
Genome-Wide Analysis of lncRNA-mRNA Co-Expression Networks in CD133+/CD44+ Stem-like PDAC Cells

 ,
,  , ,
, ,  , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Cell Sorting
2.3. Tumorsphere Formation and Passaging
2.4. Cytotoxicity Assay
2.5. Superarray Analysis for Inflammation and EMT Markers
2.6. In Vivo Studies
2.6.1. Xenograft Mouse Models
2.6.2. Histology and Immunohistochemistry Staining
2.7. Gene Microarray Assay and Data Analysis
2.8. Bioinformatic Analysis
2.8.1. Gene Ontology (GO) and Reactome Pathway Analyses
2.8.2. Ingenuity Pathway and Network Analysis
2.8.3. In Silico Data Analysis and Clinicopathological Associations
2.9. Statistical Analysis
3. Results
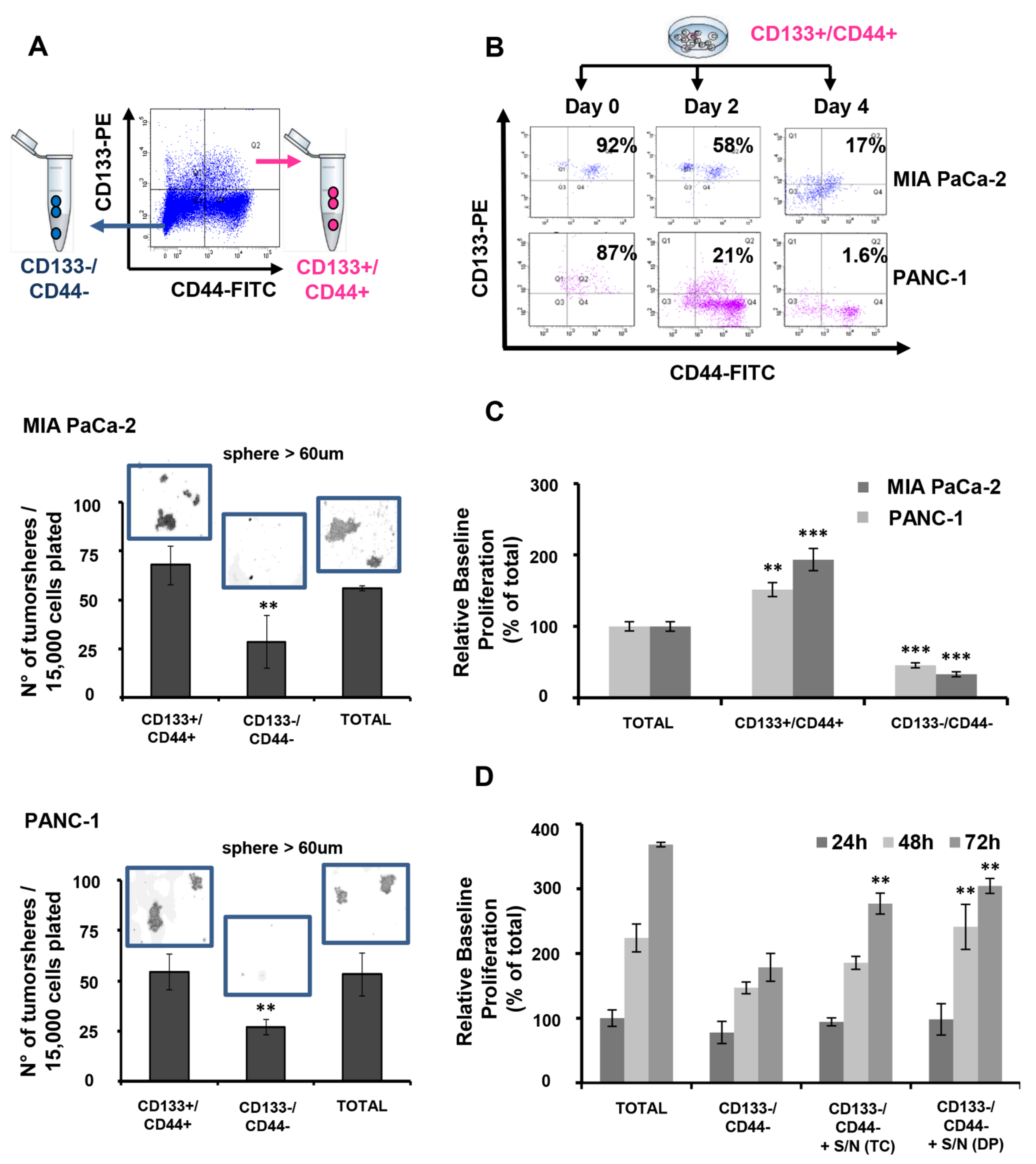
3.1. CD133+/CD44+ PDAC Cells Harbor CSC Characteristics In Vitro
3.2. CD133+/CD44+ PDAC Cell Populations Are Highly Tumorigenic In Vivo
3.3. Differentially Expressed lncRNAs and mRNAs in CD133+/CD44+ PCSCs
3.4. Gene Ontology, Pathway and Network Enrichment Analyses of Dysregulated mRNAs in CD133+/CD44+ PCSCs
3.5. Target Coding Gene Prediction; GO Analysis, Pathway, and Network Enrichment Analyses of lncRNA-Targeted mRNAs
3.6. Identification of lncRNAs Interacting with CSC-Associated mRNAs and Clinicoopathological Associations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Porta, M.; Fabregat, X.; Malats, N.; Guarner, L.; Carrato, A.; de Miguel, A.; Ruiz, L.; Jariod, M.; Costafreda, S.; Coll, S.; et al. Exocrine pancreatic cancer: Symptoms at presentation and their relation to tumour site and stage. Clin. Transl. Oncol. 2005, 7, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, X.-Q.; Wang, Z.-F.; Wu, X.-L.; Zhou, C.-H.; Yan, J.-Y.; Hu, B.-Y.; et al. The molecular biology of pancreatic adenocarcinoma: Translational challenges and clinical perspectives. Signal Transduct. Target. Ther. 2021, 6, 249. [Google Scholar] [CrossRef] [PubMed]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updat. 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Subramaniam, D.; Kaushik, G.; Dandawate, P.; Anant, S. Targeting Cancer Stem Cells for Chemoprevention of Pancreatic Cancer. Curr. Med. Chem. 2018, 25, 2585–2594. [Google Scholar] [CrossRef]
- Habib, M.; Saif, M.W. Pancreatic cancer stem cells: Their role in pancreatic cancer patient outcomes and what is future? JOP 2013, 14, 401–404. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Li, C.; Wu, J.-J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; Pasca di Magliano, M.; Simeone, D.M. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, 2218–2227.e5. [Google Scholar] [CrossRef]
- Ohara, Y.; Oda, T.; Sugano, M.; Hashimoto, S.; Enomoto, T.; Yamada, K.; Akashi, Y.; Miyamoto, R.; Kobayashi, A.; Fukunaga, K.; et al. Histological and prognostic importance of CD44(+)/CD24(+)/EpCAM(+) expression in clinical pancreatic cancer. Cancer Sci. 2013, 104, 1127–1134. [Google Scholar] [CrossRef]
- Kure, S.; Matsuda, Y.; Hagio, M.; Ueda, J.; Naito, Z.; Ishiwata, T. Expression of cancer stem cell markers in pancreatic intraepithelial neoplasias and pancreatic ductal adenocarcinomas. Int. J. Oncol. 2012, 41, 1314–1324. [Google Scholar] [CrossRef]
- Lee, C.J.; Dosch, J.; Simeone, D.M. Pancreatic cancer stem cells. J. Clin. Oncol. 2008, 26, 2806–2812. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Tang, S.-N.; Fu, J.; Nall, D.; Rodova, M.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int. J. cancer 2012, 131, 30–40. [Google Scholar] [CrossRef]
- Rodova, M.; Fu, J.; Watkins, D.N.; Srivastava, R.K.; Shankar, S. Sonic hedgehog signaling inhibition provides opportunities for targeted therapy by sulforaphane in regulating pancreatic cancer stem cell self-renewal. PLoS ONE 2012, 7, e46083. [Google Scholar] [CrossRef]
- Abel, E.V.; Kim, E.J.; Wu, J.; Hynes, M.; Bednar, F.; Proctor, E.; Wang, L.; Dziubinski, M.L.; Simeone, D.M. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS ONE 2014, 9, e91983. [Google Scholar] [CrossRef]
- Yabuuchi, S.; Pai, S.G.; Campbell, N.R.; de Wilde, R.F.; De Oliveira, E.; Korangath, P.; Streppel, M.M.; Rasheed, Z.A.; Hidalgo, M.; Maitra, A.; et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013, 335, 41–51. [Google Scholar] [CrossRef]
- Mohammed, A.; Janakiram, N.B.; Ely, M.; Lightfoot, S.; Steele, V.E.; Rao, C.V. Abstract 2839: Licofelone, a novel dual COX-LOX inhibitor prevents progression of PanIN lesions to pancreatic carcinoma by targeting miRNAs and cancer stem cells in p48Cre/+-LSL-KrasG12D/+ transgenic mice. Cancer Res. 2011, 71, 2839. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.; Carmichael, G.G. Long noncoding RNAs in mammalian cells: What, where, and why? Wiley Interdiscip. Rev. RNA 2010, 1, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Eptaminitaki, G.C.; Stellas, D.; Bonavida, B.; Baritaki, S. Long Non-coding RNAs (lncRNAs) signaling in Cancer Chemoresistance: From Prediction to Druggability. Drug Resist. Updat. 2022, 65, 100866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef]
- Gong, R.; Jiang, Y. Non-coding RNAs in Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, Y.-Y.; Xin, H.-W.; Wang, L.; Arfuso, F.; Dharmarajan, A.; Kumar, A.P.; Wang, H.; Tang, F.R.; Warrier, S.; et al. The expanding roles of long non-coding RNAs in the regulation of cancer stem cells. Int. J. Biochem. Cell Biol. 2019, 108, 17–20. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D793–D800. [Google Scholar] [CrossRef]
- Zaravinos, A.; Lambrou, G.I.; Mourmouras, N.; Katafygiotis, P.; Papagregoriou, G.; Giannikou, K.; Delakas, D.; Deltas, C. New miRNA profiles accurately distinguish renal cell carcinomas and upper tract urothelial carcinomas from the normal kidney. PLoS ONE 2014, 9, e91646. [Google Scholar] [CrossRef]
- Roufas, C.; Chasiotis, D.; Makris, A.; Efstathiades, C.; Dimopoulos, C.; Zaravinos, A. The Expression and Prognostic Impact of Immune Cytolytic Activity-Related Markers in Human Malignancies: A Comprehensive Meta-analysis. Front. Oncol. 2018, 8, 27. [Google Scholar] [CrossRef]
- Eptaminitaki, G.C.; Wolff, N.; Stellas, D.; Sifakis, K.; Baritaki, S. Long Non-Coding RNAs (lncRNAs) in Response and Resistance to Cancer Immunosurveillance and Immunotherapy. Cells 2021, 10, 3313. [Google Scholar] [CrossRef]
- Zhu, Y.-Y.; Yuan, Z. Pancreatic cancer stem cells. Am. J. Cancer Res. 2015, 5, 894–906. [Google Scholar]
- Kyriazi, A.A.; Papiris, E.; Kitsos Kalyvianakis, K.; Sakellaris, G.; Baritaki, S. Dual Effects of Non-Coding RNAs (ncRNAs) in Cancer Stem Cell Biology. Int. J. Mol. Sci. 2020, 21, 6658. [Google Scholar] [CrossRef]
- Zhou, H.; Feng, B.; Abudoureyimu, M.; Lai, Y.; Lin, X.; Tian, C.; Huang, G.; Chu, X.; Wang, R. The functional role of long non-coding RNAs and their underlying mechanisms in drug resistance of non-small cell lung cancer. Life Sci. 2020, 261, 118362. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Kalaszczynska, I.; Geng, Y.; Iino, T.; Mizuno, S.-i.; Choi, Y.; Kondratiuk, I.; Silver, D.P.; Wolgemuth, D.J.; Akashi, K.; Sicinski, P. Cyclin A Is Redundant in Fibroblasts but Essential in Hematopoietic and Embryonic Stem Cells. Cell 2009, 138, 352–365. [Google Scholar] [CrossRef]
- Banno, A.; Garcia, D.A.; Van Baarsel, E.D.; Metz, P.J.; Fisch, K.; Widjaja, C.E.; Kim, S.H.; Lopez, J.; Chang, A.N.; Geurink, P.P.; et al. Downregulation of 26S proteasome catalytic activity promotes epithelial-mesenchymal transition. Oncotarget 2016, 7, 21527–21541. [Google Scholar] [CrossRef]
- Lenos, K.J.; Vermeulen, L. Cancer stem cells don’t waste their time cleaning-low proteasome activity, a marker for cancer stem cell function. Ann. Transl. Med. 2016, 4, 519. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, Q.; Wang, Y.; You, M. 26S proteasome activity is down-regulated in lung cancer stem-like cells propagated in vitro. PLoS ONE 2010, 5, e13298. [Google Scholar] [CrossRef]
- Bradley, E.; Bieberich, E.; Mivechi, N.F.; Tangpisuthipongsa, D.; Wang, G. Regulation of embryonic stem cell pluripotency by heat shock protein 90. Stem Cells 2012, 30, 1624–1633. [Google Scholar] [CrossRef]
- De Lima Fernandes, C.F.; Iglesia, R.P.; Melo-Escobar, M.I.; Prado, M.B.; Lopes, M.H. Chaperones and beyond as key players in pluripotency maintenance. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, Y.-J.; Park, J.M.; Park, M.; Nam, K.D.; Farrand, L.; Nguyen, C.-T.; La, M.T.; Ann, J.; Lee, J.; et al. The C-terminal HSP90 inhibitor NCT-58 kills trastuzumab-resistant breast cancer stem-like cells. Cell death Discov. 2021, 7, 354. [Google Scholar] [CrossRef] [PubMed]
- Firnau, M.-B.; Brieger, A. CK2 and the Hallmarks of Cancer. Biomedicines 2022, 10, 1987. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yan, C.; Quan, X.X.; Yang, X.; Zhang, J.; Bian, Y.; Chen, Z.; Van Waes, C. CK2 Phosphorylates and Inhibits TAp73 Tumor Suppressor Function to Promote Expression of Cancer Stem Cell Genes and Phenotype in Head and Neck Cancer. Neoplasia 2014, 16, 789–800. [Google Scholar] [CrossRef]
- Sato, K.; Padgaonkar, A.A.; Baker, S.J.; Cosenza, S.C.; Rechkoblit, O.; Subbaiah, D.R.C.V.; Domingo-Domenech, J.; Bartkowski, A.; Port, E.R.; Aggarwal, A.K.; et al. Simultaneous CK2/TNIK/DYRK1 inhibition by 108600 suppresses triple negative breast cancer stem cells and chemotherapy-resistant disease. Nat. Commun. 2021, 12, 4671. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Mao, J.-H.; Hsieh, D.; Kim, I.-J.; Hu, L.-M.; Xu, Z.; Long, H.; Jablons, D.M.; You, L. Inhibition of CK2α down-regulates Hedgehog/Gli signaling leading to a reduction of a stem-like side population in human lung cancer cells. PLoS ONE 2012, 7, e38996. [Google Scholar] [CrossRef]
- Alexandre, A.; Lehninger, A.L. Bypasses of the antimycin A block of mitochondrial electron transport in relation to ubisemiquinone function. Biochim. Biophys. Acta (BBA)—Bioenerg. 1984, 767, 120–129. [Google Scholar] [CrossRef]
- García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693. [Google Scholar] [CrossRef]
- Xia, P.; Xu, X.-Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar]
- Yoon, C.; Lu, J.; Yi, B.C.; Chang, K.K.; Simon, M.C.; Ryeom, S.; Yoon, S.S. PI3K/Akt pathway and Nanog maintain cancer stem cells in sarcomas. Oncogenesis 2021, 10, 12. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Mouhieddine, T.H.; Chalhoub, R.M.; Assi, S.; Araji, T.; Chamaa, F.; Itani, M.M.; Nokkari, A.; Kobeissy, F.; Daoud, G.; et al. The Akt/mTOR pathway in cancer stem/progenitor cells is a potential therapeutic target for glioblastoma and neuroblastoma. Oncotarget 2018, 9, 33549–33561. [Google Scholar] [CrossRef]
- Fabbrizi, M.R.; Meyer, B.; Misri, S.; Raj, S.; Zobel, C.L.; Hallahan, D.E.; Sharma, G.G. Transient PP2A inhibition alleviates normal tissue stem cell susceptibility to cell death during radiotherapy. Cell Death Dis. 2018, 9, 492. [Google Scholar] [CrossRef]
- Lai, D.; Chen, M.; Su, J.; Liu, X.; Rothe, K.; Hu, K.; Forrest, D.L.; Eaves, C.J.; Morin, G.B.; Jiang, X. PP2A inhibition sensitizes cancer stem cells to ABL tyrosine kinase inhibitors in BCR-ABL+ human leukemia. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Xu, L.-Z.; Li, S.-S.; Zhou, W.; Kang, Z.-J.; Zhang, Q.-X.; Kamran, M.; Xu, J.; Liang, D.-P.; Wang, C.-L.; Hou, Z.-J.; et al. p62/SQSTM1 enhances breast cancer stem-like properties by stabilizing MYC mRNA. Oncogene 2017, 36, 304–317. [Google Scholar] [CrossRef]
- Walter, K.; Tiwary, K.; Trajkovic-Arsic, M.; Hidalgo-Sastre, A.; Dierichs, L.; Liffers, S.T.; Gu, J.; Gout, J.; Schulte, L.-A.; Münch, J.; et al. MEK Inhibition Targets Cancer Stem Cells and Impedes Migration of Pancreatic Cancer Cells In Vitro and In Vivo. Stem Cells Int. 2019, 2019, 8475389. [Google Scholar] [CrossRef]
- Ahn, H.-J.; Kim, G.; Park, K.-S. Ell3 stimulates proliferation, drug resistance, and cancer stem cell properties of breast cancer cells via a MEK/ERK-dependent signaling pathway. Biochem. Biophys. Res. Commun. 2013, 437, 557–564. [Google Scholar] [CrossRef]
- Bahari, G.; Hashemi, M.; Naderi, M.; Bojd, S.S.; Taheri, M. Long non-coding RNA PAX8-AS1 polymorphisms increase the risk of childhood acute lymphoblastic leukemia. Biomed. Rep. 2018, 8, 184–190. [Google Scholar] [CrossRef]
- Mirzazadeh, S.; Sarani, H.; Nakhaee, A.; Hashemi, S.-M.; Taheri, M.; Hashemi, M.; Bahari, G. Association between PAX8AS1 (rs4848320 C > T, rs1110839 G > T, and rs6726151 T > G) and MEG3 (rs7158663) gene polymorphisms and non-Hodgkin lymphoma risk. Nucleosides. Nucleotides Nucleic Acids 2022, 41, 1174–1186. [Google Scholar] [CrossRef]
- Han, J.; Zhou, W.; Jia, M.; Wen, J.; Jiang, J.; Shi, J.; Zhang, K.; Ma, H.; Liu, J.; Ren, J.; et al. Expression quantitative trait loci in long non-coding RNA PAX8-AS1 are associated with decreased risk of cervical cancer. Mol. Genet. Genom. 2016, 291, 1743–1748. [Google Scholar] [CrossRef]
- Shen, Y.; Tong, Z.-W.; Zhou, Y.; Sun, Y.; Xie, Y.; Li, R.; Liu, H. Inhibition of lncRNA-PAX8-AS1-N directly associated with VEGF/TGF-β1/8-OhdG enhances podocyte apoptosis in diabetic nephropathy. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6864–6872. [Google Scholar] [CrossRef]
- Huang, C.; Li, R.; Yang, C.; Ding, R.; Li, Q.; Xie, D.; Zhang, R.; Qiu, Y. PAX8-AS1 knockdown facilitates cell growth and inactivates autophagy in osteoblasts via the miR-1252-5p/GNB1 axis in osteoporosis. Exp. Mol. Med. 2021, 53, 894–906. [Google Scholar] [CrossRef]
- Venkatesha, V.A.; Parsels, L.A.; Parsels, J.D.; Zhao, L.; Zabludoff, S.D.; Simeone, D.M.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of pancreatic cancer stem cells to gemcitabine by Chk1 inhibition. Neoplasia 2012, 14, 519–525. [Google Scholar] [CrossRef]
- Di Franco, S.; Parrino, B.; Gaggianesi, M.; Pantina, V.D.; Bianca, P.; Nicotra, A.; Mangiapane, L.R.; Lo Iacono, M.; Ganduscio, G.; Veschi, V.; et al. CHK1 inhibitor sensitizes resistant colorectal cancer stem cells to nortopsentin. iScience 2021, 24, 102664. [Google Scholar] [CrossRef]
- Manic, G.; Signore, M.; Sistigu, A.; Russo, G.; Corradi, F.; Siteni, S.; Musella, M.; Vitale, S.; De Angelis, M.L.; Pallocca, M.; et al. CHK1-targeted therapy to deplete DNA replication-stressed, p53-deficient, hyperdiploid colorectal cancer stem cells. Gut 2018, 67, 903–917. [Google Scholar] [CrossRef]
- Wang, X.; Ma, Z.; Xiao, Z.; Liu, H.; Dou, Z.; Feng, X.; Shi, H. Chk1 knockdown confers radiosensitization in prostate cancer stem cells. Oncol. Rep. 2012, 28, 2247–2254. [Google Scholar] [CrossRef]
- Bartucci, M.; Svensson, S.; Romania, P.; Dattilo, R.; Patrizii, M.; Signore, M.; Navarra, S.; Lotti, F.; Biffoni, M.; Pilozzi, E.; et al. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2012, 19, 768–778. [Google Scholar] [CrossRef]
- Cavelier, C.; Didier, C.; Prade, N.; Mansat-De Mas, V.; Manenti, S.; Recher, C.; Demur, C.; Ducommun, B. Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: Potential importance for checkpoint targeting therapy. Cancer Res. 2009, 69, 8652–8661. [Google Scholar] [CrossRef]
- Wei, N.; Chao-Yang, G.; Wen-Ming, Z.; Ze-Yuan, L.; Yong-Qiang, S.; Shun-Bai, Z.; Kai, Z.; Yan-Chao, M.; Hai-Hong, Z. A ubiquitin-related gene signature for predicting prognosis and constructing molecular subtypes in osteosarcoma. Front. Pharmacol. 2022, 13, 904448. [Google Scholar] [CrossRef]
- Huang, D.; Liu, C.; Sun, X.; Sun, X.; Qu, Y.; Tang, Y.; Li, G.; Tong, T. CRL4DCAF8 and USP11 oppositely regulate the stability of myeloid leukemia factors (MLFs). Biochem. Biophys. Res. Commun. 2020, 529, 127–132. [Google Scholar] [CrossRef]
- Lau, E.; Sedy, J.; Sander, C.; Shaw, M.A.; Feng, Y.; Scortegagna, M.; Claps, G.; Robinson, S.; Cheng, P.; Srivas, R.; et al. Transcriptional repression of IFNβ1 by ATF2 confers melanoma resistance to therapy. Oncogene 2015, 34, 5739–5748. [Google Scholar] [CrossRef]
- Kim, E.S.; Sohn, Y.W.; Moon, A. TGF-β-induced transcriptional activation of MMP-2 is mediated by activating transcription factor (ATF)2 in human breast epithelial cells. Cancer Lett. 2007, 252, 147–156. [Google Scholar] [CrossRef]
- Sun, J.; Dong, Z.; Chang, Z.; Liu, H.; Jiang, Q.; Zhang, D.; Lu, S.; Jia, X.; Wu, D.; Ge, A.; et al. MARCH6 promotes hepatocellular carcinoma development through up-regulation of ATF2. BMC Cancer 2021, 21. [Google Scholar] [CrossRef]
- Han, F.; Huang, D.; Huang, X.; Wang, W.; Yang, S.; Chen, S. Exosomal microRNA-26b-5p down-regulates ATF2 to enhance radiosensitivity of lung adenocarcinoma cells. J. Cell. Mol. Med. 2020, 24, 7730–7742. [Google Scholar] [CrossRef]
- Ma, J.; Chang, K.; Peng, J.; Shi, Q.; Gan, H.; Gao, K.; Feng, K.; Xu, F.; Zhang, H.; Dai, B.; et al. SPOP promotes ATF2 ubiquitination and degradation to suppress prostate cancer progression. J. Exp. Clin. Cancer Res. 2018, 37. [Google Scholar] [CrossRef]
- Ferrandina, G.; Bonanno, G.; Pierelli, L.; Perillo, A.; Procoli, A.; Mariotti, A.; Corallo, M.; Martinelli, E.; Rutella, S.; Paglia, A.; et al. Expression of CD133-1 and CD133-2 in ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 506–514. [Google Scholar] [CrossRef]
- Florek, M.; Haase, M.; Marzesco, A.M.; Freund, D.; Ehninger, G.; Huttner, W.B.; Corbeil, D. Prominin-1/CD133, a neural and hematopoietic stem cell marker, is expressed in adult human differentiated cells and certain types of kidney cancer. Cell Tissue Res. 2005, 319, 15–26. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Sung, P.H.; Wen, J.; Bang, S.; Park, S.; Si, Y.S. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer 2009, 125, 2323–2331. [Google Scholar] [CrossRef]
- Bhatavdekar, J.M.; Patel, D.D.; Chikhlikar, P.R.; Trivedi, T.I.; Gosalia, N.M.; Ghosh, N.; Shah, N.G.; Vora, H.H.; Suthar, T.P. Overexpression of CD44: A useful independent predictor of prognosis in patients with colorectal carcinomas. Ann. Surg. Oncol. 1998, 5, 495–501. [Google Scholar] [CrossRef]
- Rall, C.J.; Rustgi, A.K. CD44 isoform expression in primary and metastatic pancreatic adenocarcinoma. Cancer Res. 1995, 55, 1831–1835. [Google Scholar] [PubMed]
- Haraguchi, N.; Ohkuma, M.; Sakashita, H.; Matsuzaki, S.; Tanaka, F.; Mimori, K.; Kamohara, Y.; Inoue, H.; Mori, M. CD133+CD44+ population efficiently enriches colon cancer initiating cells. Ann. Surg. Oncol. 2008, 15, 2927–2933. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, E.; Castelli, G.; Testa, U. Pancreatic Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Biomedicines 2017, 5, 65. [Google Scholar] [CrossRef]
- Durko, L.; Wlodarski, W.; Stasikowska-Kanicka, O.; Wagrowska-Danilewicz, M.; Danilewicz, M.; Hogendorf, P.; Strzelczyk, J.; Malecka-Panas, E. Expression and Clinical Significance of Cancer Stem Cell Markers CD24, CD44, and CD133 in Pancreatic Ductal Adenocarcinoma and Chronic Pancreatitis. Dis. Markers 2017, 2017, 3276806. [Google Scholar] [CrossRef]
- Sarkar, F.H.; Li, Y.; Wang, Z.; Kong, D. Pancreatic cancer stem cells and EMT in drug resistance and metastasis. Minerva Chir. 2009, 64, 489–500. [Google Scholar]
- Immervoll, H.; Hoem, D.; Steffensen, O.J.; Miletic, H.; Molven, A. Visualization of CD44 and CD133 in normal pancreas and pancreatic ductal adenocarcinomas: Non-overlapping membrane expression in cell populations positive for both markers. J. Histochem. Cytochem. 2011, 59, 441–455. [Google Scholar] [CrossRef]
- Poruk, K.E.; Blackford, A.L.; Weiss, M.J.; Cameron, J.L.; He, J.; Goggins, M.; Rasheed, Z.A.; Wolfgang, C.L.; Wood, L.D. Circulating tumor cells expressing markers of tumor-initiating cells predict poor survival and cancer recurrence in patients with pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2017, 23, 2681–2690. [Google Scholar] [CrossRef]
- Seiter, S.; Arch, R.; Reber, S.; Komitowski, D.; Hofmann, M.; Ponta, H.; Herrlich, P.; Matzku, S.; Zöller, M. Prevention of tumor metastasis formation by anti-variant CD44. J. Exp. Med. 1993, 177, 443–455. [Google Scholar] [CrossRef]
- Maeda, S.; Shinchi, H.; Kurahara, H.; Mataki, Y.; Maemura, K.; Sato, M.; Natsugoe, S.; Aikou, T.; Takao, S. CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. Br. J. Cancer 2008, 98, 1389–1397. [Google Scholar] [CrossRef]
- Chen, K.; Li, Z.; Jiang, P.; Zhang, X.; Zhang, Y.; Jiang, Y.; He, Y.; Li, X. Co-expression of CD133, CD44v6 and human tissue factor is associated with metastasis and poor prognosis in pancreatic carcinoma. Oncol. Rep. 2014, 32, 755–763. [Google Scholar] [CrossRef]
- Bao, B.; Azmi, A.S.; Aboukameel, A.; Ahmad, A.; Bolling-Fischer, A.; Sethi, S.; Ali, S.; Li, Y.; Kong, D.; Banerjee, S.; et al. Pancreatic cancer stem-like cells display aggressive behavior mediated via activation of FoxQ1. J. Biol. Chem. 2014, 289, 14520–14533. [Google Scholar] [CrossRef]
- Hou, Y.C.; Chao, Y.J.; Tung, H.L.; Wang, H.C.; Shan, Y.S. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer 2014, 120, 2766–2777. [Google Scholar] [CrossRef]
- Maeda, S.; Qiang, D.; Shinchi, H.; Kurahara, H.; Mataki, Y.; Maemura, K.; Natsugoe, S.; Takao, S. CD44 and CD133 Expressions in Primary Tumor Cells Correlate to Survival of Pancreatic Cancer Patients. Open Surg. Oncol. J. 2009, 1, 1–7. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Y.; Niu, M.; Shi, Y.; Liu, H.; Yang, D.; Li, F.; Lu, Y.; Bo, Y.; Zhang, R.; et al. Whole-Transcriptome Analysis of CD133+CD144+ Cancer Stem Cells Derived from Human Laryngeal Squamous Cell Carcinoma Cells. Cell. Physiol. Biochem. 2018, 47, 1696–1710. [Google Scholar] [CrossRef]
- Fang, Y.; Xiang, J.; Chen, Z.; Gu, X.; Li, Z.; Tang, F.; Zhou, Z. miRNA expression profile of colon cancer stem cells compared to non-stem cells using the SW1116 cell line. Oncol. Rep. 2012, 28, 2115–2124. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, Y.; Xu, L.; Xu, H.; Xu, Y.; Yan, N. Long non-coding RNA profile revealed by microarray indicates that lncCUEDC1 serves a negative regulatory role in breast cancer stem cells. Int. J. Oncol. 2020, 56, 807–820. [Google Scholar] [CrossRef]
- Wang, Y.; He, L.; Du, Y.; Zhu, P.; Huang, G.; Luo, J.; Yan, X.; Ye, B.; Li, C.; Xia, P.; et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell 2015, 16, 413–425. [Google Scholar] [CrossRef]
- Qian, D.; Qian, C.; Ye, B.; Xu, M.; Wu, D.; Li, J.; Li, D.; Yu, B.; Tao, Y. Development and Validation of a Novel Stemness-Index-Related Long Noncoding RNA Signature for Breast Cancer Based on Weighted Gene Co-Expression Network Analysis. Front. Genet. 2022, 13, 760514. [Google Scholar] [CrossRef]
- Li, H.; Li, H.; Hao, Y.; Jiao, Y.; Li, Z.; Yue, H.; Xu, Z.; Wang, S.; Cao, Y.; Zhao, J. Differential long non-coding RNA and mRNA expression in differentiated human glioblastoma stem cells. Mol. Med. Rep. 2016, 14, 2067–2076. [Google Scholar] [CrossRef]
- Tao, T.; Yuan, S.; Liu, J.; Shi, D.; Peng, M.; Li, C.; Wu, S. Cancer stem cell-specific expression profiles reveal emerging bladder cancer biomarkers and identify circrna_103809 as an important regulator in bladder cancer. Aging 2020, 12, 3354–3370. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eptaminitaki, G.C.; Zaravinos, A.; Stellas, D.; Panagopoulou, M.; Karaliota, S.; Baltsavia, I.; Iliopoulos, I.; Chatzaki, E.; Iliopoulos, D.; Baritaki, S. Genome-Wide Analysis of lncRNA-mRNA Co-Expression Networks in CD133+/CD44+ Stem-like PDAC Cells. Cancers 2023, 15, 1053. https://doi.org/10.3390/cancers15041053
Eptaminitaki GC, Zaravinos A, Stellas D, Panagopoulou M, Karaliota S, Baltsavia I, Iliopoulos I, Chatzaki E, Iliopoulos D, Baritaki S. Genome-Wide Analysis of lncRNA-mRNA Co-Expression Networks in CD133+/CD44+ Stem-like PDAC Cells. Cancers. 2023; 15(4):1053. https://doi.org/10.3390/cancers15041053
Chicago/Turabian StyleEptaminitaki, Giasemi C., Apostolos Zaravinos, Dimitris Stellas, Maria Panagopoulou, Sevasti Karaliota, Ismini Baltsavia, Ioannis Iliopoulos, Ekaterini Chatzaki, Dimitrios Iliopoulos, and Stavroula Baritaki. 2023. "Genome-Wide Analysis of lncRNA-mRNA Co-Expression Networks in CD133+/CD44+ Stem-like PDAC Cells" Cancers 15, no. 4: 1053. https://doi.org/10.3390/cancers15041053
APA StyleEptaminitaki, G. C., Zaravinos, A., Stellas, D., Panagopoulou, M., Karaliota, S., Baltsavia, I., Iliopoulos, I., Chatzaki, E., Iliopoulos, D., & Baritaki, S. (2023). Genome-Wide Analysis of lncRNA-mRNA Co-Expression Networks in CD133+/CD44+ Stem-like PDAC Cells. Cancers, 15(4), 1053. https://doi.org/10.3390/cancers15041053