
Integrin αvβ3 Is a Master Regulator of Resistance to TKI-Induced Ferroptosis in HER2-Positive Breast Cancer

 , , ,
, , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Stable Knockout (KO) of Integrin β3 or Ferroportin-1
2.3. Overexpression (OE) of Integrin β3 or Ferroportin-1
2.4. In Vitro Proliferation and IC50 Determination
2.5. In Vitro Colony-Formation Assay
2.6. In Vitro Migration and Adhesion Assays
2.7. Flow Cytometry
2.8. Western Blot
2.9. Phalloidin Staining
2.10. Cystine Uptake Measurement
2.11. Glutathione Measurement
2.12. Tumour Growth Assays and Neratinib Therapy
2.13. Histology and Immunohistochemistry
2.14. Statistical Methods
3. Results
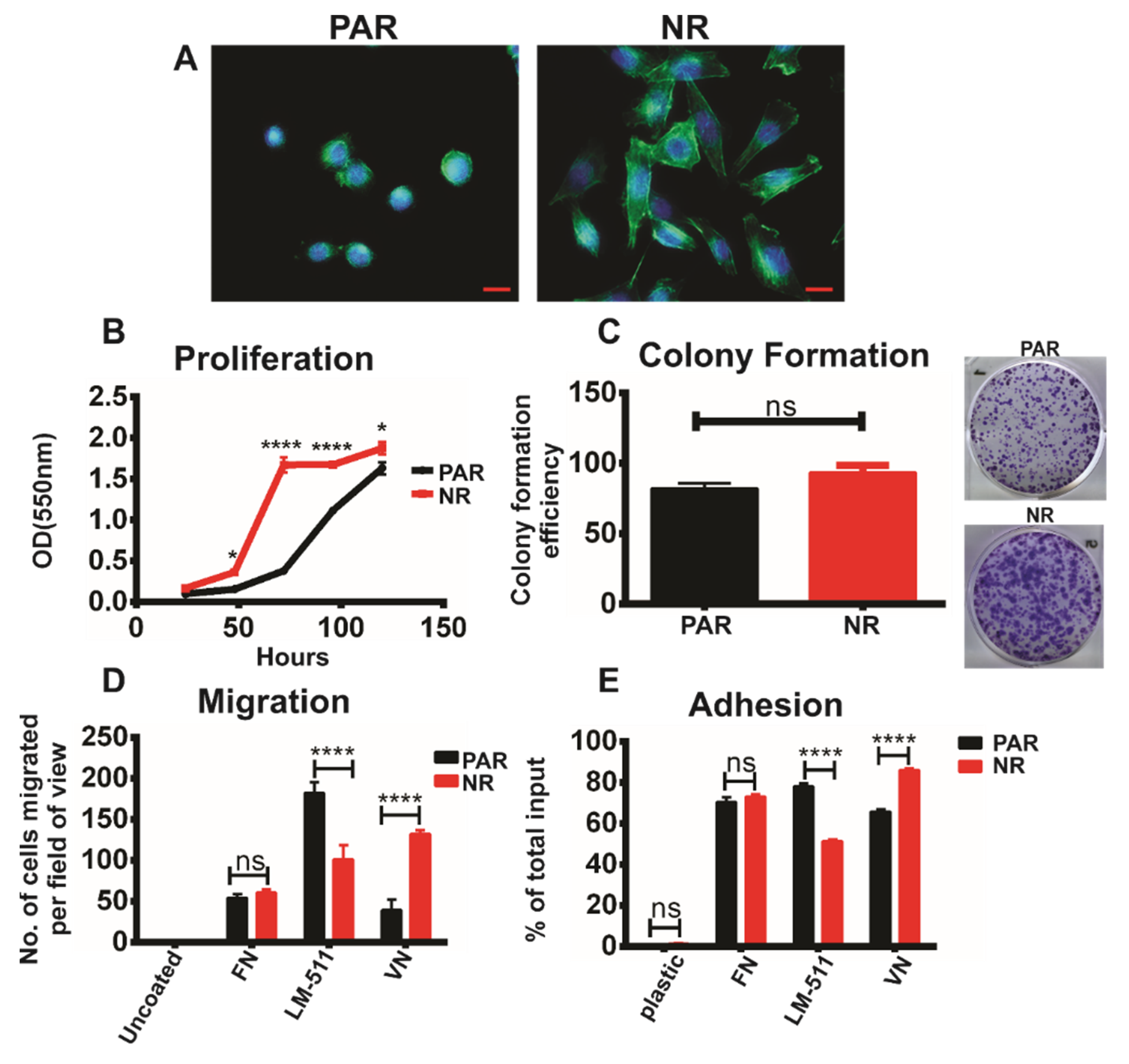
3.1. Development and Characterisation of Neratinib-Resistant Models of HER2-Positive Breast Cancer
3.2. TKI Resistance Is Associated with Increased Expression of αvβ3 Integrin
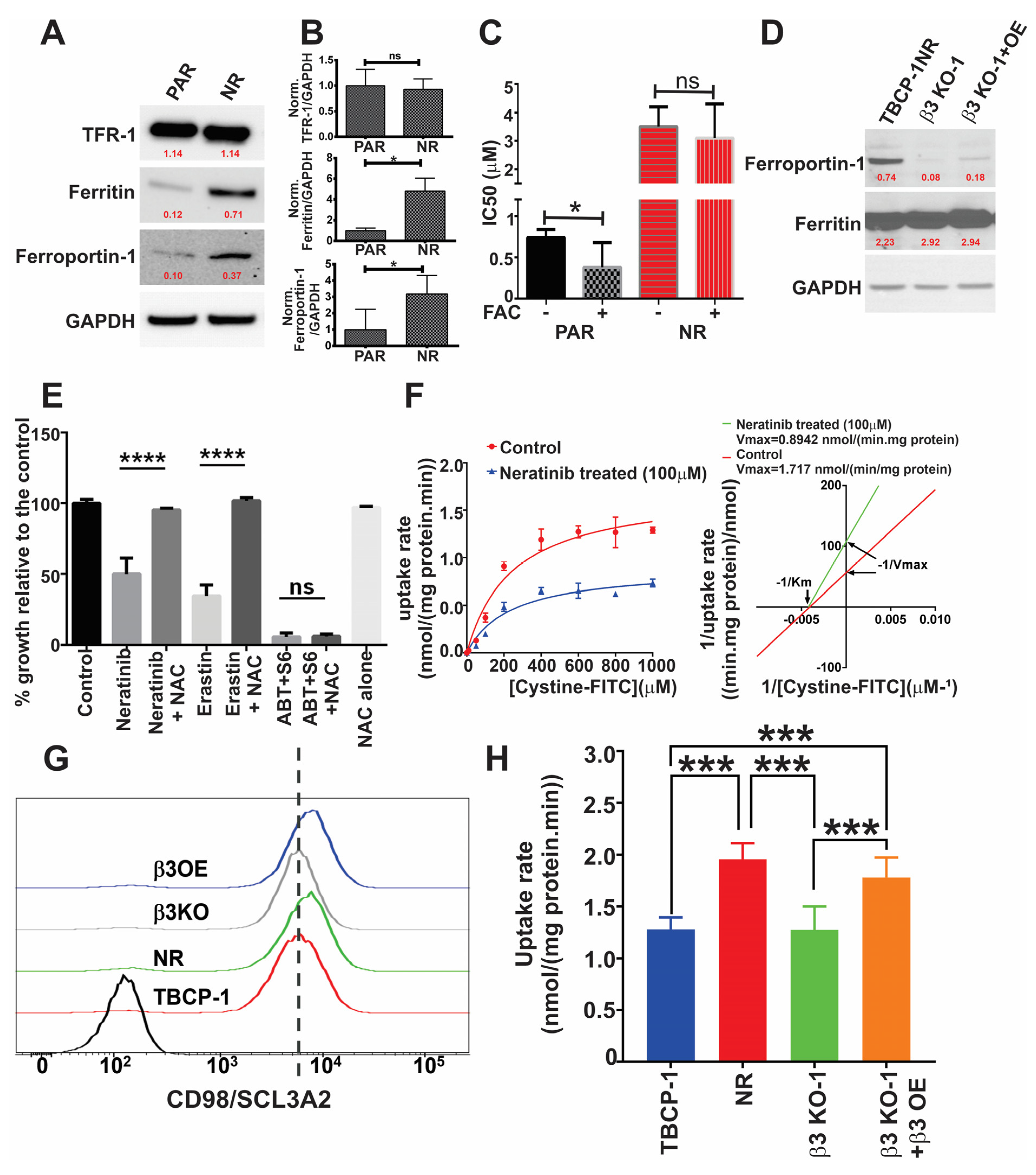
3.3. αvβ3 Integrin Functionally Regulates Resistance to TKI-Induced Ferroptosis
3.4. αvβ3 Integrin Mediates TKI Resistance through Persistent Activation of AKT Signalling
3.5. αvβ3 Integrin Mediates Resistance to Ferroptosis through Crosstalk with Iron Metabolism and Antioxidant Response Pathways
3.6. αvβ3 Integrin Mediates Neratinib Resistance In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Heitz, F.; Harter, P.; Lueck, H.J.; Fissler-Eckhoff, A.; Lorenz-Salehi, F.; Scheil-Bertram, S.; Traut, A.; du Bois, A. Triple-negative and HER2-overexpressing breast cancers exhibit an elevated risk and an earlier occurrence of cerebral metastases. Eur. J. Cancer 2009, 45, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.; André, F.; Barrios, C.; Bergh, J.; Bhattacharyya, G.; Biganzoli, L.J.A.o.O. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). J. Ann. Oncol. 2020, 31, 1623–1649. [Google Scholar] [CrossRef]
- Giordano, S.H.; Temin, S.; Chandarlapaty, S.; Crews, J.R.; Esteva, F.J.; Kirshner, J.J.; Krop, I.E.; Levinson, J.; Lin, N.U.; Modi, S.; et al. Systemic Therapy for Patients With Advanced Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 2736–2740. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Cortés, J.; Kim, S.-B.; Im, S.-A.; Hegg, R.; Im, Y.-H.; Roman, L.; Pedrini, J.L.; Pienkowski, T.; Knott, A.; et al. Pertuzumab plus Trastuzumab plus Docetaxel for Metastatic Breast Cancer. N. Engl. J. Med. 2011, 366, 109–119. [Google Scholar] [CrossRef]
- Conte, B.; Fabi, A.; Poggio, F.; Blondeaux, E.; Dellepiane, C.; D’Alonzo, A.; Buono, G.; Arpino, G.; Magri, V.; Naso, G.; et al. T-DM1 Efficacy in Patients With HER2-positive Metastatic Breast Cancer Progressing After a Taxane Plus Pertuzumab and Trastuzumab: An Italian Multicenter Observational Study. Clin. Breast Cancer 2020, 20, e181–e187. [Google Scholar] [CrossRef]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Domchek, S.M.; Burstein, H.J.; Harris, L.; Younger, J.; Kuter, I.; Bunnell, C.; Rue, M.; Gelman, R.; Winer, E.J.C. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer Cell Int. 2003, 97, 2972–2977. [Google Scholar] [CrossRef]
- Brufsky, A.M.; Mayer, M.; Rugo, H.S.; Kaufman, P.A.; Tan-Chiu, E.; Tripathy, D.; Tudor, I.C.; Wang, L.I.; Brammer, M.G.; Shing, M.J.C.C.R. Central nervous system metastases in patients with HER2-positive metastatic breast cancer: Incidence, treatment, and survival in patients from registHER. Clin. Cancer Res. 2011, 17, 4834–4843. [Google Scholar] [CrossRef]
- Lin, N.U.; Pegram, M.; Sahebjam, S.; Ibrahim, N.; Fung, A.; Cheng, A.; Nicholas, A.; Kirschbrown, W.; Kumthekar, P. Pertuzumab Plus High-Dose Trastuzumab in Patients With Progressive Brain Metastases and HER2-Positive Metastatic Breast Cancer: Primary Analysis of a Phase II Study. J. Clin. Oncol. 2021, 39, 2667–2675. [Google Scholar] [CrossRef]
- Schlam, I.; Swain, S.M. HER2-positive breast cancer and tyrosine kinase inhibitors: The time is now. npj Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef]
- Lin, N.U.; Carey, L.A.; Liu, M.C.; Younger, J.; Come, S.E.; Ewend, M.; Harris, G.J.; Bullitt, E.; Van den Abbeele, A.D.; Henson, J.W. Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2–positive breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 1993. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.A.; Gelman, R.S.; Anders, C.K.; Melisko, M.E.; Parsons, H.A.; Cropp, A.M.; Silvestri, K.; Cotter, C.M.; Componeschi, K.P.; Marte, J.M.; et al. TBCRC 022: A Phase II Trial of Neratinib and Capecitabine for Patients With Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer and Brain Metastases. J. Clin. Oncol. 2019, 37, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 12. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system xc− and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Sui, S.; Zhang, J.; Xu, S.; Wang, Q.; Wang, P.; Pang, D. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 2019, 10, 331. [Google Scholar] [CrossRef]
- Geng, N.; Shi, B.J.; Li, S.L.; Zhong, Z.Y.; Li, Y.C.; Xua, W.L.; Zhou, H.; Cai, J.H. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3826–3836. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, A.; Redvers, R.P.; Ling, X.; Ayton, S.; Fuentes, M.; Tavancheh, E.; Diala, I.; Lalani, A.; Loi, S.; David, S.; et al. Neoadjuvant neratinib promotes ferroptosis and inhibits brain metastasis in a novel syngeneic model of spontaneous HER2(+ve) breast cancer metastasis. Breast Cancer Res. 2019, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [PubMed]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar]
- Song, X.; Wang, X.; Liu, Z.; Yu, Z. Role of GPX4-Mediated Ferroptosis in the Sensitivity of Triple Negative Breast Cancer Cells to Gefitinib. Front. Oncol. 2020, 10, 597434. [Google Scholar] [CrossRef]
- Cruz da Silva, E.; Dontenwill, M.; Choulier, L.; Lehmann, M. Role of Integrins in Resistance to Therapies Targeting Growth Factor Receptors in Cancer. Cancers 2019, 11, 692. [Google Scholar] [CrossRef] [PubMed]
- Lorger, M.; Krueger, J.S.; O’Neal, M.; Staflin, K.; Felding-Habermann, B.J.P. Activation of tumor cell integrin αvβ3 controls angiogenesis and metastatic growth in the brain. Proc. Natl. Acad. Sci. USA 2009, 106, 10666–10671. [Google Scholar] [CrossRef]
- Lal, S.; Kersch, C.; Beeson, K.A.; Wu, Y.J.; Muldoon, L.L.; Neuwelt, E.A. Interactions between αv-Integrin and HER2 and Their Role in the Invasive Phenotype of Breast Cancer Cells In Vitro and in Rat Brain. PloS ONE 2015, 10, e0131842. [Google Scholar] [CrossRef]
- Fan, J.; Cai, B.; Zeng, M.; Hao, Y.; Giancotti, F.G.; Fu, B.M. Integrin β4 signaling promotes mammary tumor cell adhesion to brain microvascular endothelium by inducing ErbB2-mediated secretion of VEGF. Ann. Biomed. Eng. 2011, 39, 2223–2241. [Google Scholar] [CrossRef]
- Carter, R.Z.; Micocci, K.C.; Natoli, A.; Redvers, R.P.; Paquet-Fifield, S.; Martin, A.C.B.M.; Denoyer, D.; Ling, X.; Kim, S.H.; Tomasin, R.J.T.J.o.p. Tumour but not stromal expression of β 3 integrin is essential, and is required early, for spontaneous dissemination of bone-metastatic breast cancer. J. Pathol. 2015, 235, 760–772. [Google Scholar] [CrossRef]
- Sloan, E.K.; Pouliot, N.; Stanley, K.L.; Chia, J.; Moseley, J.M.; Hards, D.K.; Anderson, R.L. Tumor-specific expression of alphavbeta3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006, 8, R20. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, L.; Taubenberger, A.V.; Theodoropoulos, C.; Holzapfel, B.M.; Ramuz, O.; Straub, M.; Hutmacher, D.W. New mechanistic insights of integrin β1 in breast cancer bone colonization. Oncotarget 2014, 6, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Miskin, R.P.; Warren, J.S.A.; Ndoye, A.; Wu, L.; Lamar, J.M.; DiPersio, C.M. Integrin α3β1 Promotes Invasive and Metastatic Properties of Breast Cancer Cells through Induction of the Brn-2 Transcription Factor. Cancers 2021, 13, 480. [Google Scholar] [CrossRef]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, e2337–e2346. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, F.; Asselin-Labat, M.-L.; Shackleton, M.; Forrest, N.C.; Lindeman, G.J.; Visvader, J.E. The Mammary Progenitor Marker CD61/β3 Integrin Identifies Cancer Stem Cells in Mouse Models of Mammary Tumorigenesis. Cancer Res. 2008, 68, 7711–7717. [Google Scholar] [CrossRef] [PubMed]
- Aoudjit, F.; Vuori, K. Integrin signaling in cancer cell survival and chemoresistance. Chemother Res. Pract. 2012, 2012, 283181. [Google Scholar] [CrossRef] [PubMed]
- Chekenya, M.; Krakstad, C.; Svendsen, A.; Netland, I.A.; Staalesen, V.; Tysnes, B.B.; Selheim, F.; Wang, J.; Sakariassen, P.Ø.; Sandal, T.; et al. The progenitor cell marker NG2/MPG promotes chemoresistance by activation of integrin-dependent PI3K/Akt signaling. Oncogene 2008, 27, 5182–5194. [Google Scholar] [CrossRef]
- Palmieri, D.; Bronder, J.L.; Herring, J.M.; Yoneda, T.; Weil, R.J.; Stark, A.M.; Kurek, R.; Vega-Valle, E.; Feigenbaum, L.; Halverson, D.J.C.r. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007, 67, 4190–4198. [Google Scholar] [CrossRef] [PubMed]
- Murrell, D.H.; Hamilton, A.M.; Mallett, C.L.; van Gorkum, R.; Chambers, A.F.; Foster, P.J. Understanding Heterogeneity and Permeability of Brain Metastases in Murine Models of HER2-Positive Breast Cancer Through Magnetic Resonance Imaging: Implications for Detection and Therapy. Transl. Oncol. 2015, 8, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Ramos, O.H.; Kauskot, A.; Cominetti, M.R.; Bechyne, I.; Salla Pontes, C.L.; Chareyre, F.; Manent, J.; Vassy, R.; Giovannini, M.; Legrand, C.; et al. A novel alpha(v)beta (3)-blocking disintegrin containing the RGD motive, DisBa-01, inhibits bFGF-induced angiogenesis and melanoma metastasis. Clin. Exp. Metastasis 2008, 25, 53–64. [Google Scholar] [CrossRef]
- Kim, S.-H.; Redvers, R.P.; Chi, L.H.; Ling, X.; Lucke, A.J.; Reid, R.C.; Fairlie, D.P.; Martin, A.C.B.M.; Anderson, R.L.; Denoyer, D.J.D.m.; et al. Identification of brain metastasis genes and therapeutic evaluation of histone deacetylase inhibitors in a clinically relevant model of breast cancer brain metastasis. Dis. Model. Mech. 2018, 11, DMM034850. [Google Scholar] [CrossRef] [PubMed]
- Prichard, M.N.; Prichard, L.E.; Baguley, W.A.; Nassiri, M.R.; Shipman, C., Jr. Three-dimensional analysis of the synergistic cytotoxicity of ganciclovir and zidovudine. Antimicrob Agents Chemother 1991, 35, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Kusuma, N.; Denoyer, D.; Eble, J.; Redvers, R.; Parker, B.; Pelzer, R.; Anderson, R.; Pouliot, N. Integrin-dependent response to laminin-511 regulates breast tumor cell invasion and metastasis. Int. J. Cancer 2012, 130, 555–566. [Google Scholar] [CrossRef]
- Kamencic, H.; Lyon, A.; Paterson, P.G.; Juurlink, B.H. Monochlorobimane fluorometric method to measure tissue glutathione. Anal. Biochem. 2000, 286, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Longmate, W.; DiPersio, C.M.J.F. Beyond adhesion: Emerging roles for integrins in control of the tumor microenvironment. FResearch 2017, 6, 1612. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef]
- Shimada, K.; Muhlich, J.L.; Mitchison, T.J. A tool for browsing the Cancer Dependency Map reveals functional connections between genes and helps predict the efficacy and selectivity of candidate cancer drugs. bioRxiv 2019. [Google Scholar] [CrossRef]
- Rahman, R.; Pal, R. Analyzing drug sensitivity prediction based on dose response curve characteristics. In Proceedings of the 2016 IEEE-EMBS International Conference on Biomedical and Health Informatics (BHI), Las Vegas, NV, USA, 24–27 February 2016; pp. 140–143. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef]
- Reardon, D.A.; Cheresh, D. Cilengitide: A prototypic integrin inhibitor for the treatment of glioblastoma and other malignancies. Genes Cancer 2011, 2, 1159–1165. [Google Scholar] [CrossRef]
- Eliceiri, B.P. Integrin and Growth Factor Receptor Crosstalk. Circ. Res. 2001, 89, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Alexi, X.; Berditchevski, F.; Odintsova, E. The effect of cell-ECM adhesion on signalling via the ErbB family of growth factor receptors. Biochem. Soc. Trans. 2011, 39, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.-S.; Lv, Q.-M.; Zhang, K.-R.; Tang, Y.-B.; Zhang, Y.-F.; Shen, Y.; Lei, H.-M.; Zhu, L. NRF2-GPX4/SOD2 axis imparts resistance to EGFR-tyrosine kinase inhibitors in non-small-cell lung cancer cells. Acta Pharmacol. Sin. 2021, 42, 613–623. [Google Scholar] [CrossRef]
- Feral, C.C.; Nishiya, N.; Fenczik, C.A.; Stuhlmann, H.; Slepak, M.; Ginsberg, M.H. CD98hc (SLC3A2) mediates integrin signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Kabir-Salmani, M.; Fukuda, M.N.; Kanai-Azuma, M.; Ahmed, N.; Shiokawa, S.; Akimoto, Y.; Sakai, K.; Nagamori, S.; Kanai, Y.; Sugihara, K.; et al. The Membrane-Spanning Domain of CD98 Heavy Chain Promotes αvβ3 Integrin Signals in Human Extravillous Trophoblasts. Mol. Endocrinol. 2008, 22, 707–715. [Google Scholar] [CrossRef]
- Breslin, S.; Lowry, M.C.; O’Driscoll, L. Neratinib resistance and cross-resistance to other HER2-targeted drugs due to increased activity of metabolism enzyme cytochrome P4503A4. Br. J. Cancer 2017, 116, 620–625. [Google Scholar] [CrossRef]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.J.N.c.b. An integrin β 3–KRAS–RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457. [Google Scholar] [CrossRef]
- Zhu, X.; Tao, X.; Lu, W.; Ding, Y.; Tang, Y.J.C.C. Blockade of integrin β3 signals to reverse the stem-like phenotype and drug resistance in melanoma. Cancer Chemother. Pharmacol. 2019, 83, 615–624. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, Y.; Lei, Z.; Liu, T.; Cai, T.; Wang, A.; Du, W.; Zeng, Y.; Zhu, J.; Liu, Z.; et al. Abnormally activated OPN/integrin αVβ3/FAK signalling is responsible for EGFR-TKI resistance in EGFR mutant non-small-cell lung cancer. J. Hematol. Oncol. 2020, 13, 169. [Google Scholar] [CrossRef]
- Endo, Y.; Shen, Y.; Youssef, L.A.; Mohan, N.; Wu, W.J. T-DM1-resistant cells gain high invasive activity via EGFR and integrin cooperated pathways. mAbs 2018, 10, 1003–1017. [Google Scholar] [CrossRef]
- Fox, G.C.; Su, X.; Davis, J.L.; Xu, Y.; Kwakwa, K.A.; Ross, M.H.; Fontana, F.; Xiang, J.; Esser, A.K.; Cordell, E.; et al. Targeted Therapy to β3 Integrin Reduces Chemoresistance in Breast Cancer Bone Metastases. Mol. Cancer Ther. 2021, 20, 1183–1198. [Google Scholar] [CrossRef] [PubMed]
- Nair, M.G.; Desai, K.; Prabhu, J.S.; Hari, P.S.; Remacle, J.; Sridhar, T.S. β3 integrin promotes chemoresistance to epirubicin in MDA-MB-231 through repression of the pro-apoptotic protein, BAD. Exp. Cell Res. 2016, 346, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Maubant, S.; Cruet-Hennequart, S.; Poulain, L.; Carreiras, F.; Sichel, F.; Luis, J.; Staedel, C.; Gauduchon, P. Altered adhesion properties and alphav integrin expression in a cisplatin-resistant human ovarian carcinoma cell line. Int J. Cancer 2002, 97, 186–194. [Google Scholar] [CrossRef]
- Eskens, F.A.; Dumez, H.; Hoekstra, R.; Perschl, A.; Brindley, C.; Böttcher, S.; Wynendaele, W.; Drevs, J.; Verweij, J.; van Oosterom, A.T. Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur. J. Cancer 2003, 39, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, S.; Gustafson, D.; Holden, S.; McConkey, D.; Davis, D.; Morrow, M.; Basche, M.; Gore, L.; Zang, C.; O’Bryant, C.L.; et al. Assessment of the biological and pharmacological effects of the alpha nu beta3 and alpha nu beta5 integrin receptor antagonist, cilengitide (EMD 121974), in patients with advanced solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2007, 18, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef]
- Van Agthoven, J.F.; Xiong, J.-P.; Alonso, J.L.; Rui, X.; Adair, B.D.; Goodman, S.L.; Arnaout, M.A. Structural basis for pure antagonism of integrin αVβ3 by a high-affinity form of fibronectin. Nat. Struct Mol. Biol. 2014, 21, 383–388. [Google Scholar] [CrossRef]
- Li, J.; Fukase, Y.; Shang, Y.; Zou, W.; Muñoz-Félix, J.M.; Buitrago, L.; van Agthoven, J.; Zhang, Y.; Hara, R.; Tanaka, Y.; et al. Novel Pure αVβ3 Integrin Antagonists That Do Not Induce Receptor Extension, Prime the Receptor, or Enhance Angiogenesis at Low Concentrations. ACS Pharmacol. Transl. Sci. 2019, 2, 387–401. [Google Scholar] [CrossRef]
- Danilucci, T.M.; Santos, P.K.; Pachane, B.C.; Pisani, G.F.D.; Lino, R.L.B.; Casali, B.C.; Altei, W.F.; Selistre-de-Araujo, H.S. Recombinant RGD-disintegrin DisBa-01 blocks integrin α(v)β(3) and impairs VEGF signaling in endothelial cells. Cell Commun. Signal. 2019, 17, 27. [Google Scholar] [CrossRef]
- Lino, R.L.B.; Dos Santos, P.K.; Pisani, G.F.D.; Altei, W.F.; Cominetti, M.R.; Selistre-de-Araújo, H.S. Alphavbeta3 integrin blocking inhibits apoptosis and induces autophagy in murine breast tumor cells. Biochim. Et Biophys. Acta. Mol. Cell Res. 2019, 1866, 118536. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Gianni, T.; Leoni, V.; Chesnokova, L.S.; Hutt-Fletcher, L.M. Campadelli-Fiume, G. αvβ3-integrin is a major sensor and activator of innate immunity to herpes simplex virus-1. Proc. Natl. Acad. Sci. USA 2012, 109, 19792–19797. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Esser, A.K.; Amend, S.R.; Xiang, J.; Xu, Y.; Ross, M.H.; Fox, G.C.; Kobayashi, T.; Steri, V.; Roomp, K.; et al. Antagonizing Integrin β3 Increases Immunosuppression in Cancer. Cancer Res. 2016, 76, 3484–3495. [Google Scholar] [CrossRef]
- Havaki, S.; Kouloukoussa, M.; Amawi, K.; Drosos, Y.; Arvanitis, L.D.; Goutas, N.; Vlachodimitropoulos, D.; Vassilaros, S.D.; Katsantoni, E.Z.; Voloudakis-Baltatzis, I.; et al. Altered expression pattern of integrin alphavbeta3 correlates with actin cytoskeleton in primary cultures of human breast cancer. Cancer Cell Int. 2007, 7, 16. [Google Scholar] [CrossRef]
- Jin, H.; He, Y.; Zhao, P.; Hu, Y.; Tao, J.; Chen, J.; Huang, Y. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics 2019, 9, 265–278. [Google Scholar] [CrossRef]
- Lesniak, D.; Sabri, S.; Xu, Y.; Graham, K.; Bhatnagar, P.; Suresh, M.; Abdulkarim, B.J.P.O. Spontaneous epithelial-mesenchymal transition and resistance to HER-2-targeted therapies in HER-2-positive luminal breast cancer. PLoS ONE 2013, 8, e71987. [Google Scholar] [CrossRef]
- Oliveras-Ferraros, C.; Corominas-Faja, B.; Cufí, S.; Vazquez-Martin, A.; Martin-Castillo, B.; Iglesias, J.M.; López-Bonet, E.; Martin, Á.G.; Menendez, J.A. Epithelial-to-mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin). Cell Cycle 2012, 11, 4020–4032. [Google Scholar] [CrossRef]
- Vesuna, F.; van Diest, P.; Chen, J.H.; Raman, V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem. Biophys. Res. Commun. 2008, 367, 235–241. [Google Scholar] [CrossRef]
- Phillips, S.; Kuperwasser, C. SLUG: Critical regulator of epithelial cell identity in breast development and cancer. Cell Adhes. Migr. 2014, 8, 578–587. [Google Scholar] [CrossRef]
- Kaufhold, S.; Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.T.; Yang, M.H. Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells 2020, 9, 623. [Google Scholar] [CrossRef]
- Soung, Y.-H.; Clifford, J.L.; Chung, J. Crosstalk between integrin and receptor tyrosine kinase signaling in breast carcinoma progression. BMB Rep. 2010, 43, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Im, H.; Lee, J.; Ryu, K.-Y.; Yi, J. Integrin αvβ3-Akt signaling plays a role in radioresistance of melanoma. Exp. Dermatol. 2020, 29, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-S.; Wang, Z.-F.; Huang, X.-D.; Dai, L.-M.; Cao, C.-J.; Li, Z.-Q. Involvement of ROS-alpha v beta 3 integrin-FAK/Pyk2 in the inhibitory effect of melatonin on U251 glioma cell migration and invasion under hypoxia. J. Transl. Med. 2015, 13, 95. [Google Scholar] [CrossRef]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef]
- Ma, C.X.; Suman, V.; Goetz, M.P.; Northfelt, D.; Burkard, M.E.; Ademuyiwa, F.; Naughton, M.; Margenthaler, J.; Aft, R.; Gray, R.; et al. A Phase II Trial of Neoadjuvant MK-2206, an AKT Inhibitor, with Anastrozole in Clinical Stage II or III <em>PIK3CA</em>-Mutant ER-Positive and HER2-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 6823–6832. [Google Scholar] [CrossRef]
- Turner, N.C.; Alarcón, E.; Armstrong, A.C.; Philco, M.; López Chuken, Y.A.; Sablin, M.P.; Tamura, K.; Gómez Villanueva, A.; Pérez-Fidalgo, J.A.; Cheung, S.Y.A.; et al. BEECH: A dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA mutant sub-population. Ann. Oncol. 2019, 30, 774–780. [Google Scholar] [CrossRef]
- Wisinski, K.B.; Tevaarwerk, A.J.; Burkard, M.E.; Rampurwala, M.; Eickhoff, J.; Bell, M.C.; Kolesar, J.M.; Flynn, C.; Liu, G. Phase I Study of an AKT Inhibitor (MK-2206) Combined with Lapatinib in Adult Solid Tumors Followed by Dose Expansion in Advanced HER2+ Breast Cancer. Clin. Cancer Res. 2016, 22, 2659–2667. [Google Scholar] [CrossRef]
- Hudis, C.; Swanton, C.; Janjigian, Y.Y.; Lee, R.; Sutherland, S.; Lehman, R.; Chandarlapaty, S.; Hamilton, N.; Gajria, D.; Knowles, J.; et al. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013, 15, 110. [Google Scholar] [CrossRef]
- Guerin, M.; Rezai, K.; Isambert, N.; Campone, M.; Autret, A.; Pakradouni, J.; Provansal, M.; Camerlo, J.; Sabatier, R.; Bertucci, F.; et al. PIKHER2: A phase IB study evaluating buparlisib in combination with lapatinib in trastuzumab-resistant HER2-positive advanced breast cancer. Eur. J. Cancer 2017, 86, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Prager, G.W.; Féral, C.C.; Kim, C.; Han, J.; Ginsberg, M.H. CD98hc (SLC3A2) interaction with the integrin beta subunit cytoplasmic domain mediates adhesive signaling. J. Biol. Chem. 2007, 282, 24477–24484. [Google Scholar] [CrossRef]
- Brozovic, A.; Majhen, D.; Roje, V.; Mikac, N.; Jakopec, S.; Fritz, G.; Osmak, M.; Ambriovic-Ristov, A. alpha(v)beta(3) Integrin-mediated drug resistance in human laryngeal carcinoma cells is caused by glutathione-dependent elimination of drug-induced reactive oxidative species. Mol. Pharmacol. 2008, 74, 298–306. [Google Scholar] [CrossRef]
- Reddy, N.M.; Kleeberger, S.R.; Yamamoto, M.; Kensler, T.W.; Scollick, C.; Biswal, S.; Reddy, S.P. Genetic dissection of the Nrf2-dependent redox signaling-regulated transcriptional programs of cell proliferation and cytoprotection. Physiol. Genom. 2007, 32, 74–81. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2017, 29, 1756–1773. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Hadian, K. Ferroptosis Suppressor Protein 1 (FSP1) and Coenzyme Q10 Cooperatively Suppress Ferroptosis. Biochemistry 2020, 59, 637–638. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TBCP-1 | SKBR3 | BT474 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| DRUG * | PAR | NR | FOLD DIFFERENCE | PAR | NR | FOLD DIFFERENCE | PAR | NR | FOLD DIFFERENCE |
| Neratinib | 0.21 ± 0.05 | 2.92 ± 1.01 | 13.9 | 0.009 ± 0.009 | 0.1 ± 0.07 | 11.1 | 0.003 ± 0.001 | 0.062 ± 0.065 | 20.6 |
| Lapatinib | 0.58 ± 0.08 | >16 | >27.6 | 0.15 ± 0.10 | >4 | >26.6 | 0.064 ± 0.001 | >4 | >62.5 |
| Tucatinib | 0.36 ± 0.07 | >16 | >44.4 | 0.02 ± 0.014 | >4 | >200 | 0.023 ± 0.018 | >4 | >173.9 |
| Erastin | 2.37 ± 0.79 | 6.34 ± 3.34 | 2.67 | 2.54 ± 1.73 | 6.38 ± 5.11 | 2.51 | >10 | >10 | NA |
| RSL3 | 1.75 ± 0.04 | >4 | >2.29 | 1.39 ± 0.52 | 3.42 ± 1.21 | 2.45 | 1.71 ± 0.4 | >10 | >5.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagpal, A.; Needham, K.; Lane, D.J.R.; Ayton, S.; Redvers, R.P.; John, M.; Selistre-de-Araujo, H.S.; Denoyer, D.; Pouliot, N. Integrin αvβ3 Is a Master Regulator of Resistance to TKI-Induced Ferroptosis in HER2-Positive Breast Cancer. Cancers 2023, 15, 1216. https://doi.org/10.3390/cancers15041216
Nagpal A, Needham K, Lane DJR, Ayton S, Redvers RP, John M, Selistre-de-Araujo HS, Denoyer D, Pouliot N. Integrin αvβ3 Is a Master Regulator of Resistance to TKI-Induced Ferroptosis in HER2-Positive Breast Cancer. Cancers. 2023; 15(4):1216. https://doi.org/10.3390/cancers15041216
Chicago/Turabian StyleNagpal, Aadya, Kristen Needham, Darius J. R. Lane, Scott Ayton, Richard P. Redvers, Melissa John, Heloisa S. Selistre-de-Araujo, Delphine Denoyer, and Normand Pouliot. 2023. "Integrin αvβ3 Is a Master Regulator of Resistance to TKI-Induced Ferroptosis in HER2-Positive Breast Cancer" Cancers 15, no. 4: 1216. https://doi.org/10.3390/cancers15041216
APA StyleNagpal, A., Needham, K., Lane, D. J. R., Ayton, S., Redvers, R. P., John, M., Selistre-de-Araujo, H. S., Denoyer, D., & Pouliot, N. (2023). Integrin αvβ3 Is a Master Regulator of Resistance to TKI-Induced Ferroptosis in HER2-Positive Breast Cancer. Cancers, 15(4), 1216. https://doi.org/10.3390/cancers15041216