



Multi-Fold Computational Analysis to Discover Novel Putative Inhibitors of Isethionate Sulfite-Lyase (Isla) from Bilophila wadsworthia: Combating Colorectal Cancer and Inflammatory Bowel Diseases

 ,
,  ,
,  ,
,  ,
, 

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Crystal Coordinates Selection
2.2. Molecular Docking and ADME Prediction
2.3. Molecular Dynamic Simulations
2.4. Post Dynamic Evaluation
2.5. Assessment of Structural Dynamic Changes
2.6. Principal Component Analysis
2.7. Hydrogen Bonds Analysis
2.8. Binding Free Energy Calculation
2.9. Data Analysis
3. Results and Discussion
3.1. Structure of Isethionate Sulfite-Lyase and Molecular Docking
3.2. ADMET Analysis
3.3. Molecular Dynamic Simulations
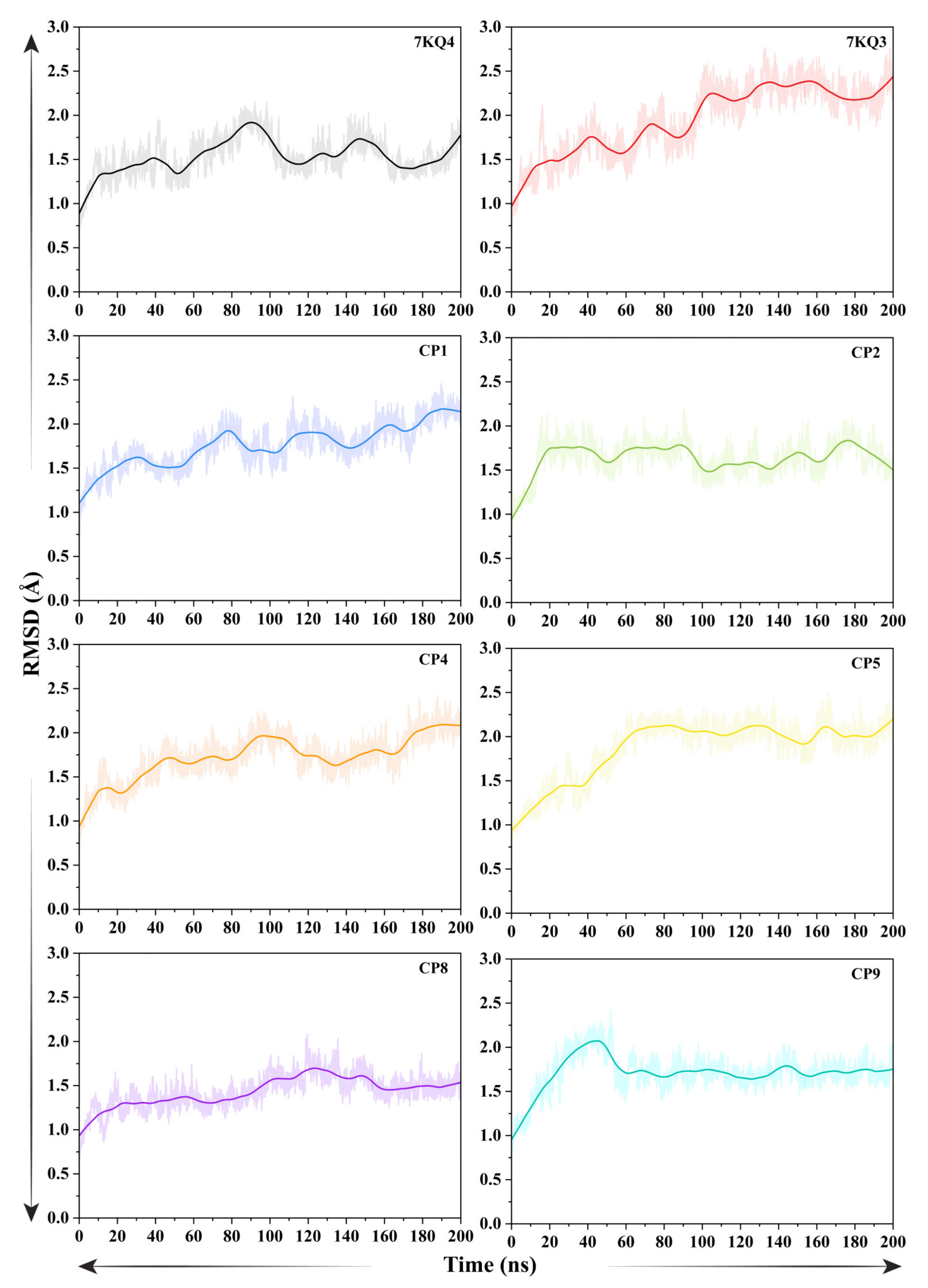
3.3.1. Evaluation of Structural Stability Via Root Mean Square Deviation
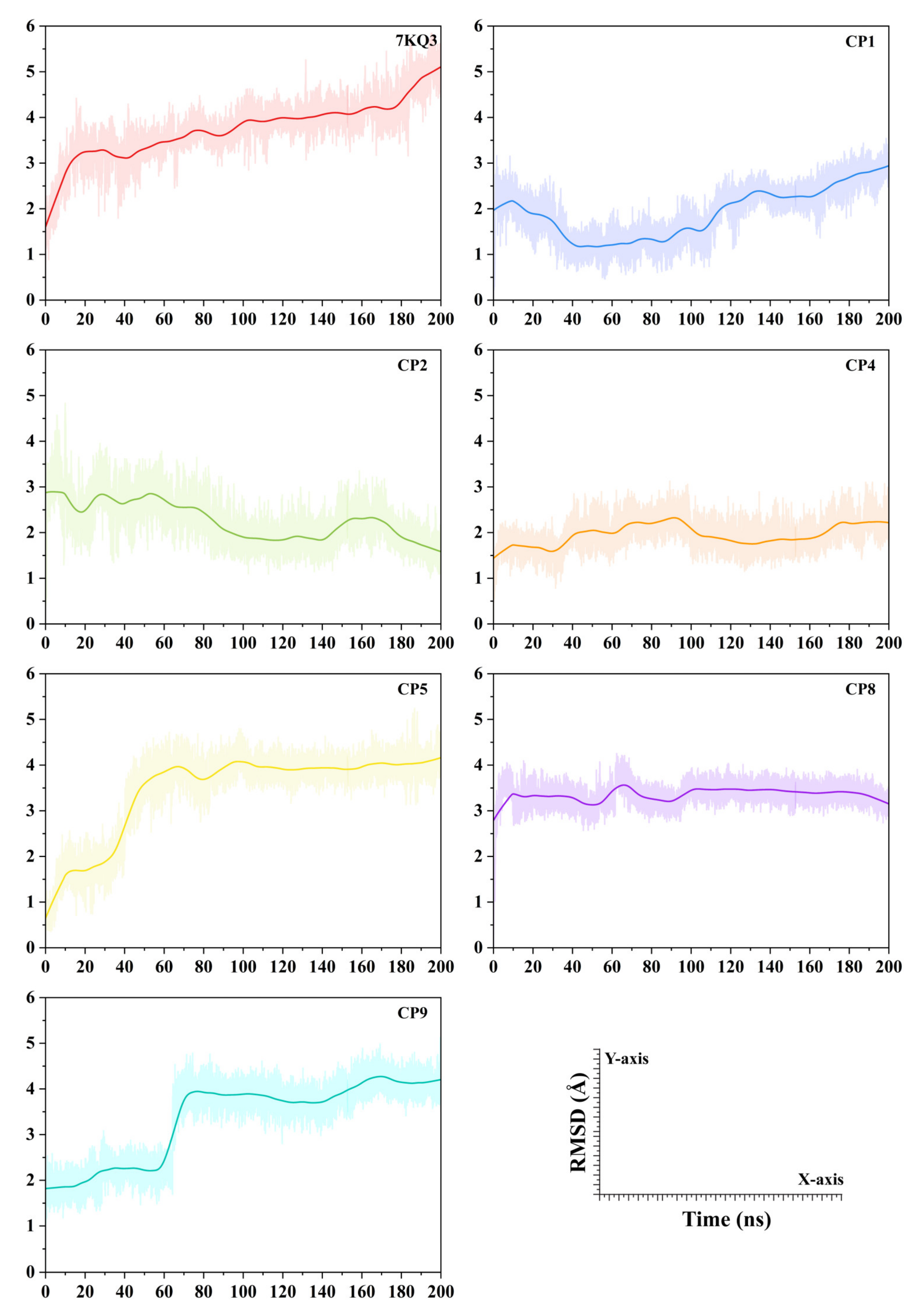
3.3.2. Evaluation of Ligand Stability in the Active Site of Isla
3.3.3. Thermal Stability Analysis of the Protein
3.3.4. Residual Flexibility of Isla
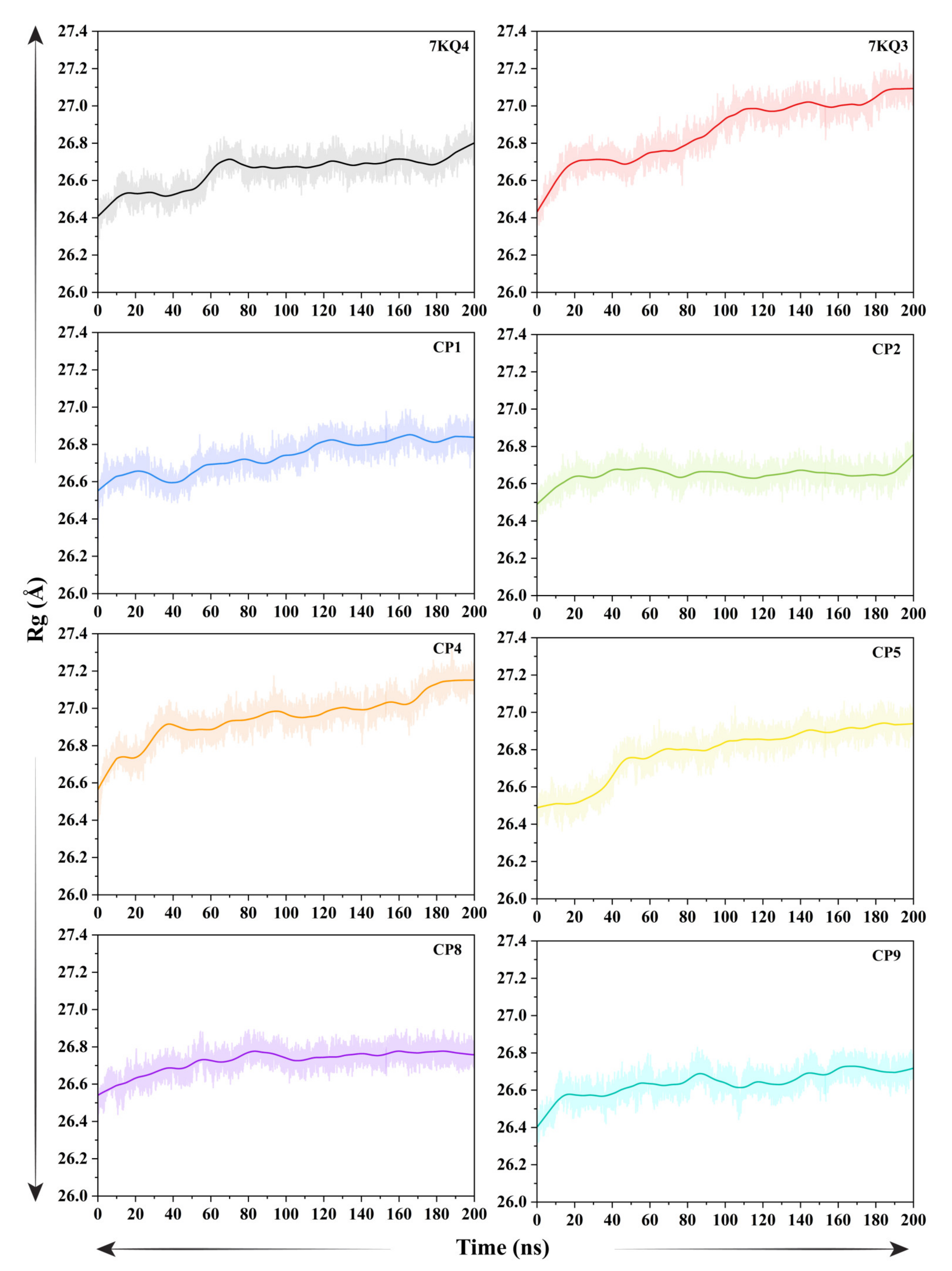
3.3.5. Analysis of Protein Compactness
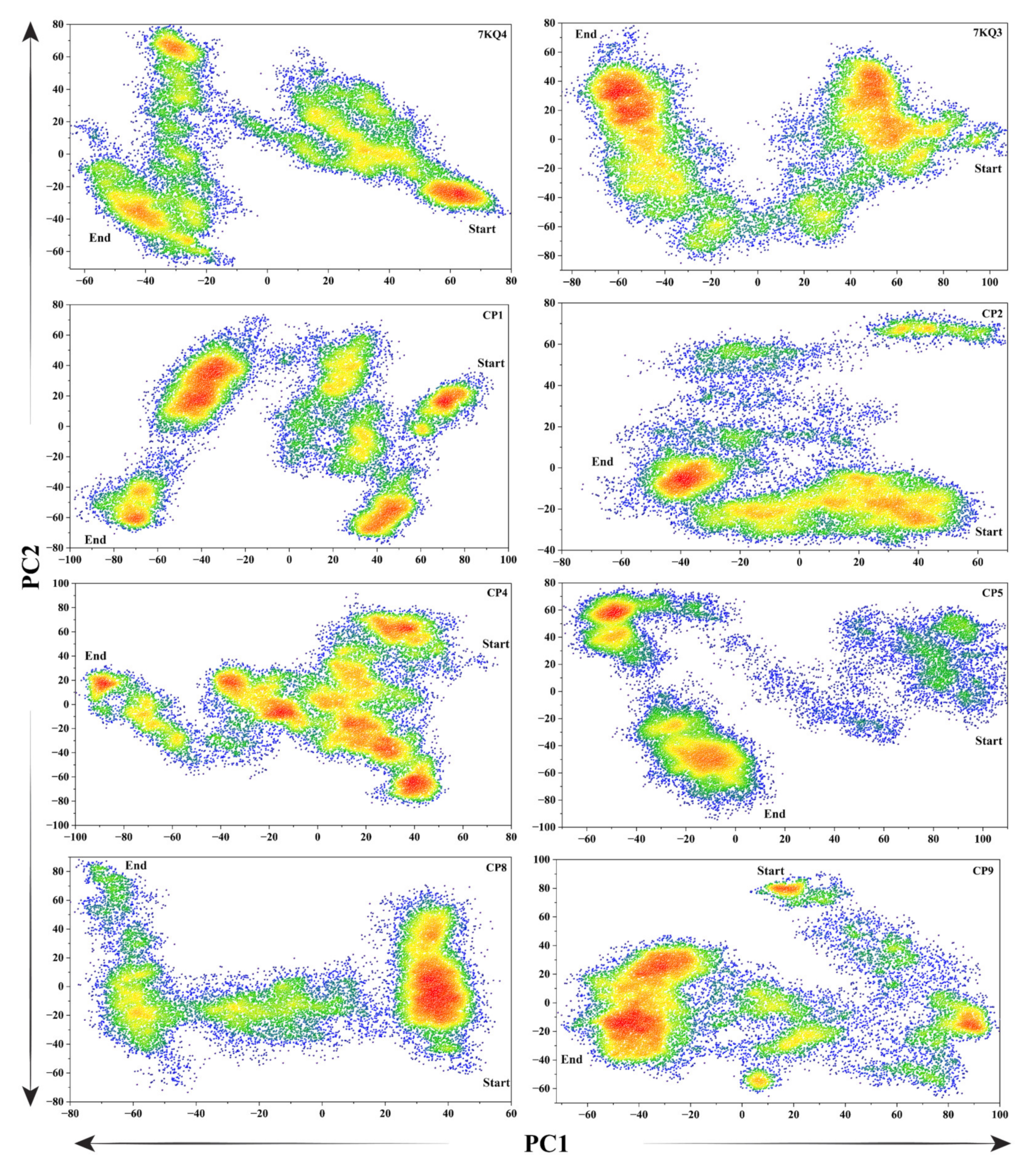
3.3.6. Protein Principal Movements
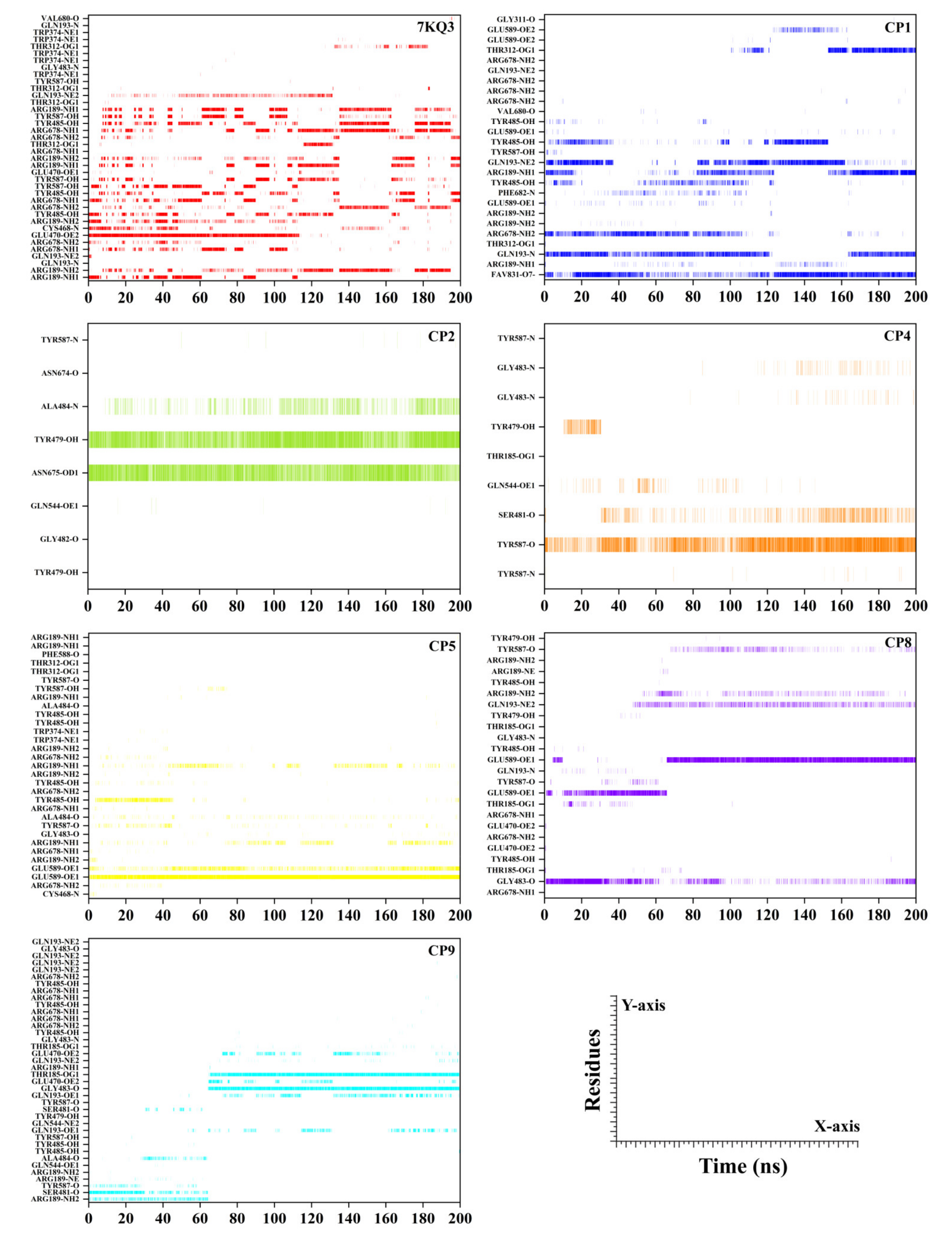
3.3.7. Hydrogen Bond Analysis
3.3.8. Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernard, D.; Verschraegen, G.; Claeys, G.; Lauwers, S.; Rosseel, P. Bilophila wadsworthia bacteremia in a patient with gangrenous appendicitis. Clin. Infect. Dis. 1994, 18, 1023–1024. [Google Scholar] [CrossRef] [PubMed]
- Baron, E.J. Bilophila wadsworthia: A unique Gram-negative anaerobic rod. Anaerobe 1997, 3, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.J.; Rosenblatt, J.E.; Gustafson, D.R. Bilophila wadsworthia bacteremia in two patients with hepatic abscesses. J. Clin. Microbiol. 1992, 30, 2502–2503. [Google Scholar] [CrossRef] [PubMed]
- Ijssennagger, N.; Belzer, C.; Hooiveld, G.J.; Dekker, J.; van Mil, S.W.; Muller, M.; Kleerebezem, M.; van der Meer, R. Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc. Natl. Acad. Sci. USA 2015, 112, 10038–10043. [Google Scholar] [CrossRef] [PubMed]
- Ijssennagger, N.; van der Meer, R.; van Mil, S.W. Sulfide as a mucus barrier-breaker in inflammatory bowel disease? Trends Mol. Med. 2016, 22, 190–199. [Google Scholar] [CrossRef]
- Schulz, M.D.; Atay, C.; Heringer, J.; Romrig, F.K.; Schwitalla, S.; Aydin, B.; Ziegler, P.K.; Varga, J.; Reindl, W.; Pommerenke, C.; et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 2014, 514, 508–512. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Warren, R.L.; Freeman, D.J.; Pleasance, S.; Watson, P.; Moore, R.A.; Cochrane, K.; Allen-Vercoe, E.; Holt, R.A. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome 2013, 1, 16. [Google Scholar] [CrossRef]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Bohm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Zackular, J.P.; Rogers, M.A.; Ruffin IV, M.T.; Schloss, P.D. The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev. Res. 2014, 7, 1112–1121. [Google Scholar] [CrossRef]
- O’Keefe, S.J.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef]
- Deplancke, B.; Gaskins, H.R. Hydrogen sulfide induces serum-independent cell cycle entry in nontransformed rat intestinal epithelial cells. FASEB J. 2003, 17, 1310–1312. [Google Scholar] [CrossRef]
- Attene-Ramos, M.S.; Wagner, E.D.; Plewa, M.J.; Gaskins, H.R. Evidence that hydrogen sulfide is a genotoxic agent. Mol. Cancer Res. 2006, 4, 9–14. [Google Scholar] [CrossRef]
- Attene-Ramos, M.S.; Wagner, E.D.; Gaskins, H.R.; Plewa, M.J. Hydrogen sulfide induces direct radical-associated DNA damage. Mol. Cancer Res. 2007, 5, 455–459. [Google Scholar] [CrossRef]
- Dawson, C.D.; Irwin, S.M.; Backman, L.R.F.; Le, C.; Wang, J.X.; Vennelakanti, V.; Yang, Z.; Kulik, H.J.; Drennan, C.L.; Balskus, E.P. Molecular basis of C-S bond cleavage in the glycyl radical enzyme isethionate sulfite-lyase. Cell Chem. Biol. 2021, 28, 1333–1346.e7. [Google Scholar] [CrossRef]
- Peck, S.C.; Denger, K.; Burrichter, A.; Irwin, S.M.; Balskus, E.P.; Schleheck, D. A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila wadsworthia. Proc. Natl. Acad. Sci. USA 2019, 116, 3171–3176. [Google Scholar] [CrossRef] [PubMed]
- Carbonero, F.; Benefiel, A.C.; Alizadeh-Ghamsari, A.H.; Gaskins, H.R. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 2012, 3, 448. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Lin, H.C. Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms 2015, 3, 866–889. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.A.; Macfarlane, G.T. Enumeration of amino acid fermenting bacteria in the human large intestine: Effects of pH and starch on peptide metabolism and dissimilation of amino acids. FEMS Microbiol. Ecol. 1998, 25, 355–368. [Google Scholar] [CrossRef]
- Backman, L.R.; Huang, Y.Y.; Andorfer, M.C.; Gold, B.; Raines, R.T.; Balskus, E.P.; Drennan, C.L. Molecular basis for catabolism of the abundant metabolite trans-4-hydroxy-l-proline by a microbial glycyl radical enzyme. eLife 2020, 9, 1609. [Google Scholar] [CrossRef]
- Xing, M.; Wei, Y.; Zhou, Y.; Zhang, J.; Lin, L.; Hu, Y.; Hua, G.; Nanjaraj Urs, A.N.; Liu, D.; Wang, F.; et al. Radical-mediated C-S bond cleavage in C2 sulfonate degradation by anaerobic bacteria. Nat. Commun. 2019, 10, 1609. [Google Scholar] [CrossRef]
- Lourenco, R.; Camilo, M.E. Taurine: A conditionally essential amino acid in humans? An overview in health and disease. Nutr. Hosp. 2002, 17, 262–270. [Google Scholar]
- Ripps, H.; Shen, W. Taurine: A “very essential” amino acid. Mol. Vis. 2012, 18, 2673. [Google Scholar]
- Halim, S.A.; Waqas, M.; Asim, A.; Khan, M.; Khan, A.; Al-Harrasi, A. Discovering novel inhibitors of P2Y(12) receptor using structure-based virtual screening, molecular dynamics simulation and MMPBSA approaches. Comput. Biol. Med. 2022, 147, 105743. [Google Scholar] [CrossRef]
- Khan, A.; Waqas, M.; Khan, M.; Halim, S.A.; Rehman, N.U.; Al-Harrasi, A. Identification of novel prolyl oligopeptidase inhibitors from resin of Boswellia papyrifera (Del.) Hochst. and their mechanism: Virtual and biochemical studies. Int. J. Biol. Macromol. 2022, 213, 751–767. [Google Scholar] [CrossRef]
- Asmari, M.; Waqas, M.; Ibrahim, A.E.; Halim, S.A.; Khan, A.; Al-Harrasi, A.; Watzig, H.; El Deeb, S. Microscale Thermophoresis and Molecular Modelling to Explore the Chelating Drug Transportation in the Milk to Infant. Molecules 2022, 27, 4604. [Google Scholar] [CrossRef]
- Halim, S.A.; Waqas, M.; Khan, A.; Al-Harrasi, A. In silico prediction of novel inhibitors of SARS-CoV-2 main protease through structure-based virtual screening and molecular dynamic simulation. Pharmaceuticals 2021, 14, 896. [Google Scholar] [CrossRef]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE), version 2022.02; Chemical Computing Group ULC: Montreal, QC, Canada, 2022. [Google Scholar]
- Gerber, P.R.; Muller, K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput. Aided Mol. Des. 1995, 9, 251–268. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Case, D.; Aktulga, H.; Belfon, K.; Ben-Shalom, I.; Berryman, J.; Brozell, S.; Cerutti, D.; Cheatham, T.; Cisneros, G.; Cruzeiro, V.; et al. Amber2022, version 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Vassetti, D.; Pagliai, M.; Procacci, P. Assessment of GAFF2 and OPLS-AA General Force Fields in Combination with the Water Models TIP3P, SPCE, and OPC3 for the Solvation Free Energy of Druglike Organic Molecules. J. Chem. Theory Comput. 2019, 15, 1983–1995. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Press, W.H.; Flannery, B.P.; Teukolsky, S.A.; Vetterling, W.T.; Kramer, P.B. Numerical recipes: The art of scientific computing. Phys. Today 1987, 40, 120. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Kim, S.; Voter, A.F.; Roitberg, A.E. Bad Seeds Sprout Perilous Dynamics: Stochastic Thermostat Induced Trajectory Synchronization in Biomolecules. J. Chem. Theory Comput. 2009, 5, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Atilgan, A.R.; Demirel, M.C.; Erman, B. Vibrational dynamics of folded proteins: Significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 1998, 80, 2733. [Google Scholar] [CrossRef]
- Wen, J.; Scoles, D.R.; Facelli, J.C. Molecular dynamics analysis of the aggregation propensity of polyglutamine segments. PLoS ONE 2017, 12, e0178333. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Yang, T.; Wu, J.C.; Yan, C.; Wang, Y.; Luo, R.; Gonzales, M.B.; Dalby, K.N.; Ren, P. Virtual screening using molecular simulations. Proteins 2011, 79, 1940–1951. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schrödinger, L. The PyMOL Molecular Graphics System, version 1.8; Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Community, B.O. Blender—A 3D Modelling and Rendering Package. 2018. Available online: https://www.blender.org/ (accessed on 28 January 2023).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Name | Docking Score | Ligand Atoms | Receptor Residues | Bonds | Distance (Å) | Energy (kcal/mol) |
|---|---|---|---|---|---|---|---|
| 7KQ3 | 2-hydroxyethylsulfonic acid | −5.55 | O6 | OE2-GLU470 | HBD | 2.31 | −0.9 |
| O4 | NH1-ARG189 | HBA | 2.68 | −7.0 | |||
| O4 | N-GLN193 | HBA | 3.41 | −0.8 | |||
| O5 | NH2-ARG189 | HBA | 2.76 | −7.2 | |||
| O5 | NH2-ARG678 | HBA | 3.09 | −5.5 | |||
| O65 | N-CYS468 | HBA | 2.98 | −1.9 | |||
| O76 | NE2GLN193 | HBA | 2.94 | −3.9 | |||
| O76 | NH1-ARG678 | HBA | 3.16 | −4.9 | |||
| O43 | NH1-ARG189 | Ionic | 2.68 | −7.0 | |||
| O43 | NH2-ARG189 | Ionic | 3.43 | −2.2 | |||
| O54 | NH1-ARG189 | Ionic | 3.69 | −1.2 | |||
| O54 | NH2-ARG189 | Ionic | 2.76 | −6.3 | |||
| O54 | NH2-ARG678 | Ionic | 3.09 | −3.9 | |||
| O76 | NH1-ARG678 | Ionic | 3.16 | −3.5 | |||
| CP1 | 3-carbamoyl-5-fluoropyrazin-2-olate | −6.62 | N6 | OG1-THR312 | HBD | 2.88 | −4.6 |
| N3 | NH2-ARG678 | HBA | 2.89 | −3.7 | |||
| O7 | NH1-ARG189 | HBA | 2.76 | −2.3 | |||
| O7 | N-GLN193 | HBA | 3.06 | −1.9 | |||
| O10 | NH2-ARG189 | HBA | 2.76 | −3.1 | |||
| CP2 | 5-(1H-indol-2-yl)-1,3,4-oxadiazole-2(3H)-thione | −6.35 | N7 | OE2-GLU470 | HBD | 2.98 | −2.2 |
| N13 | OE1-GLN544 | HBD | 2.71 | −6.6 | |||
| S15 | N-ALA484 | HBA | 3.33 | −1.9 | |||
| 5-ring | NH2-ARG678 | π-cation | 3.43 | −0.5 | |||
| CP4 | (E)-2-(1-(2,5-dimethylthiophen-3-yl)ethylidene)-N-ethylhydrazine-1-carbothioamide | −6.24 | N14 | O-TYR587 | HBD | 2.67 | −1.8 |
| N3 | NH2-ARG189 | HBA | 2.91 | −1.1 | |||
| S13 | CE1-TYR485 | HBA | 3.40 | −0.5 | |||
| CP5 | (E)-2-(4-nitrobenzylidene)hydrazine-1-carbothioamide | −6.23 | N4 | OG1-THR185 | HBD | 2.95 | −2.0 |
| N7 | O-TYR587 | HBD | 2.92 | −1.2 | |||
| N3 | ANH2-RG189 | HBA | 2.87 | −6.2 | |||
| S6 | N-GLY483 | HBA | 3.20 | −2.0 | |||
| S6 | N-TYR587 | HBA | 3.23 | −2.9 | |||
| O14 | NH1-ARG678 | HBA | 2.78 | −1.9 | |||
| O14 | NH2-ARG678 | HBA | 3.18 | −0.7 | |||
| O15 | N-CYS468 | HBA | 2.82 | −2.4 | |||
| O15 | SG-CYS468 | HBA | 3.04 | −0.8 | |||
| CP8 | 4-amino-5-(pyridin-4-yl)-2,4-dihydro-3H-1,2,4-triazole-3-thione | −5.58 | N2 | N-CYS468 | HBA | 3.41 | −2.5 |
| S13 | N-TYR587 | HBA | 3.58 | −0.9 | |||
| 5-ring | NH2-ARG189 | π-cation | 3.12 | −0.6 | |||
| CP9 | 4-amino-5-(pyridin-3-yl)-2,4-dihydro-3H-1,2,4-triazole-3-thione | −5.35 | N10 | O-TYR587 | HBD | 2.93 | −2.7 |
| S13 | CA-GLY586 | HBA | 3.50 | −1.6 | |||
| S13 | N-TYR587 | HBA | 3.03 | −3.4 |
| ZINC ID | ΔEvdW (kcal/mol) | ΔEelec (kcal/mol) | ΔEGB (kcal/mol) | ΔESURF (Å2) | ΔGTOTAL (kcal/mol) |
|---|---|---|---|---|---|
| 7KQ3 | −10.52 ± 0.03 | 65.22 ± 0.18 | −67.00 ± 0.14 | −3.23 ± 0.01 | −15.53 ± 0.08 |
| CP1 | −23.91 ± 0.03 | −48.61 ± 0.07 | 48.90 ± 0.03 | −3.23 ± 0.01 | −26.85 ± 0.05 |
| CP2 | −30.88 ± 0.04 | −35.00 ± 0.17 | 39.83 ± 0.16 | −4.11 ± 0.01 | −30.17 ± 0.04 |
| CP4 | −45.56 ± 0.03 | −14.34 ± 0.01 | 31.14 ± 0.04 | −5.50 ± 0.01 | −34.27 ± 0.03 |
| CP5 | −36.43 ± 0.04 | −57.58 ± 0.07 | 67.78 ± 0.06 | −4.60 ± 0.01 | −30.83 ± 0.04 |
| CP8 | −30.86 ± 0.03 | −35.60 ± 0.04 | 48.71 ± 0.03 | −3.79 ± 0.01 | −21.54 ± 0.04 |
| CP9 | −31.46 ± 0.03 | −36.74 ± 0.06 | 48.98 ± 0.04 | −4.00 ± 0.01 | −23.22 ± 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waqas, M.; Halim, S.A.; Ullah, A.; Ali, A.A.M.; Khalid, A.; Abdalla, A.N.; Khan, A.; Al-Harrasi, A. Multi-Fold Computational Analysis to Discover Novel Putative Inhibitors of Isethionate Sulfite-Lyase (Isla) from Bilophila wadsworthia: Combating Colorectal Cancer and Inflammatory Bowel Diseases. Cancers 2023, 15, 901. https://doi.org/10.3390/cancers15030901
Waqas M, Halim SA, Ullah A, Ali AAM, Khalid A, Abdalla AN, Khan A, Al-Harrasi A. Multi-Fold Computational Analysis to Discover Novel Putative Inhibitors of Isethionate Sulfite-Lyase (Isla) from Bilophila wadsworthia: Combating Colorectal Cancer and Inflammatory Bowel Diseases. Cancers. 2023; 15(3):901. https://doi.org/10.3390/cancers15030901
Chicago/Turabian StyleWaqas, Muhammad, Sobia Ahsan Halim, Atta Ullah, Assim Alaa Mohammed Ali, Asaad Khalid, Ashraf N. Abdalla, Ajmal Khan, and Ahmed Al-Harrasi. 2023. "Multi-Fold Computational Analysis to Discover Novel Putative Inhibitors of Isethionate Sulfite-Lyase (Isla) from Bilophila wadsworthia: Combating Colorectal Cancer and Inflammatory Bowel Diseases" Cancers 15, no. 3: 901. https://doi.org/10.3390/cancers15030901
APA StyleWaqas, M., Halim, S. A., Ullah, A., Ali, A. A. M., Khalid, A., Abdalla, A. N., Khan, A., & Al-Harrasi, A. (2023). Multi-Fold Computational Analysis to Discover Novel Putative Inhibitors of Isethionate Sulfite-Lyase (Isla) from Bilophila wadsworthia: Combating Colorectal Cancer and Inflammatory Bowel Diseases. Cancers, 15(3), 901. https://doi.org/10.3390/cancers15030901