


Syngeneic N1-S1 Orthotopic Hepatocellular Carcinoma in Sprague Dawley Rat for the Development of Interventional Oncology-Based Immunotherapy: Survival Assay and Tumor Immune Microenvironment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Cell Line
2.2. N1-S1 Cell Implantation in Sprague Dawley Rats
2.3. Monitoring Tumor Growth
2.4. Flow Cytometry Analysis
2.5. Histology Analysis
2.6. Statistical Analysis
3. Results and Discussion
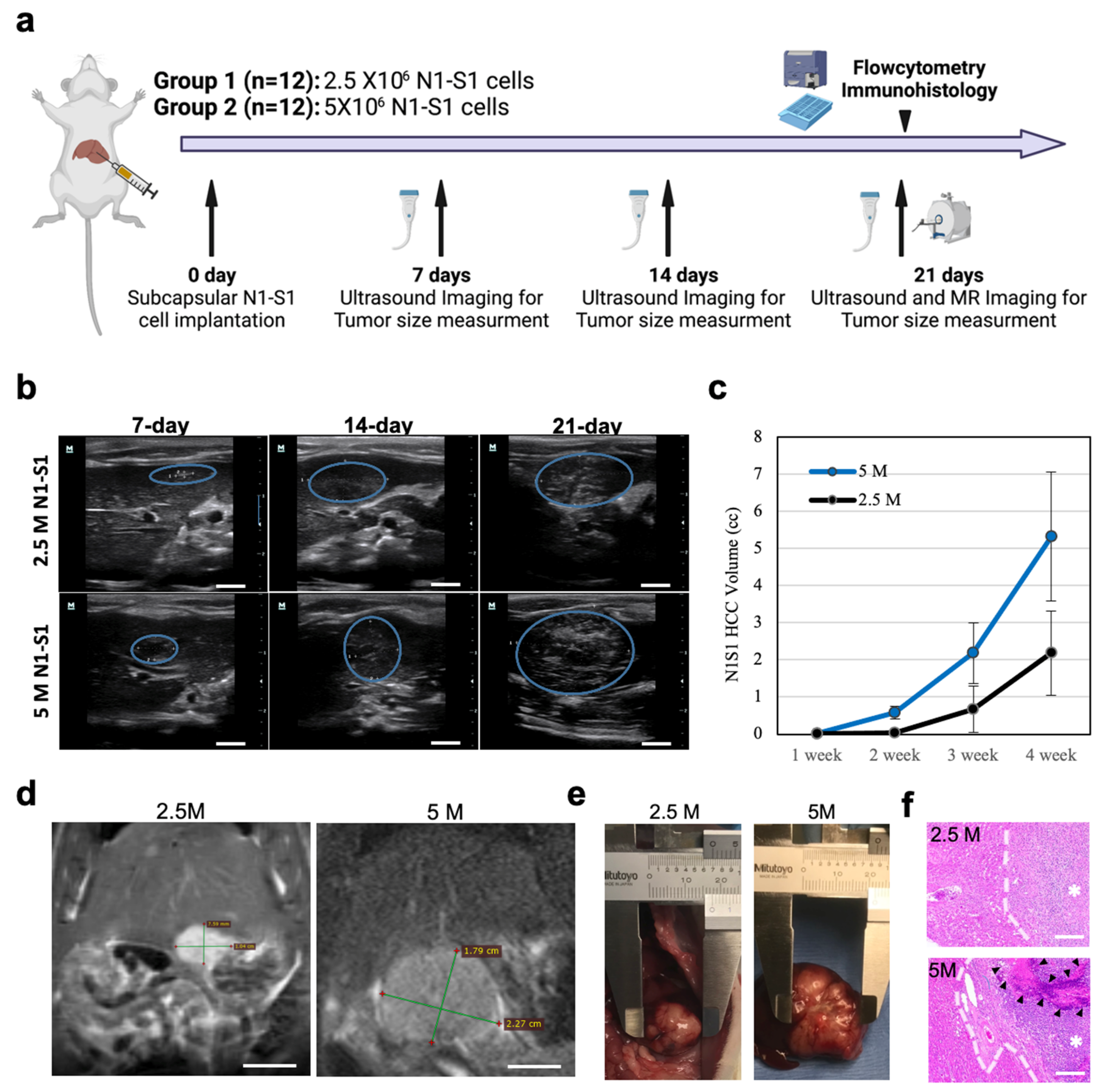
3.1. N1-S1 Cell Implantation and HCC growth in Sprague Dawley Rats
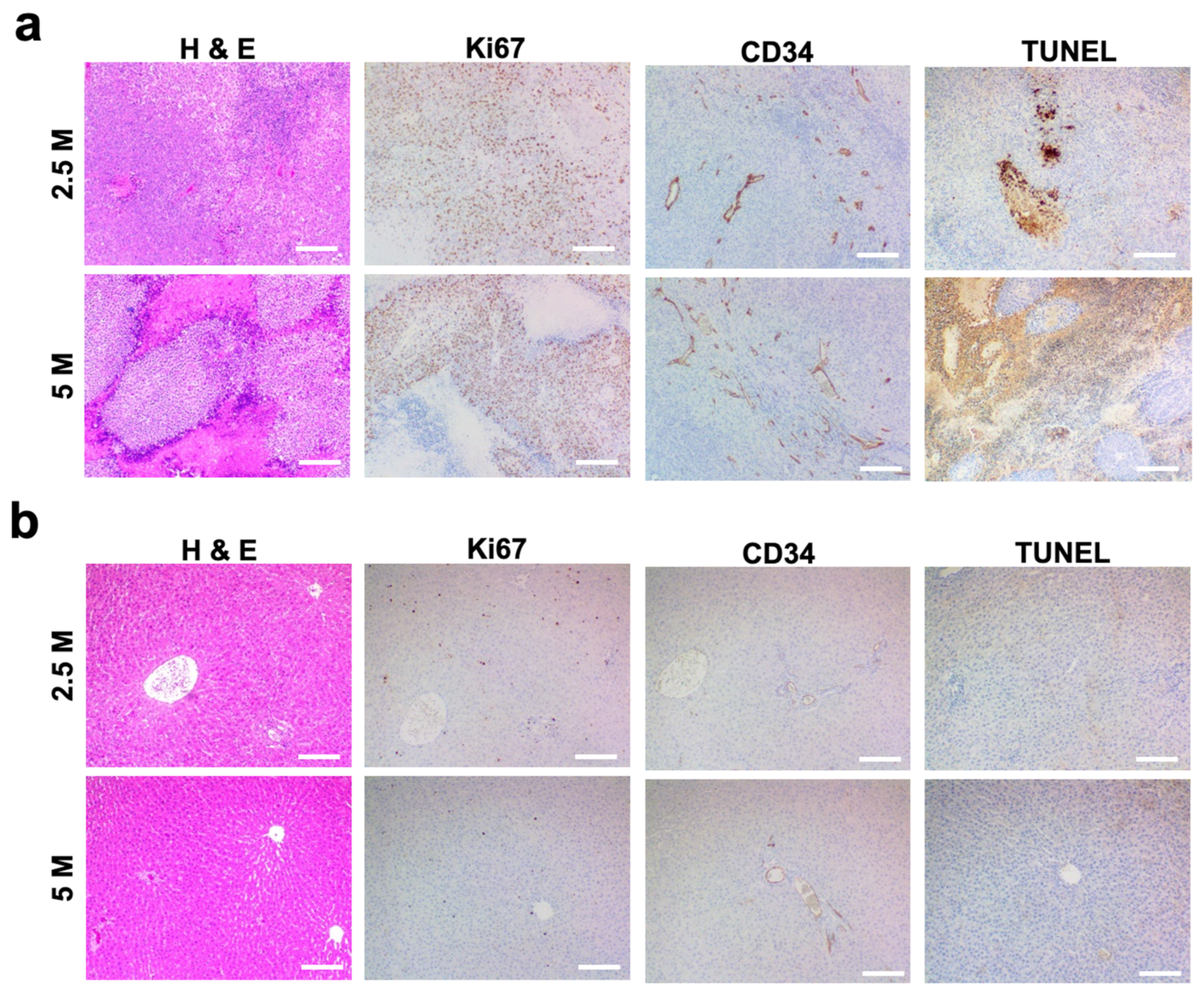
3.2. Histological Analysis of N1-S1 HCC Rats
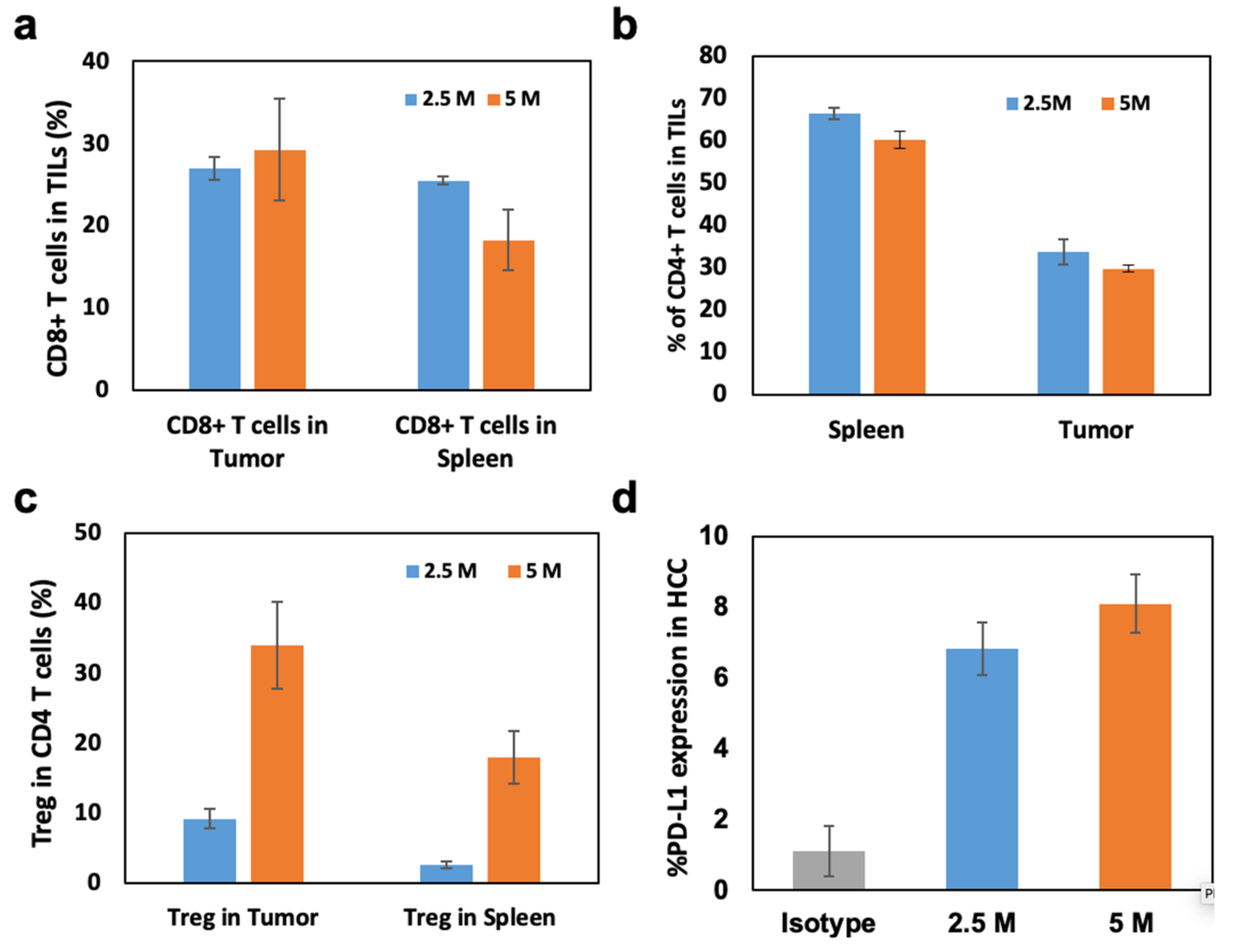
3.3. Immunological Analysis of N1-S1 HCC Rats
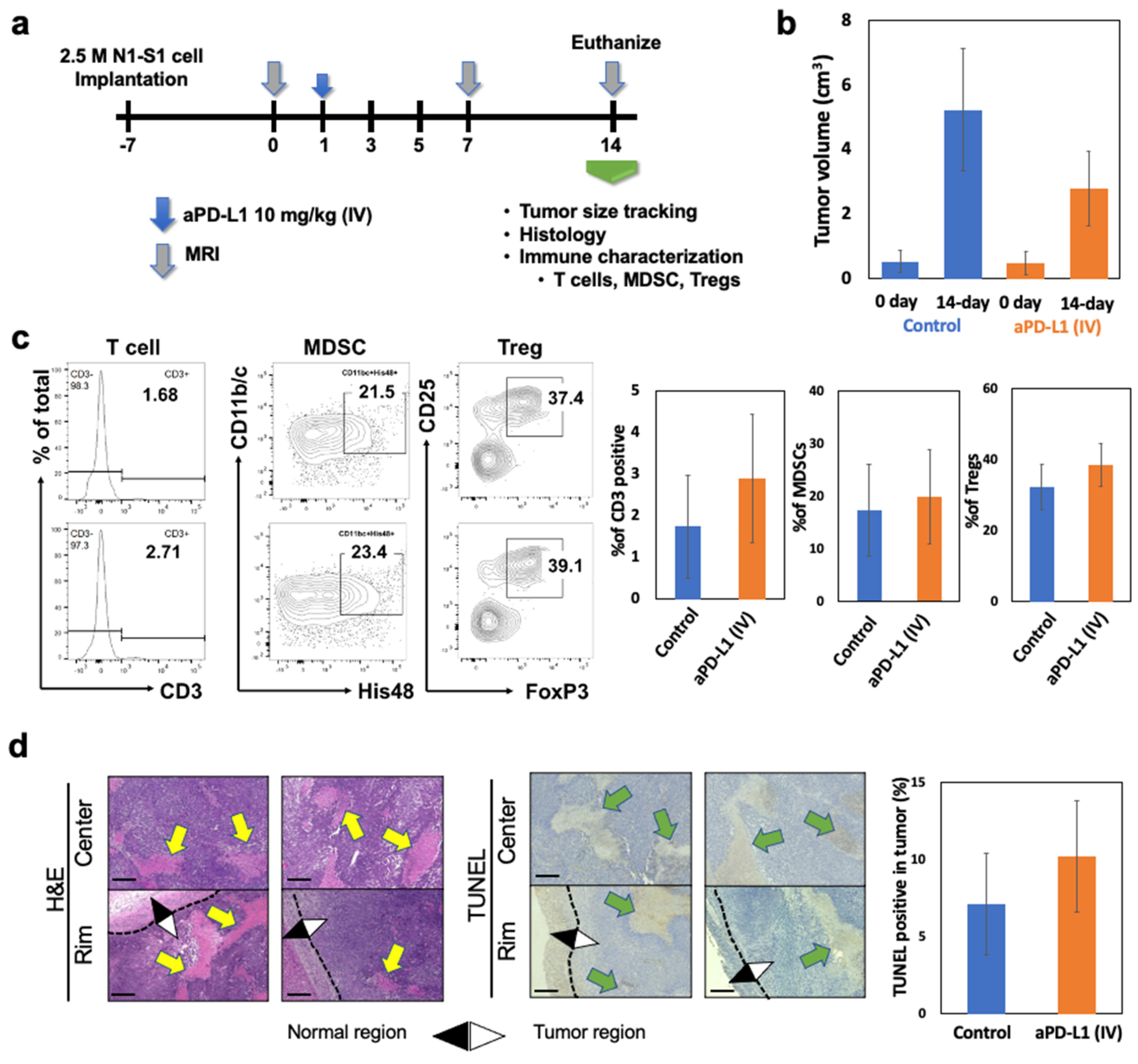
3.4. Tumor Microenvironment and HCC Tumor Response to aPD-L1 Immunotherapy in N1-S1 HCC Rat Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gardini, A.C.; Marisi, G.; Canale, M.; Foschi, F.G.; Donati, G.; Ercolani, G.; Valgiusti, M.; Passardi, A.; Frassineti, G.L.; Scarpi, E. Radiofrequency ablation of hepatocellular carcinoma: A meta-analysis of overall survival and recurrence-free survival. Oncotargets Ther. 2018, 11, 6555–6567. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Kirkwood, A.; Roughton, M.; Beare, S.; Tsochatzis, E.; Yu, D.; Davies, N.; Williams, E.; Pereira, S.; Hochhauser, D.; et al. A randomised phase ii/iii trial of 3-weekly cisplatin-based sequential transarterial chemoembolisation vs embolisation alone for hepatocellular carcinoma. Br. J. Cancer 2013, 108, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Marelli, L.; Stigliano, R.; Triantos, C.; Senzolo, M.; Cholongitas, E.; Davies, N.; Tibballs, J.; Meyer, T.; Patch, D.W.; Burroughs, A.K. Transarterial therapy for hepatocellular carcinoma: Which technique is more effective? A systematic review of cohort and randomized studies. Cardiovasc. Interv. Radiol. 2007, 30, 6–25. [Google Scholar] [CrossRef] [PubMed]
- Biolato, M.; Marrone, G.; Racco, S.; Di Stasi, C.; Miele, L.; Gasbarrini, G.; Landolfi, R.; Grieco, A. Transarterial Chemoembolization (Tace) for Unresectable HCC: A New Life Begins? In Eur. Rev. Med. Pharmacol. Sci.; 2010; 14, pp. 356–362. Available online: http://www.ncbi.nlm.nih.gov/pubmed/20496548 (accessed on 1 May 2022).
- Llovet, J.M.; De Baere, T.; Kulik, L.; Haber, P.K.; Greten, T.F.; Meyer, T.; Lencioni, R. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 293–313. [Google Scholar] [CrossRef]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2021, 19, 151–172. [Google Scholar] [CrossRef]
- Leuchte, K.; Staib, E.; Thelen, M.; Gödel, P.; Lechner, A.; Zentis, P.; Garcia-Marquez, M.; Waldschmidt, D.; Datta, R.R.; Wahba, R.; et al. Microwave ablation enhances tumor-specific immune response in patients with hepatocellular carcinoma. Cancer Immunol. Immunother. 2021, 70, 893–907. [Google Scholar] [CrossRef]
- Leppelmann, K.S.; Mooradian, M.J.; Ganguli, S.; Uppot, R.N.; Yamada, K.; Irani, Z.; Wehrenberg-Klee, E.P.; Zubiri, L.; Reynolds, K.L.; Arellano, R.S.; et al. Thermal ablation, embolization, and selective internal radiation therapy combined with checkpoint inhibitor cancer immunotherapy: Safety analysis. J. Vasc. Interv. Radiol. 2021, 32, 187–195. [Google Scholar] [CrossRef]
- Kim, D.-H. Combination of interventional oncology local therapies and immunotherapy for the treatment of hepatocellular carcinoma. J. Liver Cancer 2022, 22, 93–102. [Google Scholar] [CrossRef]
- Choi, B.; Choi, H.; Kim, H.; Choi, A.; Kwon, S.-W.; Mouli, S.K.; Lewandowski, R.J.; Kim, D.-H. Z-domain protein nano-bio interfaced mri visible anti-program death ligand-1 nanoconjugates for enhanced local immune checkpoint inhibitor immunotherapy. Nano Today 2022, 45, 101552. [Google Scholar] [CrossRef]
- Lu, J.Q.; Liu, X.S.; Liao, Y.P.; Salazar, F.; Sun, B.B.; Jiang, W.; Chang, C.H.; Jiang, J.H.; Wang, X.; Wu, A.M.; et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat. Commun. 2017, 8, 1811. [Google Scholar] [CrossRef]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (checkmate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Choi, B.; Kim, D.-H. Multifunctional nanocarriers-mediated synergistic combination of immune checkpoint inhibitor cancer immunotherapy and interventional oncology therapy. Adv. NanoBiomed Res. 2021, 1, 2100010. [Google Scholar] [CrossRef]
- Park, W.; Cho, S.; Ji, J.; Lewandowski, R.J.; Larson, A.C.; Kim, D.-H. Development and validation of sorafenib-eluting microspheres to enhance therapeutic efficacy of transcatheter arterial chemoembolization in a rat model of hepatocellular carcinoma. Radiol. Imaging Cancer 2021, 3, e200006. [Google Scholar] [CrossRef]
- Choi, B.; Choi, H.; Yu, B.; Kim, D.-H. Synergistic local combination of radiation and anti-programmed death ligand 1 immunotherapy using radiation-responsive splintery metallic nanocarriers. ACS Nano 2020, 14, 13115–13126. [Google Scholar] [CrossRef]
- Kim, D.-H.; Chen, J.; Omary, R.A.; Larson, A.C. Mri visible drug eluting magnetic microspheres for transcatheter intra-arterial delivery to liver tumors. Theranostics 2015, 5, 477–488. [Google Scholar] [CrossRef]
- Morris, H.P. Studies on the development, biochemistry, and biology of experimental hepatomas. Adv. Cancer Res. 1965, 9, 227–302. [Google Scholar]
- Novikoff, A.B. A transplantable rat liver tumor induced by 4-dimethylaminoazobenzene. Cancer Res. 1957, 17, 1010–1027. [Google Scholar]
- Gade, T.P.; Hunt, S.J.; Harrison, N.; Nadolski, G.J.; Weber, C.; Pickup, S.; Furth, E.E.; Schnall, M.D.; Soulen, M.C.; Simon, M.C. Segmental transarterial embolization in a translational rat model of hepatocellular carcinoma. J. Vasc. Interv. Radiol. 2015, 26, 1229–1237. [Google Scholar] [CrossRef]
- Cho, H.R.; Choi, J.W.; Kim, H.C.; Song, Y.S.; Kim, G.M.; Son, K.R.; Chung, J.W. Sprague-dawley rats bearing mca-rh7777 cells for study of hepatoma and transarterial chemoembolization. Anticancer. Res. 2013, 33, 223–230. [Google Scholar] [PubMed]
- Choi, J.W.; Kim, J.H.; Kim, H.-C.; Choi, W.S.; Baek, S.Y.; Lee, K.; Chung, J.W. Comparison of tumor vascularity and hemodynamics in three rat hepatoma models. Abdom. Radiol. 2016, 41, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.M.; Callstrom, M.R.; Knudsen, B.; Anderson, J.L.; Carter, R.E.; Grande, J.P.; Roberts, L.R.; Woodrum, D.A. Development and preliminary testing of a translational model of hepatocellular carcinoma for mr imaging and interventional oncologic investigations. J. Vasc. Interv. Radiol. 2012, 23, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Garin, E.; Denizot, B.; Roux, J.; Noiret, N.; Lepareur, N.; Moreau, M.; Mesba, H.; Laurent, J.F.; Herry, J.Y.; Bourguet, P. Description and technical pitfalls of a hepatoma model and of intra-arterial injection of radiolabelled lipiodol in the rat. Lab. Anim. 2005, 39, 314–320. [Google Scholar] [CrossRef]
- Kim, D.-H.; Rozhkova, E.A.; Ulasov, I.; Bader, S.D.; Rajh, T.; Lesniak, M.S.; Novosad, V. Biofunctionalized magnetic-vortex microdiscs for targeted cancer-cell destruction. Nat. Mater. 2010, 9, 165–171. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. Pd-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of tim-3 and pd-1 expression is associated with tumor antigen-specific cd8(+) t cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Targeting the pd-1/b7-h1(pd-l1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012, 24, 207–212. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-pd-l1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Sangro, B.; Sarobe, P.; Hervás-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef]
- Pe, J.; Choi, B.; Choi, H.; Kwon, S.W.; Kim, D.-H. Preclinical therapeutic evaluation of lenvatinib-eluting microspheres for transcatheter arterial chemoembolization of hepatocellular carcinoma. CardioVascular Interv. Radiol. 2022, 45, 1834–1841. [Google Scholar] [CrossRef]
- Sim, T.; Choi, B.; Kwon, S.W.; Kim, K.-S.; Choi, H.; Ross, A.; Kim, D.-H. Magneto-activation and magnetic resonance imaging of natural killer cells labeled with magnetic nanocomplexes for the treatment of solid tumors. ACS Nano 2021, 15, 12780–12793. [Google Scholar] [CrossRef]
- Cho, S.; Min, N.G.; Park, W.; Kim, S.H.; Kim, D.H. Janus microcarriers for magnetic field-controlled combination chemotherapy of hepatocellular carcinoma. Adv. Funct. Mater. 2019, 29, 1901384. [Google Scholar] [CrossRef]
- Kudo, M. Immune checkpoint blockade in hepatocellular carcinoma. Liver Cancer 2015, 4, 201–207. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Melero, I.; Crocenzi, T.S.; Welling, T.H.; Yau, T.C.; Yeo, W.N.; Chopra, A.; Grosso, J.; Lang, L.X.; Anderson, J.; et al. Phase i/ii safety and antitumor activity of nivolumab in patients with advanced hepatocellular carcinoma (hcc): Ca209-040. J. Clin. Oncol. 2015, 33, LBA101. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Mazières, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-pd-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (checkmate 063): A phase 2, single-arm trial. Lancet Oncol. 2015, 16, 257–265. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in keynote-240: A randomized, double-blind, phase iii trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.H.; Harding, J.J.; Merle, P.; et al. Checkmate 459: A randomized, multi-center phase iii study of nivolumab (nivo) vs sorafenib (sor) as first-line (1l) treatment in patients (pts) with advanced hepatocellular carcinoma (ahcc). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Prieto, J.; Melero, I.; Sangro, B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 681. [Google Scholar] [CrossRef]
- Desrichard, A.; Snyder, A.; Chan, T.A. Cancer neoantigens and applications for immunotherapy. Clin. Cancer Res. 2016, 22, 807–812. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; June, C.H.; Haas, N.; Vapiwala, N. Radiation and immunotherapy: A synergistic combination. J. Clin. Investig. 2013, 123, 2756–2763. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Jain, R.K.; Duda, D.G. The current landscape of immune checkpoint blockade in hepatocellular carcinoma: A review. JAMA Oncol. 2020, 7, 113–123. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, B.; Pe, J.; Yu, B.; Kim, D.-H. Syngeneic N1-S1 Orthotopic Hepatocellular Carcinoma in Sprague Dawley Rat for the Development of Interventional Oncology-Based Immunotherapy: Survival Assay and Tumor Immune Microenvironment. Cancers 2023, 15, 913. https://doi.org/10.3390/cancers15030913
Choi B, Pe J, Yu B, Kim D-H. Syngeneic N1-S1 Orthotopic Hepatocellular Carcinoma in Sprague Dawley Rat for the Development of Interventional Oncology-Based Immunotherapy: Survival Assay and Tumor Immune Microenvironment. Cancers. 2023; 15(3):913. https://doi.org/10.3390/cancers15030913
Chicago/Turabian StyleChoi, Bongseo, Jason Pe, Bo Yu, and Dong-Hyun Kim. 2023. "Syngeneic N1-S1 Orthotopic Hepatocellular Carcinoma in Sprague Dawley Rat for the Development of Interventional Oncology-Based Immunotherapy: Survival Assay and Tumor Immune Microenvironment" Cancers 15, no. 3: 913. https://doi.org/10.3390/cancers15030913
APA StyleChoi, B., Pe, J., Yu, B., & Kim, D.-H. (2023). Syngeneic N1-S1 Orthotopic Hepatocellular Carcinoma in Sprague Dawley Rat for the Development of Interventional Oncology-Based Immunotherapy: Survival Assay and Tumor Immune Microenvironment. Cancers, 15(3), 913. https://doi.org/10.3390/cancers15030913