

Unlocking the Resistance to Anti-HER2 Treatments in Breast Cancer: The Issue of HER2 Spatial Distribution

 , ,
, ,  , ,
, ,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Overview of the Current Management of HER2-Positive Breast Cancer
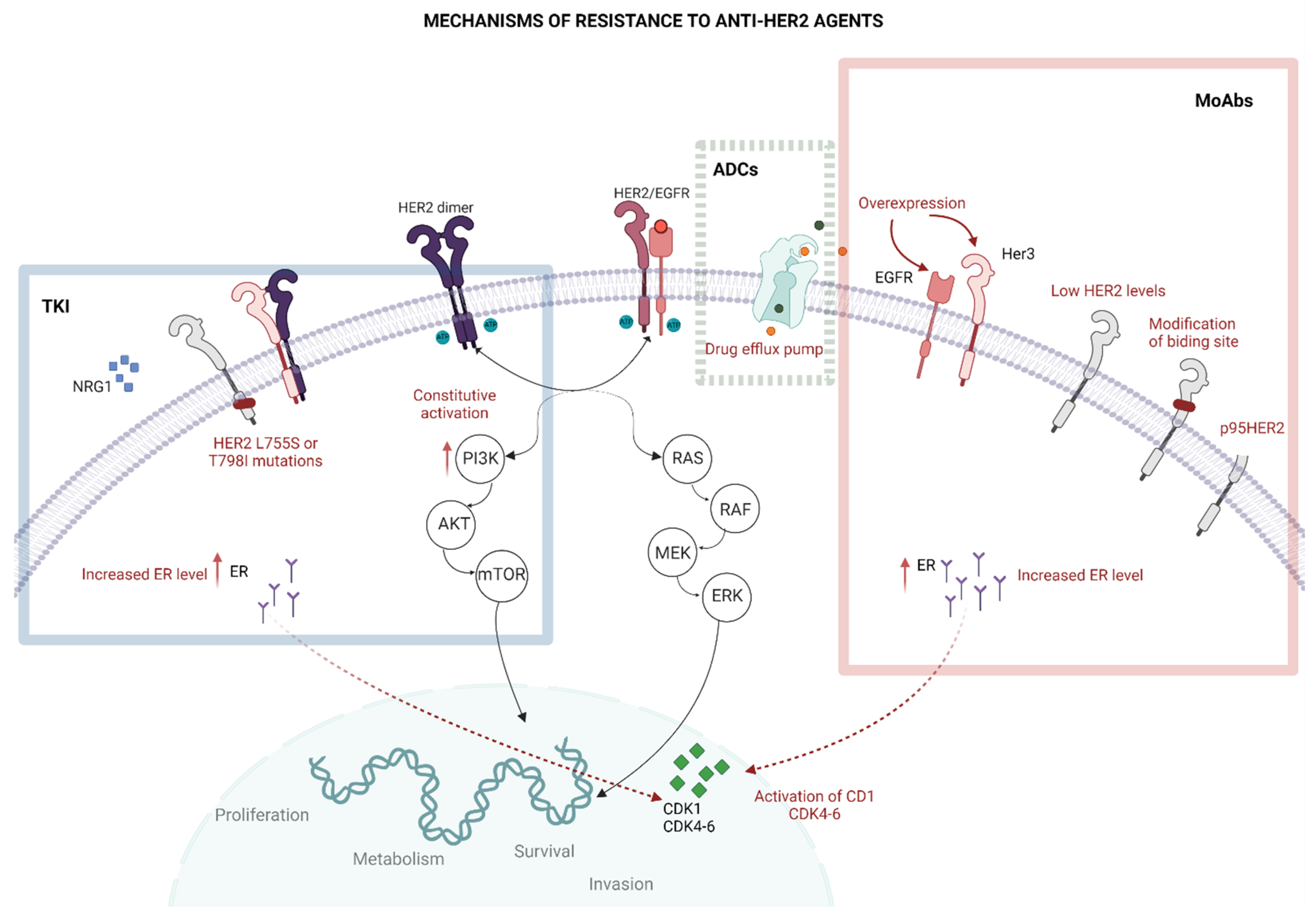
3. The Resistance to Anti-HER2 Treatments
3.1. Impaired Binding to HER2
3.2. HER2 Mutations
3.3. Altered Intracellular Signalling
3.4. Other Mechanisms of Resistance
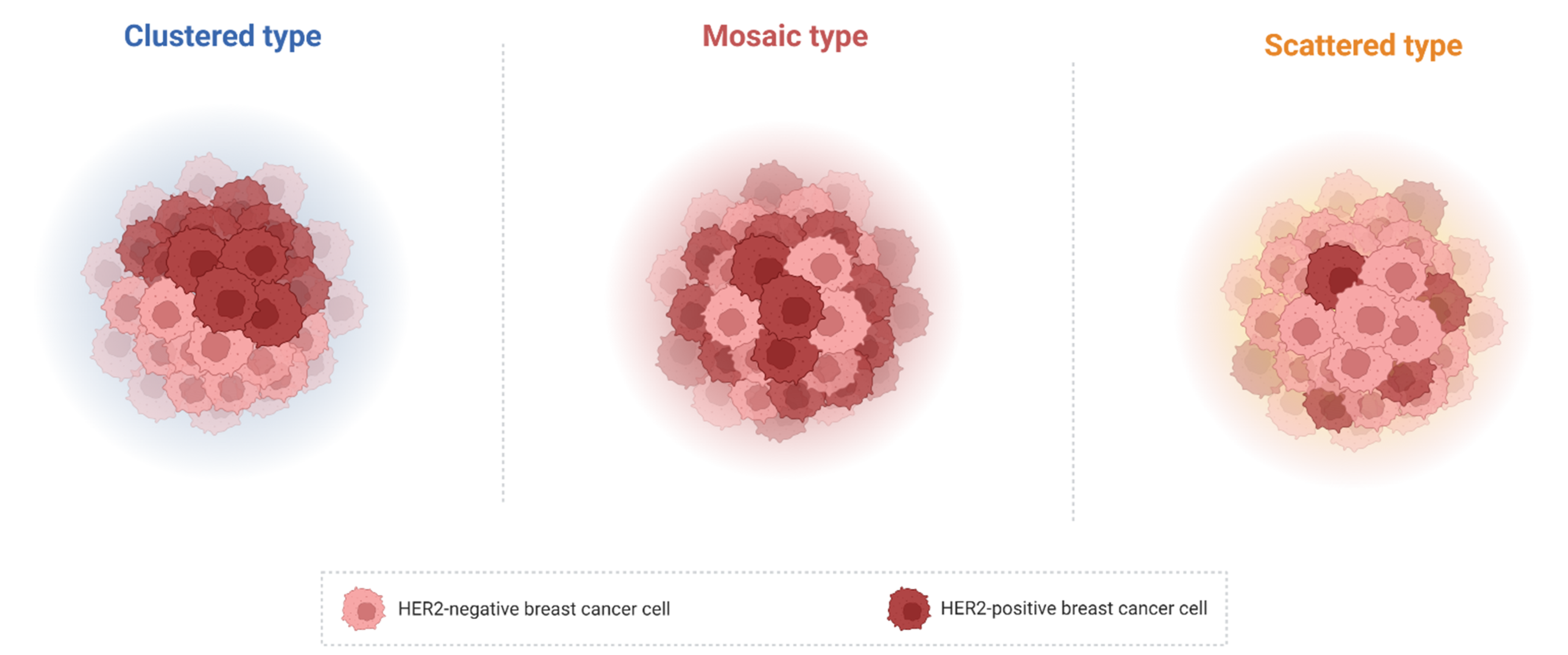
4. HER Heterogeneity, HER2 Spatial Distribution and Anti-HER2 Therapy Resistance
5. HER2 Heterogeneity and Response to Different Anti-HER2 Treatments
6. Further Directions
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schalper, K.A.; Kumar, S.; Hui, P.; Rimm, D.L.; Gershkovich, P. A retrospective population-based comparison of HER2 immunohistochemistry and fluorescence in situ hybridization in breast carcinomas: Impact of 2007 American Society of Clinical Oncology/College of American Pathologists criteria. Arch. Pathol. Lab. Med. 2014, 138, 213–219. [Google Scholar] [CrossRef]
- Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [CrossRef]
- Gennari, A.; André, F.; Barrios, C.H.; Cortés, J.; de Azambuja, E.; DeMichele, A.; Dent, R.; Fenlon, D.; Gligorov, J.; Hurvitz, S.A.; et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. 2021, 32, 1475–1495. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.S.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W.; et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. Arch. Pathol. Lab. Med. 2018, 142, 1364–1382. [Google Scholar] [CrossRef]
- Marchiò, C.; Annaratone, L.; Marques, A.; Casorzo, L.; Berrino, E.; Sapino, A. Evolving concepts in HER2 evaluation in breast cancer: Heterogeneity, HER2-low carcinomas and beyond. Semin. Cancer Biol. 2020, 72, 123–135. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Hayes, D.F. HER2 and Breast Cancer-A Phenomenal Success Story. N. Engl. J. Med. 2019, 381, 1284–1286. [Google Scholar] [CrossRef]
- Society, A.C. Breast Cancer Survival Rates by Stage. Available online: http://www.cancer.org/cancer/breastcancer/detailedguide/breast-cancer-survival-by-stage (accessed on 12 January 2023).
- Mendes, D.; Alves, C.; Afonso, N.; Cardoso, F.; Passos-Coelho, J.L.; Costa, L.; Andrade, S.; Batel-Marques, F. The benefit of HER2-targeted therapies on overall survival of patients with metastatic HER2-positive breast cancer–a systematic review. Breast Cancer Res. 2015, 17, 140. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E.; ESMO Guidelines Committee. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef]
- Schlam, I.; Tarantino, P.; Tolaney, S.M. Overcoming Resistance to HER2-Directed Therapies in Breast Cancer. Cancers 2022, 14, 3996. [Google Scholar] [CrossRef]
- De Melo Gagliato, D.; Leonardo Fontes Jardim, D.; Marchesi, M.S.P.; Hortobagyi, G.N. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget 2016, 7, 64431–64446. [Google Scholar] [CrossRef] [PubMed]
- Pernas, S.; Tolaney, S.M. Targeting HER2 heterogeneity in early-stage breast cancer. Curr. Opin. Oncol. 2020, 32, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Filho, O.M.; Viale, G.; Stein, S.; Trippa, L.; Yardley, D.A.; Mayer, I.A.; Abramson, V.G.; Arteaga, C.L.; Spring, L.M.; Waks, A.G.; et al. Impact of HER2 Heterogeneity on Treatment Response of Early-Stage HER2-Positive Breast Cancer: Phase II Neoadjuvant Clinical Trial of T-DM1 Combined with Pertuzumab. Cancer Discov. 2021, 11, 2474–2487. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Rye, I.H.; Trinh, A.; Sætersdal, A.B.; Nebdal, D.; Lingjærde, O.C.; Almendro, V.; Polyak, K.; Børresen-Dale, A.L.; Helland, Å.; Markowetz, F.; et al. Intratumor heterogeneity defines treatment-resistant HER2+ breast tumors. Mol. Oncol. 2018, 12, 1838–1855. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Barry, W.T.; Dang, C.T.; Yardley, D.A.; Moy, B.; Marcom, P.K.; Albain, K.S.; Rugo, H.S.; Ellis, M.; Shapira, I.; et al. Adjuvant Paclitaxel and Trastuzumab for Node-Negative, HER2-Positive Breast Cancer. N. Engl. J. Med. 2015, 372, 134–141. [Google Scholar] [CrossRef]
- Burstein, H.J.; Curigliano, G.; Thürlimann, B.; Weber, W.P.; Poortmans, P.; Regan, M.M.; Senn, H.J.; Winer, E.P.; Gnant, M.; Aebi, S.; et al. Customizing local and systemic therapies for women with early breast cancer: The St. Gallen International Consensus Guidelines for treatment of early breast cancer 2021. Ann. Oncol. 2021, 32, 1216–1235. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef]
- Swain, S.M.; Miles, D.; Kim, S.B.; Im, Y.H.; Im, S.A.; Semiglazov, V.; Ciruelos, E.; Schneeweiss, A.; Loi, S.; Monturus, E.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): End-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 519–530. [Google Scholar] [CrossRef]
- Rimawi, M.; Ferrero, J.M.; de la Haba-Rodriguez, J.; Poole, C.; De Placido, S.; Osborne, C.K.; Hegg, R.; Easton, V.; Wohlfarth, C.; Arpino, G.; et al. First-Line Trastuzumab Plus an Aromatase Inhibitor, with or without Pertuzumab, in Human Epidermal Growth Factor Receptor 2-Positive and Hormone Receptor-Positive Metastatic or Locally Advanced Breast Cancer (PERTAIN): A Randomized, Open-Label Phase II Trial. J. Clin. Oncol. 2018, 36, 2826–2835. [Google Scholar] [CrossRef]
- Cortés, J.; Kim, S.-B.; Chung, W.-P.; Im, S.-A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.-M.; Petry, V.; Chung, C.-F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N. Engl. J. Med. 2022, 386, 1143–1154. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Hegg, R.; Chung, W.-P.; Im, S.-A.; Jacot, W.; Ganju, V.; Chiu, J.W.Y.; Xu, B.; Hamilton, E.; Madhusudan, S.; et al. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: Updated results from DESTINY-Breast03, a randomised, open-label, phase 3 trial. Lancet 2023, 401, 105–117. [Google Scholar] [CrossRef]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2019, 382, 597–609. [Google Scholar] [CrossRef]
- Saura, C.; Oliveira, M.; Feng, Y.-H.; Dai, M.-S.; Chen, S.-W.; Hurvitz, S.A.; Kim, S.-B.; Moy, B.; Delaloge, S.; Gradishar, W.; et al. Neratinib Plus Capecitabine Versus Lapatinib Plus Capecitabine in HER2-Positive Metastatic Breast Cancer Previously Treated with ≥ 2 HER2-Directed Regimens: Phase III NALA Trial. J. Clin. Oncol. 2020, 38, 3138–3149. [Google Scholar] [CrossRef]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2022, 22, 101–126. [Google Scholar] [CrossRef]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef]
- Vernieri, C.; Milano, M.; Brambilla, M.; Mennitto, A.; Maggi, C.; Cona, M.S.; Prisciandaro, M.; Fabbroni, C.; Celio, L.; Mariani, G.; et al. Resistance mechanisms to anti-HER2 therapies in HER2-positive breast cancer: Current knowledge, new research directions and therapeutic perspectives. Crit. Rev. Oncol. Hematol. 2019, 139, 53–66. [Google Scholar] [CrossRef]
- Tarantino, P.; Hamilton, E.; Tolaney, S.M.; Cortes, J.; Morganti, S.; Ferraro, E.; Marra, A.; Viale, G.; Trapani, D.; Cardoso, F.; et al. HER2-Low Breast Cancer: Pathological and Clinical Landscape. J. Clin. Oncol. 2020, 38, 1951–1962. [Google Scholar] [CrossRef]
- Scaltriti, M.; Rojo, F.; Ocaña, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J.; et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Pályi-Krekk, Z.; Barok, M.; Isola, J.; Tammi, M.; Szöllosi, J.; Nagy, P. Hyaluronan-induced masking of ErbB2 and CD44-enhanced trastuzumab internalisation in trastuzumab resistant breast cancer. Eur. J. Cancer 2007, 43, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Tomasello, G.; Barni, S.; Lonati, V.; Passalacqua, R.; Ghidini, M. Clinical and pathological characterization of HER2 mutations in human breast cancer: A systematic review of the literature. Breast Cancer Res. Treat. 2017, 166, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Gaibar, M.; Beltrán, L.; Romero-Lorca, A.; Fernández-Santander, A.; Novillo, A. Somatic Mutations in HER2 and Implications for Current Treatment Paradigms in HER2-Positive Breast Cancer. J. Oncol. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Loi, S.; Michiels, S.; Lambrechts, D.; Fumagalli, D.; Claes, B.; Kellokumpu-Lehtinen, P.L.; Bono, P.; Kataja, V.; Piccart, M.J.; Joensuu, H.; et al. Somatic mutation profiling and associations with prognosis and trastuzumab benefit in early breast cancer. J. Natl. Cancer Inst. 2013, 105, 960–967. [Google Scholar] [CrossRef]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013, 3, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Guedes, A.; Gomes, C.A.; Martins, S.; Cruz, R. Pharmacogenomic Application in HER2-positive Breast Cancer. Int. J. Clin. Exp. Med. Res. 2021, 5, 490–497. [Google Scholar] [CrossRef]
- Xu, X.; De Angelis, C.; Burke, K.A.; Nardone, A.; Hu, H.; Qin, L.; Veeraraghavan, J.; Sethunath, V.; Heiser, L.M.; Wang, N.; et al. HER2 Reactivation through Acquisition of the HER2 L755S Mutation as a Mechanism of Acquired Resistance to HER2-targeted Therapy in HER2. Clin. Cancer Res. 2017, 23, 5123–5134. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef]
- Bose, R.; Ma, C.X. Breast Cancer, HER2 Mutations, and Overcoming Drug Resistance. N. Engl. J. Med. 2021, 385, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Razavi, P.; Johnson, J.L.; Shao, H.; Reznik, E.; Smith, M.L.; Sebra, R.; Cantley, L.C.; Scaltriti, M.; Baselga, J. Double PIK3CA mutations in cis enhance PI3Kα oncogene activation and sensitivity to PI3Kα inhibitors in breast cancer. Ann. Oncol. 2019, 30, iii1. [Google Scholar] [CrossRef]
- Smyth, L.M.; Saura, C.; Piha-Paul, S.A.; Lu, J.; Mayer, I.A.; Brufksy, A.M.; Spanggaard, I.; Arnedos, M.; Cutler, R.E.; Hyman, D.M. Update on the phase II SUMMIT trial: Neratinib + fulvestrant for HER2-mutant, HR-positive, metastatic breast cancer. Ann. Oncol. 2019, 30, iii10–iii11. [Google Scholar] [CrossRef]
- Smyth, L.M.; Piha-Paul, S.A.; Won, H.H.; Schram, A.M.; Saura, C.; Loi, S.; Lu, J.; Shapiro, G.I.; Juric, D.; Mayer, I.A.; et al. Efficacy and Determinants of Response to HER Kinase Inhibition in. Cancer Discov. 2020, 10, 198–213. [Google Scholar] [CrossRef]
- Jhaveri, K.L.; Goldman, J.W.; Hurvitz, S.A.; Guerrero-Zotano, A.; Unni, N.; Brufsky, A.; Park, H.; Waisman, J.R.; Yang, E.S.-H.; Spanggaard, I.; et al. Neratinib plus fulvestrant plus trastzuzumab (N+F+T) for hormone receptor-positive (HR+), HER2-negative, HER2-mutant metastatic breast cancer (MBC): Outcomes and biomarker analysis from the SUMMIT trial. J. Clin. Oncol. 2022, 40, 1028. [Google Scholar] [CrossRef]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef]
- Loibl, S.; Majewski, I.; Guarneri, V.; Nekljudova, V.; Holmes, E.; Bria, E.; Denkert, C.; Schem, C.; Sotiriou, C.; Loi, S.; et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: Pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Ann. Oncol. 2016, 27, 1519–1525. [Google Scholar] [CrossRef]
- Morganti, S.; Ivanova, M.; Ferraro, E.; Ascione, L.; Vivanet, G.; Bonizzi, G.; Curigliano, G.; Fusco, N.; Criscitiello, C. Loss of HER2 in breast cancer: Biological mechanisms and technical pitfalls. Cancer Drug Resist. 2022, 5, 971–980. [Google Scholar] [CrossRef]
- Venetis, K.; Pepe, F.; Munzone, E.; Sajjadi, E.; Russo, G.; Pisapia, P.; Ivanova, M.; Bonizzi, G.; Vacirca, D.; Rappa, A.; et al. Analytical Performance of Next-Generation Sequencing and RT-PCR on Formalin-Fixed Paraffin-Embedded Tumor Tissues for PIK3CA Testing in HR+/HER2- Breast Cancer. Cells 2022, 11, 3545. [Google Scholar] [CrossRef]
- Smith, A.E.; Ferraro, E.; Safonov, A.; Morales, C.B.; Lahuerta, E.J.A.; Li, Q.; Kulick, A.; Ross, D.; Solit, D.B.; De Stanchina, E.; et al. HER2 + breast cancers evade anti-HER2 therapy via a switch in driver pathway. Nat. Commun. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Schettini, F.; Buono, G.; Cardalesi, C.; Desideri, I.; De Placido, S.; Del Mastro, L. Hormone Receptor/Human Epidermal Growth Factor Receptor 2-positive breast cancer: Where we are now and where we are going. Cancer Treat. Rev. 2016, 46, 20–26. [Google Scholar] [CrossRef]
- Zhang, S.; Huang, W.C.; Li, P.; Guo, H.; Poh, S.B.; Brady, S.W.; Xiong, Y.; Tseng, L.M.; Li, S.H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Bon, G.; Pizzuti, L.; Laquintana, V.; Loria, R.; Porru, M.; Marchiò, C.; Krasniqi, E.; Barba, M.; Maugeri-Saccà, M.; Gamucci, T.; et al. Loss of HER2 and decreased T-DM1 efficacy in HER2 positive advanced breast cancer treated with dual HER2 blockade: The SePHER Study. J. Exp. Clin. Cancer Res. 2020, 39, 279. [Google Scholar] [CrossRef] [PubMed]
- Kute, T.E.; Savage, L.; Stehle, J.R.; Kim-Shapiro, J.W.; Blanks, M.J.; Wood, J.; Vaughn, J.P. Breast tumor cells isolated from in vitro resistance to trastuzumab remain sensitive to trastuzumab anti-tumor effects in vivo and to ADCC killing. Cancer Immunol. Immunother. 2009, 58, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Fan, X.; Meng, W.; Deng, H.; Zhang, N.; An, Z. Engagement of immune effector cells by trastuzumab induces HER2/ERBB2 downregulation in cancer cells through STAT1 activation. Breast Cancer Res. 2014, 16, R33. [Google Scholar] [CrossRef]
- Tarantino, P.; Morganti, S.; Uliano, J.; Giugliano, F.; Crimini, E.; Curigliano, G. Margetuximab for the treatment of HER2-positive metastatic breast cancer. Expert Opin. Biol. Ther. 2021, 21, 127–133. [Google Scholar] [CrossRef]
- Vance, G.H.; Barry, T.S.; Bloom, K.J.; Fitzgibbons, P.L.; Hicks, D.G.; Jenkins, R.B.; Persons, D.L.; Tubbs, R.R.; Hammond, M.E. Genetic heterogeneity in HER2 testing in breast cancer: Panel summary and guidelines. Arch. Pathol. Lab. Med. 2009, 133, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, B.; Chiaravalli, A.M.; Finzi, G.; Milani, K.; Tibiletti, M.G. Genetic heterogeneity in HER2 testing may influence therapy eligibility. Breast Cancer Res. Treat. 2012, 133, 161–168. [Google Scholar] [CrossRef]
- Bartlett, J.M.S.; Starczynski, J.; Atkey, N.; Kay, E.; O’Grady, A.; Gandy, M.; Ibrahim, M.; Jasani, B.; Ellis, I.O.; Pinder, S.E.; et al. HER2 testing in the UK: Recommendations for breast and gastric in-situ hybridisation methods. J. Clin. Pathol. 2011, 64, 649–653. [Google Scholar] [CrossRef]
- Venetis, K.; Crimini, E.; Sajjadi, E.; Corti, C.; Guerini-Rocco, E.; Viale, G.; Curigliano, G.; Criscitiello, C.; Fusco, N. HER2 Low, Ultra-low, and Novel Complementary Biomarkers: Expanding the Spectrum of HER2 Positivity in Breast Cancer. Front. Mol. Biosci. 2022, 9, 834651. [Google Scholar] [CrossRef]
- Caswell-Jin, J.L.; McNamara, K.; Reiter, J.G.; Sun, R.; Hu, Z.; Ma, Z.; Ding, J.; Suarez, C.J.; Tilk, S.; Raghavendra, A.; et al. Clonal replacement and heterogeneity in breast tumors treated with neoadjuvant HER2-targeted therapy. Nat. Commun. 2019, 10, 657. [Google Scholar] [CrossRef] [PubMed]
- Brasó-Maristany, F.; Griguolo, G.; Pascual, T.; Paré, L.; Nuciforo, P.; Llombart-Cussac, A.; Bermejo, B.; Oliveira, M.; Morales, S.; Martínez, N.; et al. Phenotypic changes of HER2-positive breast cancer during and after dual HER2 blockade. Nat. Commun. 2020, 11, 385. [Google Scholar] [CrossRef]
- Sajjadi, E.; Venetis, K.; Ivanova, M.; Fusco, N. Improving HER2 testing reproducibility in HER2-low breast cancer. Cancer Drug Resist. 2022, 5, 882–888. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, A.N.; Kim, E.J.; Jang, M.H.; Suh, K.J.; Ryu, H.S.; Kim, Y.J.; Kim, J.H.; Im, S.A.; Gong, G.; et al. HER2 heterogeneity affects trastuzumab responses and survival in patients with HER2-positive metastatic breast cancer. Am. J. Clin. Pathol. 2014, 142, 755–766. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Perez, E.A.; Barrios, C.; Eiermann, W.; Toi, M.; Im, Y.H.; Conte, P.; Martin, M.; Pienkowski, T.; Pivot, X.; Burris, H.; et al. Trastuzumab Emtansine with or without Pertuzumab Versus Trastuzumab Plus Taxane for Human Epidermal Growth Factor Receptor 2-Positive, Advanced Breast Cancer: Primary Results From the Phase III MARIANNE Study. J. Clin. Oncol. 2017, 35, 141–148. [Google Scholar] [CrossRef]
- Perez, E.A.; de Haas, S.L.; Eiermann, W.; Barrios, C.H.; Toi, M.; Im, Y.H.; Conte, P.F.; Martin, M.; Pienkowski, T.; Pivot, X.B.; et al. Relationship between tumor biomarkers and efficacy in MARIANNE, a phase III study of trastuzumab emtansine ± pertuzumab versus trastuzumab plus taxane in HER2-positive advanced breast cancer. BMC Cancer 2019, 19, 517. [Google Scholar] [CrossRef]
- Hou, Y.; Nitta, H.; Wei, L.; Banks, P.M.; Portier, B.; Parwani, A.V.; Li, Z. HER2 intratumoral heterogeneity is independently associated with incomplete response to anti-HER2 neoadjuvant chemotherapy in HER2-positive breast carcinoma. Breast Cancer Res. Treat. 2017, 166, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Martin, M.; Jung, K.H.; Huang, C.-S.; Harbeck, N.; Valero, V.; Stroyakovskiy, D.; Wildiers, H.; Campone, M.; Boileau, J.-F.; et al. Neoadjuvant Trastuzumab Emtansine and Pertuzumab in Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer: Three-Year Outcomes From the Phase III KRISTINE Study. J. Clin. Oncol. 2019, 37, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, F.; Corti, C.; Tarantino, P.; Michelini, F.; Curigliano, G. Bystander effect of antibody-drug conjugates: Fact or fiction? Curr. Oncol. Rep. 2022, 24, 809–817. [Google Scholar] [CrossRef]
- Diéras, V.; Deluche, E.; Lusque, A.; Pistilli, B.; Bachelot, T.; Pierga, J.-Y.; Viret, F.; Levy, C.; Salabert, L.; Du, F.L.; et al. Abstract PD8-02: Trastuzumab deruxtecan (T-DXd) for advanced breast cancer patients (ABC), regardless HER2 status: A phase II study with biomarkers analysis (DAISY). Cancer Res. 2022, 82 (Suppl. S4), PD8-02. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Mamounas, E.P.; Untch, M.; Mano, M.S.; Huang, C.-S.; Geyer Jr, C.E.; Von Minckwitz, G.; Wolmark, N.; Pivot, X.; Kuemmel, S.; Digiovanna, M.P.; et al. Adjuvant T-DM1 versus trastuzumab in patients with residual invasive disease after neoadjuvant therapy for HER2-positive breast cancer: Subgroup analyses from KATHERINE. Ann. Oncol. 2021, 32, 1005–1014. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 382, 610–621. [Google Scholar] [CrossRef]
- Saura Manich, C.; O’Shaughnessy, J.; Aftimos, P.G.; Van Den Tweel, E.; Oesterholt, M.; Escrivá-De-Romaní, S.I.; Quenel Tueux, N.; Tan, T.J.; Lim, J.S.; Ladoire, S.; et al. LBA15 Primary outcome of the phase III SYD985.002/TULIP trial comparing [vic-]trastuzumab duocarmazine to physician’s choice treatment in patients with pre-treated HER2-positive locally advanced or metastatic breast cancer. Ann. Oncol. 2021, 32, S1288. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Park, H.; Frentzas, S.; Shannon, C.M.; Cuff, K.; Eek, R.W.; Budd, G.T.; McCartney, A.; O’Shaughnessy, J.; Lu, J.M.; et al. Safety and unique pharmacokinetic profile of ARX788, a site-specific ADC, in heavily pretreated patients with HER2-overexpresing solid tumors: Results from two phase 1 clinical trials. J. Clin. Oncol. 2021, 39 (Suppl. S15), 1038. [Google Scholar] [CrossRef]
- Mosele, M.F.; Lusque, A.; Dieras, V.; Deluche, E.; Ducoulombier, A.; Pistilli, B.; Bachelot, T.; Viret, F.; Levy, C.; Signolle, N.; et al. LBA1 Unraveling the mechanism of action and resistance to trastuzumab deruxtecan (T-DXd): Biomarker analyses from patients from DAISY trial. Ann. Oncol. 2022, 33, S123. [Google Scholar] [CrossRef]
- Mosele, M.F.; Lusque, A.; Dieras, V.C.; Deluche, E.; Ducoulombier, A.; Pistilli, B.; Bachelot, T.; Viret, F.; Levy, C.; Pradat, Y.C.; et al. LBA72 Unraveling the mechanism of action and resistance to trastuzumab deruxtecan (T-DXd): Biomarker analyses from patients from DAISY trial. Ann. Oncol. 2022, 33, S1440. [Google Scholar] [CrossRef]
- Yardley, D.A.; Kaufman, P.A.; Huang, W.; Krekow, L.; Savin, M.; Lawler, W.E.; Zrada, S.; Starr, A.; Einhorn, H.; Schwartzberg, L.S.; et al. Quantitative measurement of HER2 expression in breast cancers: Comparison with ‘real-world’ routine HER2 testing in a multicenter Collaborative Biomarker Study and correlation with overall survival. Breast Cancer Res. 2015, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.; Larsson, L.; Stenbeck, L.; Salmén, F.; Ehinger, A.; Wu, S.Z.; Al-Eryani, G.; Roden, D.; Swarbrick, A.; Borg, Å.; et al. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- McNamara, K.L.; Caswell-Jin, J.L.; Joshi, R.; Ma, Z.; Kotler, E.; Bean, G.R.; Kriner, M.; Zhou, Z.; Hoang, M.; Beechem, J.; et al. Spatial proteomic characterization of HER2-positive breast tumors through neoadjuvant therapy predicts response. Nat. Cancer 2021, 2, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.; Hobbs, N.; Lopez-Ramos, D.; Cole, J.; Patel, S. 45P Spatially-resolved single-cell HER2 tumor heterogeneity captured by biophysical modeling. Ann. Oncol. 2022, 33, S142. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| HER2 Status | Score IHC (Definition) | ISH (Definition) °^ |
|---|---|---|
| HER2-positive | 3+ (Circumferential membrane staining, complete, intense, in > 10% of TCs) | Amplified (Average HER2 copy number ≥ 6.0 signals/cell) |
| 2+ * (Weak/moderate complete membrane staining in > 10% of TCs) | ||
| HER2-negative | 2+ # (Weak/moderate complete membrane staining in > 10% of TCs) | Not Amplified (Average HER2 copy number < 4.0 signals/cell) |
| 1+ (Incomplete membrane staining, faint/barely perceptible in > 10% of TCs) | ||
| 0 (No staining or incomplete membrane staining, faint/barely perceptible in ≤10% of TCs) |
| Drug | Antibody | Linker | Payload | Bystander Effect | Indication |
|---|---|---|---|---|---|
| Trastuzumab emtansine | Trastuzumab | Non-cleavable | DM1 (microtubule inhibitor) | No | HER2-positive BC |
| Trastuzumab deruxtecan | Trastuzumab | Cleavable | DXd (topoisomerase-1 inhibitor) | Yes | HER2-positive BC HER2-low BC |
| HER2 Expression | Treatment | Trial | Phase | Setting | Primary Endpoint | Ref. |
|---|---|---|---|---|---|---|
| HER2-negative (HER2 IHC: 0) | T-DXd | DAISY (NCT04132960) | 2 | Advanced | BOR 30.6% (95%CI: 22.7–45.4) | [73] |
| HER2-Low (HER2 IHC 1+ OR HER2 IHC 2+ ISH non ampl) | T-DXd | DAISY (NCT04132960) | 2 | Advanced | BOR 33.3% (95%CI: 56.7–79.8) | [73] |
| T-DXd vs. TPC | DESTINY-Breast06 (NCT04494425) | 3 | Advanced | Ongoing | // | |
| T-DXd vs. TPC | DESTINY- Breast04 (NCT03734029) | 3 | Advanced (>1–2 line) | mPFS * 9.9mo vs. 4.9mo HR 0.50 (95% CI: 0.40–0.63; p < 0.0001) | [74] | |
| HER2-positive (HER2 IHC 2+ and ISH ampl OR HER2 IHC: 3+) | T-DM1 vs. lapatinib + capecitabine | EMILIA (NCT00829166) | 3 | Advanced (2nd line) | mPFS 9.6 mo vs. 6.4 mo; HR: 0.65 (95% CI, 0.55–0.77, p < 0.001) | [75] |
| T-DM1 vs. TTZ + taxane | MARIANNE (NCT01120184) | 3 | Advanced (1st line) | mPFS 14.1 mo vs. 13.7 mo (non-inferiority) HR: 0.91 (97.5% CI, 0.73–1.13) | [68] | |
| T-DM1 vs. TTZ | KATHERINE (NCT01772472) | 3 | Post-neoadjuvant | 13y-iDFS 88.3% vs. 77%; HR: 0.50 (95% CI, 0.39–0.64; p < 0.001) | [21,76] | |
| T-Dxd | DAISY (NCT04132960) | 2 | advanced | BOR 69.1% (95% CI: 39.1–54.2) | [73] | |
| Combinations of T-DXd and other agents | DESTINY-Breast07 (NCT04538742) | 1b-2 | Advanced (1st line) | ongoing | // | |
| T-DXd vs. T-DM1 | DESTINY-Breast03 | 3 | Advanced (2nd line) | mPFS 28.8 vs. 6.8 mo; HR: 0.33 (95% CI, p = <0.0001) | [24,25] | |
| T-DXd | DESTINY-Breast01 (NCT03248492) | 2 | Advanced (≥3 line) | ORR 60.9% (95% CI, 53.4–68) | [77] | |
| Trastuzumab Duocarmazine | TULIP (NCT03262935) | 3 | Advanced (≥2 line) | mPFS 7 mo (95% CI, 5.4–7.2) vs. 4.9 mo TPC (4–5.5); HR: 0.64 (95% CI, 0.49–0.84, p = 0.002) | [78] | |
| ARX788 | ACE-Breast01 and ACE-Pantumour01 (NCT03255070) | 1 | Advanced | ORR 74% (14/19) ACE-Breast-01 67% (2/3) ACE-Pan tumour-01 | [79] | |
| T-DM1 + pertuzumab | NA | 2 | neoadjuvant | pCR 55% in the non heterogeneous subgroup and 0% in the heterogeneous group (p < 0.0001) # | [15] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giugliano, F.; Carnevale Schianca, A.; Corti, C.; Ivanova, M.; Bianco, N.; Dellapasqua, S.; Criscitiello, C.; Fusco, N.; Curigliano, G.; Munzone, E. Unlocking the Resistance to Anti-HER2 Treatments in Breast Cancer: The Issue of HER2 Spatial Distribution. Cancers 2023, 15, 1385. https://doi.org/10.3390/cancers15051385
Giugliano F, Carnevale Schianca A, Corti C, Ivanova M, Bianco N, Dellapasqua S, Criscitiello C, Fusco N, Curigliano G, Munzone E. Unlocking the Resistance to Anti-HER2 Treatments in Breast Cancer: The Issue of HER2 Spatial Distribution. Cancers. 2023; 15(5):1385. https://doi.org/10.3390/cancers15051385
Chicago/Turabian StyleGiugliano, Federica, Ambra Carnevale Schianca, Chiara Corti, Mariia Ivanova, Nadia Bianco, Silvia Dellapasqua, Carmen Criscitiello, Nicola Fusco, Giuseppe Curigliano, and Elisabetta Munzone. 2023. "Unlocking the Resistance to Anti-HER2 Treatments in Breast Cancer: The Issue of HER2 Spatial Distribution" Cancers 15, no. 5: 1385. https://doi.org/10.3390/cancers15051385
APA StyleGiugliano, F., Carnevale Schianca, A., Corti, C., Ivanova, M., Bianco, N., Dellapasqua, S., Criscitiello, C., Fusco, N., Curigliano, G., & Munzone, E. (2023). Unlocking the Resistance to Anti-HER2 Treatments in Breast Cancer: The Issue of HER2 Spatial Distribution. Cancers, 15(5), 1385. https://doi.org/10.3390/cancers15051385