Endocrinol. Metab. 2015, 100, E360–E364. [Google Scholar] [CrossRef]
- Pandya, C.; Uzilov, A.V.; Bellizzi, J.; Lau, C.Y.; Moe, A.S.; Strahl, M.; Hamou, W.; Newman, L.C.; Fink, M.Y.; Antipin, Y.; et al. Genomic profiling reveals mutational landscape in parathyroid carcinomas. JCI Insight 2017, 2, e92061. [Google Scholar] [CrossRef]
- Machida, T.; Fujita, T.; Ooo, M.L.; Ohira, M.; Isogai, E.; Mihara, M.; Hirato, J.; Tomotsune, D.; Hirata, T.; Fujimori, M.; et al. Increased expression of proapoptotic BMCC1, a novel gene with the BNIP2 and Cdc42GAP homology (BCH) domain, is associated with favorable prognosis in human neuroblastomas. Oncogene 2006, 25, 1931–1942. [Google Scholar] [CrossRef]
- Li, T.; Huang, S.; Yan, W.; Zhang, Y.; Guo, Q. PRUNE2 inhibits progression of colorectal cancer in vitro and in vivo. Exp. Ther. Med. 2021, 23, 169. [Google Scholar] [CrossRef]
- Salameh, A.; Lee, A.K.; Cardó-Vila, M.; Nunes, D.N.; Efstathiou, E.; Staquicini, F.I.; Dobroff, A.S.; Marchiò, S.; Navone, N.M.; Hosoya, H.; et al. PRUNE2 is a human prostate cancer suppressor regulated by the intronic long noncoding RNA PCA3. Proc. Natl. Acad. Sci. USA 2015, 112, 8403–8408. [Google Scholar] [CrossRef]
- Duffy, M.J. Biomarkers for prostate cancer: Prostate-specific antigen and beyond. Clin. Chem. Lab. Med. 2020, 58, 326–339. [Google Scholar] [CrossRef]
- Filella, X.; Foj, L. Novel biomarkers for prostate cancer detection and prognosis. Adv. Exp. Med. Biol. 2018, 1095, 15–39. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Baniahmad, A.; Taheri, M.; Rashnoo, F. A review on the role of PCA3 lncRNA in carcinogenesis with an especial focus on prostate cancer. Pathol. Res. Pract. 2022, 231, 153800. [Google Scholar] [CrossRef] [PubMed]
- Kääriäinen, H.; Muilu, J.; Perola, M.; Kristiansson, K. Genetics in an isolated population like Finland: A different basis for genomic medicine? J. Community Genet. 2017, 8, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Katainen, R.; Donner, I.; Cajuso, T.; Kaasinen, E.; Palin, K.; Mäkinen, V.; Aaltonen, L.A.; Pitkänen, E. Discovery of potential causative mutations in human coding and noncoding genome with the interactive software BasePlayer. Nat. Protoc. 2018, 13, 2580–2600. [Google Scholar] [CrossRef] [PubMed]
- Kouvaraki, M.A.; Lee, J.E.; Shapiro, S.E.; Gagel, R.F.; Sherman, S.I.; Sellin, R.V.; Cote, G.J.; Evans, D.B. Genotype-phenotype analysis in multiple endocrine neoplasia type 1. Arch. Surg. 2002, 137, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, L.; Pickel, J.; Zinner, K.; Hering, U.; Höfler, M.; Goretzki, P.E.; Spelsberg, F.; Raue, F.; Von Zur Mühlen, A.; Gerl, H.; et al. Developing effective screening strategies in multiple endocrine neoplasia type 1 (MEN 1) on the basis of clinical and sequencing data of German patients with MEN 1. Exp. Clin. Endocrinol. Diabetes 2007, 115, 509–517. [Google Scholar] [CrossRef] [PubMed]
- DeRycke, M.S.; Gunawardena, S.; Balcom, J.R.; Pickart, A.M.; Waltman, L.A.; French, A.J.; McDonnell, S.; Riska, S.M.; Fogarty, Z.C.; Larson, M.C.; et al. Targeted sequencing of 36 known or putative colorectal cancer susceptibility genes. Mol. Genet. Genom. Med. 2017, 5, 553–569. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J. Genes in Patients with Suspected Lynch Syndrome. Gastroenterology 2015, 149, 604–613.e20. [Google Scholar] [CrossRef]
- Chubb, D.; Broderick, P.; Frampton, M.; Kinnersley, B.; Sherborne, A.; Penegar, S.; Lloyd, A.; Ma, Y.P.; Dobbins, S.E.; Houlston, R.S. Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. J. Clin. Oncol. 2015, 33, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.; Maia, S.; Brandão, A.; Sahasrabudhe, R.; Lott, P.; Belter, N.; Carvajal-Carmona, L.G.; Paulo, P.; Teixeira, M.R. Exome sequencing of affected duos and trios uncovers PRUNE2 as a novel prostate cancer predisposition gene. Br. J. Cancer, 2022; online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Marini, F.; Giusti, F.; Palmini, G.; Perigli, G.; Santoro, R.; Brandi, M.L. Genetics and Epigenetics of Parathyroid Carcinoma. Front. Endocrinol. 2022, 13, 834362. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Karczewski, K.J.; Kerminen, S.; Kurki, M.I.; Sarin, A.P.; Artomov, M.; Eriksson, J.G.; Esko, T.; Genovese, G.; Havulinna, A.S.; et al. Haplotype Sharing Provides Insights into Fine-Scale Population History and Disease in Finland. Am. J. Hum. Genet. 2018, 102, 760–775. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Vitali, E.; Boemi, I.; Rosso, L.; Cambiaghi, V.; Novellis, P.; Mantovani, G.; Spada, A.; Alloisio, M.; Veronesi, G.; Ferrero, S.; et al. FLNA is implicated in pulmonary neuroendocrine tumors aggressiveness and progression. Oncotarget 2017, 8, 77330–77340. [Google Scholar] [CrossRef]
- Hjälm, G.; MacLeod, R.J.; Kifor, O.; Chattopadhyay, N.; Brown, E.M. Filamin-A Binds to the Carboxyl-terminal Tail of the Calcium-sensing Receptor, an Interaction that Participates in CaR-mediated Activation of Mitogen-activated Protein Kinase. J. Biol. Chem. 2001, 276, 34880–34887. [Google Scholar] [CrossRef]
- Awata, H.; Huang, C.; Handlogten, M.E.; Miller, R.T. Interaction of the Calcium-sensing Receptor and Filamin, a Potential Scaffolding Protein. J. Biol. Chem. 2001, 276, 34871–34879. [Google Scholar] [CrossRef]
- Mingione, A.; Verdelli, C.; Ferrero, S.; Vaira, V.; Guarnieri, V.; Scillitani, A.; Vicentini, L.; Balza, G.; Beretta, E.; Terranegra, A.; et al. Filamin A is reduced and contributes to the CASR sensitivity in human parathyroid tumors. J. Mol. Endocrinol. 2017, 58, 91–103. [Google Scholar] [CrossRef]
- Storvall, S.; Leijon, H.; Ryhänen, E.M.; Vesterinen, T.; Heiskanen, I.; Schalin-Jäntti, C.; Arola, J. Filamin A and parafibromin expression in parathyroid carcinoma. Eur. J. Endocrinol. 2021, 185, 803–812. [Google Scholar] [CrossRef]
- Rosano, D.; Di Croce, L.; Futreal, P.A.; The, B.T.; Tonon, G.; Segalla, S.; Frenquelli, M.; Antonini, E.; Huang, D.; Lazarevic, D.; et al. Histone demethylase JARID1C inactivation triggers genomic instability in sporadic renal cancer. J. Clin. Investig. 2016, 126, 4387. [Google Scholar] [CrossRef]
- Fang, X.; Svitkina, T.M. Adenomatous Polyposis Coli (APC) in cell migration. Eur. J. Cell Biol. 2022, 101, 151228. [Google Scholar] [CrossRef] [PubMed]
- Perrier, N.D.; Arnold, A.; Costa-guda, J.; Busaidy, N.L.; Nguyen, H.; Chuang, H.H. Thematic review, Hereditary endocrine tumours: Current state-of-the-art New and future perspectives for parathyroid carcinoma. Endocr. Relat. Cancer 2020, 27, T53–T63. [Google Scholar] [CrossRef] [PubMed]
- Walls, G.V.; Stevenson, M.; Lines, K.E.; Newey, P.J.; Reed, A.A.C.; Bowl, M.R.; Jeyabalan, J.; Harding, B.; Bradley, K.J.; Manek, S.; et al. Mice deleted for cell division cycle 73 gene develop parathyroid and uterine tumours: Model for the hyperparathyroidism-jaw tumour syndrome. Oncogene 2017, 36, 4025–4036. [Google Scholar] [CrossRef] [PubMed]
- Harari, A.; Waring, A.; Fernandez-Ranvier, G.; Hwang, J.; Suh, I.; Mitmaker, E.; Shen, W.; Gosnell, J.; Duh, Q.Y.; Clark, O. Parathyroid carcinoma: A 43-year outcome and survival analysis. J. Clin. Endocrinol. Metab. 2011, 96, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Villar Del Moral, J.; Jiménez-García, A.; Salvador-Egea, P.; Martos-Martínez, J.M.; Nuño-Vázquez-Garza, J.M.; Serradilla-Martín, M.; Gómez-Palacios, A.; Moreno-Llorente, P.; Ortega-Serrano, J.; De La Quintana-Basarrate, A. Prognostic factors and staging systems in parathyroid cancer: A multicenter cohort study. Surgery 2014, 156, 1132–1144. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).