
Performance Metrics of the Scoring System for the Diagnosis of the Beckwith–Wiedemann Spectrum (BWSp) and Its Correlation with Cancer Development

 ,
,  , , , , , , , , ,
, , , , , , , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mussa, A.; Russo, S.; De Crescenzo, A.; Chiesa, N.; Molinatto, C.; Selicorni, A.; Richiardi, L.; Larizza, L.; Silengo, M.C.; Riccio, A.; et al. Prevalence of Beckwith-Wiedemann syndrome in North West of Italy. Am. J. Med. Genet. A 2013, 161, 2481–2486. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Kirby, G.; Hardy, C.; Dias, R.P.; Tee, L.; Lim, D.; Berg, J.; MacDonald, F.; Nightingale, P.; Maher, E.R. Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1000 subjects. Clin. Epigenetics 2014, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Weksberg, R.; Nishikawa, J.; Caluseriu, O.; Fei, Y.L.; Shuman, C.; Wei, C.; Steele, L.; Cameron, J.; Smith, A.; Ambus, I.; et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum. Mol. Genet. 2001, 10, 2989–3000. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Russo, S.; De Crescenzo, A.; Freschi, A.; Calzari, L.; Maitz, S.; Macchiaiolo, M.; Molinatto, C.; Baldassarre, G.; Mariani, M.; et al. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2016, 24, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.M.; Vansenne, F.; Kadouch, D.J.; Ibrahim, A.; Bliek, J.; Hopman, S.; Mannens, M.M.; Merks, J.H.; Maher, E.R.; Hennekam, R.C. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am. J. Med. Genet. A 2016, 170, 2248–2260. [Google Scholar] [CrossRef]
- Brioude, F.; Lacoste, A.; Netchine, I.; Vazquez, M.P.; Auber, F.; Audry, G.; Gauthier-Villars, M.; Brugieres, L.; Gicquel, C.; Le Bouc, Y.; et al. Beckwith-Wiedemann syndrome: Growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm. Res. Paediatr. 2013, 80, 457–465. [Google Scholar] [CrossRef]
- Shuman, C.; Beckwith, J.B.; Weksberg, R. Beckwith-Wiedemann Syndrome. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Chiesa, N.; De Crescenzo, A.; Mishra, K.; Perone, L.; Carella, M.; Palumbo, O.; Mussa, A.; Sparago, A.; Cerrato, F.; Russo, S.; et al. The KCNQ1OT1 imprinting control region and non-coding RNA: New properties derived from the study of Beckwith-Wiedemann syndrome and Silver-Russell syndrome cases. Hum. Mol. Genet. 2012, 21, 10–25. [Google Scholar] [CrossRef]
- Eggermann, T.; Maher, E.R.; Kratz, C.P.; Prawitt, D. Molecular Basis of Beckwith-Wiedemann Syndrome Spectrum with Associated Tumors and Consequences for Clinical Practice. Cancers 2022, 14, 3083. [Google Scholar] [CrossRef]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef]
- Kalish, J.M.; Biesecker, L.G.; Brioude, F.; Deardorff, M.A.; Di Cesare-Merlone, A.; Druley, T.; Ferrero, G.B.; Lapunzina, P.; Larizza, L.; Maas, S.; et al. Nomenclature and definition in asymmetric regional body overgrowth. Am. J. Med. Genet. A 2017, 173, 1735–1738. [Google Scholar] [CrossRef]
- Duffy, K.A.; Sajorda, B.J.; Yu, A.C.; Hathaway, E.R.; Grand, K.L.; Deardorff, M.A.; Kalish, J.M. Beckwith-Wiedemann syndrome in diverse populations. Am. J. Med. Genet. A 2019, 179, 525–533. [Google Scholar] [CrossRef]
- Baker, S.W.; Duffy, K.A.; Richards-Yutz, J.; Deardorff, M.A.; Kalish, J.M.; Ganguly, A. Improved molecular detection of mosaicism in Beckwith-Wiedemann Syndrome. J. Med. Genet. 2021, 58, 178–184. [Google Scholar] [CrossRef]
- Elliott, M.; Bayly, R.; Cole, T.; Temple, I.K.; Maher, E.R. Clinical features and natural history of Beckwith-Wiedemann syndrome: Presentation of 74 new cases. Clin. Genet. 1994, 46, 168–174. [Google Scholar] [CrossRef]
- DeBaun, M.R.; Tucker, M.A. Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J. Pediatr. 1998, 132, 398–400. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Mena, R.; Martin, L.J.; Steele, P.; Tinkle, B.T.; Hopkin, R.J. Experience with hemihyperplasia and Beckwith-Wiedemann syndrome surveillance protocol. Am. J. Med. Genet. A 2009, 149, 1691–1697. [Google Scholar] [CrossRef]
- Gaston, V.; Le Bouc, Y.; Soupre, V.; Burglen, L.; Donadieu, J.; Oro, H.; Audry, G.; Vazquez, M.P.; Gicquel, C. Analysis of the methylation status of the KCNQ1OT and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2001, 9, 409–418. [Google Scholar] [CrossRef]
- Mussa, A.; Di Candia, S.; Russo, S.; Catania, S.; De Pellegrin, M.; Di Luzio, L.; Ferrari, M.; Tortora, C.; Meazzini, M.C.; Brusati, R.; et al. Recommendations of the Scientific Committee of the Italian Beckwith-Wiedemann Syndrome Association on the diagnosis, management and follow-up of the syndrome. Eur. J. Med. Genet. 2016, 59, 52–64. [Google Scholar] [CrossRef]
- Priolo, M.; Sparago, A.; Mammì, C.; Cerrato, F.; Laganà, C.; Riccio, A. MS-MLPA is a specific and sensitive technique for detecting all chromosome 11p15.5 imprinting defects of BWS and SRS in a single-tube experiment. Eur. J. Hum. Genet. 2008, 16, 565–571. [Google Scholar] [CrossRef]
- Russo, S.; Calzari, L.; Mussa, A.; Mainini, E.; Cassina, M.; Di Candia, S.; Clementi, M.; Guzzetti, S.; Tabano, S.; Miozzo, M.; et al. A multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying Silver-Russell and Beckwith-Wiedemann syndromes. Clin. Epigenetics 2016, 8, 23. [Google Scholar] [CrossRef]
- Sparago, A.; Cerrato, F.; Vernucci, M.; Ferrero, G.B.; Silengo, M.C.; Riccio, A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat. Genet. 2004, 36, 958–960. [Google Scholar] [CrossRef]
- Brioude, F.; Netchine, I.; Praz, F.; Le Jule, M.; Calmel, C.; Lacombe, D.; Edery, P.; Catala, M.; Odent, S.; Isidor, B.; et al. Mutations of the Imprinted CDKN1C Gene as a Cause of the Overgrowth Beckwith-Wiedemann Syndrome: Clinical Spectrum and Functional Characterization. Hum. Mutat. 2015, 36, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Defabianis, P.; Mussa, A.; Ninivaggi, R.; Carli, D.; Romano, F. Maxillo-Facial Morphology in Beckwith-Wiedemann Syndrome: A Preliminary Study on (epi)Genotype-Phenotype Association in Caucasians. Int. J. Environ. Res. Public Health 2022, 19, 2448. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Peruzzi, L.; Chiesa, N.; De Crescenzo, A.; Russo, S.; Melis, D.; Tarani, L.; Baldassarre, G.; Larizza, L.; Riccio, A.; et al. Nephrological findings and genotype-phenotype correlation in Beckwith-Wiedemann syndrome. Pediatr. Nephrol. 2012, 27, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Duffy, K.A.; Cielo, C.M.; Cohen, J.L.; Gonzalez-Gandolfi, C.X.; Griff, J.R.; Hathaway, E.R.; Kupa, J.; Taylor, J.A.; Wang, K.H.; Ganguly, A.; et al. Characterization of the Beckwith-Wiedemann spectrum: Diagnosis and management. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 693–708. [Google Scholar] [CrossRef]
- Cooper, W.N.; Luharia, A.; Evans, G.A.; Raza, H.; Haire, A.C.; Grundy, R.; Bowdin, S.C.; Riccio, A.; Sebastio, G.; Bliek, J.; et al. Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2005, 13, 1025–1032. [Google Scholar] [CrossRef]
- Bliek, J.; Gicquel, C.; Maas, S.; Gaston, V.; Le Bouc, Y.; Mannens, M. Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith-Wiedemann syndrome (BWS). J. Pediatr. 2004, 145, 796–799. [Google Scholar] [CrossRef]
- Engel, J.R.; Smallwood, A.; Harper, A.; Higgins, M.J.; Oshimura, M.; Reik, W.; Schofield, P.N.; Maher, E.R. Epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. J. Med. Genet. 2000, 37, 921–926. [Google Scholar] [CrossRef]
- Mussa, A.; Ferrero, G.B.; Ceoloni, B.; Basso, E.; Chiesa, N.; De Crescenzo, A.; Pepe, E.; Silengo, M.; de Sanctis, L. Neonatal hepatoblastoma in a newborn with severe phenotype of Beckwith-Wiedemann syndrome. Eur. J. Pediatr. 2011, 170, 1407–1411. [Google Scholar] [CrossRef]
- Mussa, A.; Russo, S.; Larizza, L.; Riccio, A.; Ferrero, G.B. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: A paradigm for genomic medicine. Clin. Genet. 2016, 89, 403–415. [Google Scholar] [CrossRef]
- Mussa, A.; Molinatto, C.; Baldassarre, G.; Riberi, E.; Russo, S.; Larizza, L.; Riccio, A.; Ferrero, G.B. Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J. Pediatr. 2016, 176, 142–149.e1. [Google Scholar] [CrossRef]
- Cohen, J.L.; Duffy, K.A.; Sajorda, B.J.; Hathaway, E.R.; Gonzalez-Gandolfi, C.X.; Richards-Yutz, J.; Gunter, A.T.; Ganguly, A.; Kaplan, J.; Deardorff, M.A.; et al. Diagnosis and management of the phenotypic spectrum of twins with Beckwith-Wiedemann syndrome. Am. J. Med. Genet. A 2019, 179, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Duffy, K.A.; Getz, K.D.; Hathaway, E.R.; Byrne, M.E.; MacFarland, S.P.; Kalish, J.M. Characteristics Associated with Tumor Development in Individuals Diagnosed with Beckwith-Wiedemann Spectrum: Novel Tumor-(epi)Genotype-Phenotype Associations in the BWSp Population. Genes 2021, 12, 1839. [Google Scholar] [CrossRef] [PubMed]
- MacFarland, S.P.; Duffy, K.A.; Bhatti, T.R.; Bagatell, R.; Balamuth, N.J.; Brodeur, G.M.; Ganguly, A.; Mattei, P.A.; Surrey, L.F.; Balis, F.M.; et al. Diagnosis of Beckwith-Wiedemann syndrome in children presenting with Wilms tumor. Pediatr. Blood Cancer 2018, 65, e27296. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Carli, D.; Cardaropoli, S.; Ferrero, G.B.; Resta, N. Lateralized and segmental overgrowth in children. Cancers 2021, 13, 6166. [Google Scholar] [CrossRef] [PubMed]
- Mackay, D.J.G.; Bliek, J.; Lombardi, M.P.; Russo, S.; Calzari, L.; Guzzetti, S.; Izzi, C.; Selicorni, A.; Melis, D.; Temple, K.; et al. Discrepant molecular and clinical diagnoses in Beckwith-Wiedemann and Silver-Russell syndromes. Genet. Res. (Camb.) 2019, 4, 101:e3. [Google Scholar]
- Mussa, A.; Leoni, C.; Iacoviello, M.; Carli, D.; Ranieri, C.; Pantaleo, A.; Buonuomo, P.S.; Bagnulo, R.; Ferrero, G.B.; Bartuli, A.; et al. Genotypes and phenotypes heterogeneity in PIK3CA-related overgrowth spectrum and overlapping conditions: 150 novel patients and systematic review of 1007 patients with PIK3CA pathogenetic variants. J. Med. Genet. 2022, 7, 108093. [Google Scholar] [CrossRef]
- Carli, D.; De Pellegrin, M.; Franceschi, L.; Zinali, F.; Paonessa, M.; Spolaore, S.; Cardaropoli, S.; Cravino, M.; Marcucci, L.; Andreacchio, A.; et al. Evolution over Time of Leg Length Discrepancy in Patients with Syndromic and Isolated Lateralized Overgrowth. J. Pediatr. 2021, 234, 123–127. [Google Scholar] [CrossRef]
- Gazzin, A.; Carli, D.; Sirchia, F.; Molinatto, C.; Cardaropoli, S.; Palumbo, G.; Zampino, G.; Ferrero, G.B.; Mussa, A. Phenotype evolution and health issues of adults with Beckwith-Wiedemann syndrome. Am. J. Med. Genet. A 2019, 179, 1691–1702. [Google Scholar] [CrossRef]
- Mussa, A.; Molinatto, C.; Cerrato, F.; Palumbo, O.; Carella, M.; Baldassarre, G.; Carli, D.; Peris, C.; Riccio, A.; Ferrero, G.B. Assisted Reproductive Techniques and Risk of Beckwith-Wiedemann Syndrome. Pediatrics 2017, 140, e20164311. [Google Scholar] [CrossRef]
- Carli, D.; Operti, M.; Russo, S.; Cocchi, G.; Milani, D.; Leoni, C.; Prada, E.; Melis, D.; Falco, M.; Spina, J.; et al. Clinical and molecular characterization of patients affected by Beckwith-Wiedemann spectrum conceived through assisted reproduction techniques. Clin. Genet. 2022, 102, 314–323. [Google Scholar] [CrossRef]
- Carli, D.; Bertola, C.; Cardaropoli, S.; Ciuffreda, V.P.; Pieretto, M.; Ferrero, G.B.; Mussa, A. Prenatal features in Beckwith-Wiedemann syndrome and indications for prenatal testing. J. Med. Genet. 2021, 58, 842–849. [Google Scholar] [CrossRef]
- Fiala, E.M.; Ortiz, M.V.; Kennedy, J.A.; Glodzik, D.; Fleischut, M.H.; Duffy, K.A.; Hathaway, E.R.; Heaton, T.; Gerstle, J.T.; Steinherz, P.; et al. 11p15.5 epimutations in children with Wilms tumor and hepatoblastoma detected in peripheral blood. Cancer 2020, 126, 3114–3121. [Google Scholar] [CrossRef]
- Mussa, A.; Duffy, K.A.; Carli, D.; Griff, J.R.; Fagiano, R.; Kupa, J.; Brodeur, G.M.; Ferrero, G.B.; Kalish, J.M. The effectiveness of Wilms tumor screening in Beckwith-Wiedemann spectrum. J. Cancer Res. Clin. Oncol. 2019, 145, 3115–3123. [Google Scholar] [CrossRef]
- Hol, J.A.; Kuiper, R.P.; van Dijk, F.; Waanders, E.; van Peer, S.E.; Koudijs, M.J.; Bladergroen, R.; van Reijmersdal, S.V.; Morgado, L.M.; Bliek, J.; et al. Prevalence of (Epi)genetic Predisposing Factors in a 5-Year Unselected National Wilms Tumor Cohort: A Comprehensive Clinical and Genomic Characterization. J. Clin. Oncol. 2022, 40, 1892–1902. [Google Scholar] [CrossRef]
- Kalish, J.M.; Doros, L.; Helman, L.J.; Hennekam, R.C.; Kuiper, R.P.; Maas, S.M.; Maher, E.R.; Nichols, K.E.; Plon, S.E.; Porter, C.C.; et al. Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin. Cancer Res. 2017, 23, e115–e122. [Google Scholar] [CrossRef]
- Griff, J.R.; Duffy, K.A.; Kalish, J.M. Characterization and Childhood Tumor Risk Assessment of Genetic and Epigenetic Syndromes Associated With Lateralized Overgrowth. Front. Pediatr. 2020, 8, 613260. [Google Scholar] [CrossRef]
- Radley, J.A.; Connolly, M.; Sabir, A.; Kanani, F.; Carley, H.; Jones, R.L.; Hyder, Z.; Gompertz, L.; Reardon, W.; Richardson, R.; et al. Isolated- and Beckwith-Wiedemann syndrome related- lateralised overgrowth (hemihypertrophy): Clinical and molecular correlations in 94 individuals. Clin. Genet. 2021, 100, 292–297. [Google Scholar] [CrossRef]
- Mussa, A.; Ciuffreda, V.P.; Sauro, P.; Pagliardini, V.; Pagliardini, S.; Carli, D.; Kalish, J.M.; Fagioli, F.; Pavanello, E.; Ferrero, G.B. Longitudinal Monitoring of Alpha-Fetoprotein by Dried Blood Spot for Hepatoblastoma Screening in Beckwith⁻Wiedemann Syndrome. Cancers 2019, 11, 86. [Google Scholar] [CrossRef]
- Mussa, A.; Ferrero, G.B. Screening Hepatoblastoma in Beckwith-Wiedemann Syndrome: A Complex Issue. J. Pediatr. Hematol. Oncol. 2015, 37, 627. [Google Scholar] [CrossRef]
- Mussa, A.; Ferrero, G.B. Serum alpha-fetoprotein screening for hepatoblastoma in Beckwith-Wiedemann syndrome. Am. J. Med. Genet. A 2017, 173, 585–587. [Google Scholar] [CrossRef]
- Mussa, A.; Duffy, K.A.; Carli, D.; Ferrero, G.B.; Kalish, J.M. Defining an optimal time window to screen for hepatoblastoma in children with Beckwith-Wiedemann syndrome. Pediatr. Blood Cancer 2019, 66, e27492. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Major Features | Minor Features | Clinical Diagnosis of Beckwith–Wiedemann Syndrome | |
|---|---|---|---|
| Elliott et al., 1994 [14] | Abdominal wall defect Macroglossia Macrosomia a | Ear creases/pits Facial naevus simplex Lateralized overgrowth Hypoglycemia Nephromegaly | At least three major features, or two major features plus three or more minor features |
| DeBaun and Tucker 1998 [15] | Abdominal wall defect Ear creases/pits Hypoglycemia Macroglossia Macrosomia | At least two major features | |
| Weksberg et al., 2001 [3] | Abdominal wall defect Ear creases/pits Embryonal tumors Lateralized overgrowth Macroglossia Macrosomia | Hypoglycemia Organomegaly Renal malformation | At least three major features, or two major features and one or more minor features |
| Gaston et al., 2001 [17] | Abdominal wall defect Macroglossia Macrosomia Organomegaly | Ear creases/pits Facial naevus simplex Lateralized overgrowth Hypoglycemia | Complete and incomplete BWS syndrome classification. Complete—at least three major features. Incomplete—less than three major features and one or more minor features |
| Zarate et al., 2009 [16] | Abdominal wall defect Macroglossia Macrosomia | Cardiomegaly Ear creases/pits Facial naevus simplex Lateralized overgrowth Hypoglycemia Mid-face hypoplasia Polyhydramnios | At least three major features, or two major features and one or more minor features |
| Ibrahim et al., 2014 [2] | Macroglossia Omphalocele Organomegaly Macrosomia Facial naevus simplex Lateralized overgrowth Hypoglycemia | Macroglossia 2.5 pts Omphalocele 1.5 pts Organomegaly 1 pts Macrosomia 1 pts Facial naevus simplex 1 pts Lateralized overgrowth 0.5 pts Hypoglycemia 0.5 pts Clinical diagnosis if score is ≥3.5 pts | |
| Brioude et al., 2018 [10] | Macroglossia Omphalocele Lateralized overgrowth Multifocal and/or bilateral Wilms tumor or nephroblastomatosis Hyperinsulinism b Pathology findings c | Macrosomia Facial naevus simplex Polyhydramnios and/or placentomegaly Ear creases and/or pits Transient hypoglycemia d Typical BWSp tumors e Nephromegaly and/or hepatomegaly Umbilical hernia and/or diastasis recti | Clinical diagnosis if score is ≥4. Patients with a score of ≥2 merit genetic testing for investigation and diagnosis of BWS. Patients with a score of <2 do not meet the criteria for genetic testing. |
| Total | IC1-GoM | IC2-LoM | UPD(11)pat | CDKN1C | Negative | p Value | |
|---|---|---|---|---|---|---|---|
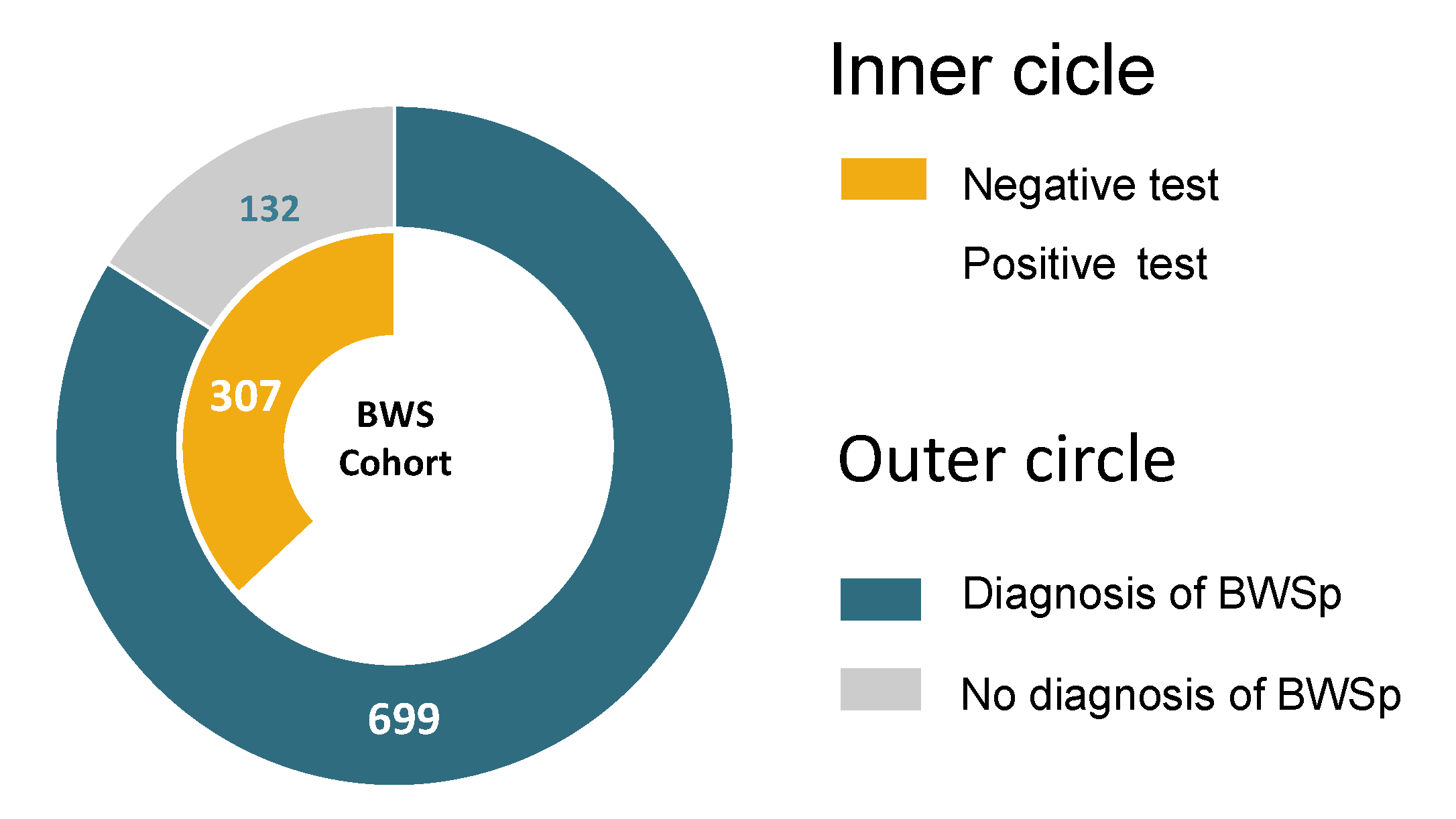
| Total | 831 | 52 (6.3%) | 322 (38.7%) | 138 (16.6%) | 12 (1.4%) | 307 (36.9%) | |
| Females | 445 | 27 (51.9%) | 187 (58.1%) | 67 (48.6%) | 6 (50%) | 158 (51.5%) | 0.321 |
| Males | 386 | 25 (48.1%) | 135 (41.9%) | 71 (51.4%) | 6 (50%) | 149 (48.5%) | 0.362 |
| Cardinal and suggestive features | |||||||
| Macroglossia | 557 | 42 (80.8%) | 276 (85.7%) | 92 (66.7%) | 10 (83.3%) | 137 (44.6%) | <0.001 |
| Abdominal wall defects | 476 | 28 (53.8%) | 214 (66.5%) | 68 (49.3%) | 10 (83.3%) | 156 (50.8%) | <0.001 |
| Umbilical hernia | 151 | 6 (11.5%) | 64 (19.9%) | 29 (21%) | 5 (41.7%) | 47 (15.3%) | 0.060 |
| Diastasis recti | 226 | 20 (38.5%) | 80 (24.8%) | 30 (21.7%) | 0 (0%) | 96 (31.3%) | 0.01 |
| Omphalocele | 99 | 2 (3.8%) | 70 (21.7%) | 9 (6.5%) | 5 (41.7%) | 13 (4.2%) | <0.001 |
| Lateralized overgrowth | 551 | 29 (55.8%) | 178 (55.3%) | 119 (86.2%) | 2 (16.7%) | 223 (72.6%) | <0.001 |
| Neonatal hyperinsulinism | 11 | 1 (1.9%) | 3 (0.9%) | 5 (3.6%) | 0 (0%) | 2 (0.7%) | 0.119 |
| Fetal overgrowth | 334 | 36 (69.2%) | 142 (44.1%) | 62 (44.9%) | 6 (50%) | 88 (28.7%) | <0.001 |
| Facial naevus simplex | 256 | 10 (19.2%) | 139 (43.2%) | 32 (23.2%) | 6 (50%) | 69 (22.5%) | <0.001 |
| Polyhydramnios | 100 | 11 (21.2%) | 55 (17.1%) | 17 (12.3%) | 1 (8.3%) | 16 (5.2%) | <0.001 |
| Placentomegaly | 7 | 0 (0%) | 5 (1.6%) | 1 (0.7%) | 1 (8.3%) | 0 (0%) | 0.011 |
| Ear anomalies | 296 | 17 (32.7%) | 142 (44.1%) | 47 (34.1%) | 6 (50%) | 84 (27.4%) | <0.001 |
| Transient hypoglycemia | 226 | 18 (34.6%) | 104 (32.3%) | 52 (37.7%) | 3 (25%) | 49 (16%) | <0.001 |
| Organomegaly | 176 | 30 (57.7%) | 75 (23.3%) | 26 (18.8%) | 1 (8.3%) | 44 (14.3%) | <0.001 |
| Typical tumors | 44 | 9 (17.3%) | 6 (1.9%) | 17 (12.3%) | 1 (8.3%) | 11 (3.6%) | <0.001 |
| Wilms tumor–multifocal | 22–4 | 8 (15.4%)–1 (1.9%) | 0 (0%) | 6–0 (4.3%) | 0 (0%) | 8 (3.6%)–3 (1%) | <0.001 |
| Hepatoblastoma | 7 | 0 (0%) | 1 (0.3%) | 6 (4.3%) | 0 (0%) | 0 (0%) | <0.001 |
| Neuroblastoma | 4 | 0 (0%) | 1 (0.3%) | 2 (1.4%) | 1 (8.3%) | 0 (0%) | <0.001 |
| Pancreatoblastoma | 2 | 1 (1.9%) | 0 (0%) | 1 (0.7%) | 0 (0%) | 0 (0%) | 0.061 |
| Rhabdomyosarcoma | 1 | 0 (0%) | 1 (0.3%) | 0 (0%) | 0 (0%) | 0 (0%) | 0.812 |
| Adrenal gland carcinoma | 4 | 0 (0%) | 2 (0.6%) | 2 (1.4%) | 0 (0%) | 1 (0.3%) | 0.656 |
| Pheochromocytoma | 1 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (0.3%) | 0.789 |
| Other features | |||||||
| Other tumors | 3 | 0 (0%) | 0 (0%) | 1 (0.7%) | 0 (0%) | 2 (0.6%) | 0.668 |
| Cleft palate | 11 | 0 (0%) | 8 (2.5%) | 0 (0%) | 1 (8.3%) | 2 (0.7%) | 0.022 |
| Assisted reproduction | 88 | 3 (5.8%) | 52 (16.1%) | 14 (10.1%) | 1 (8.3%) | 18 (5.9%) | <0.001 |
| Postnatal overgrowth | 267 | 19 (36.5%) | 130 (40.4%) | 40 (29%) | 7 (58.3%) | 71 (23.1%) | <0.001 |
| Cryptorchidism | 47 | 5 (20%) | 19 (14.1%) | 5 (7%) | 4 (66.7%) | 11 (7.4%) | <0.001 |
| Twin pregnancy | 50 | 1 (1.9%) | 22 (6.8%) | 1 (0.7%) | 0 (0%) | 12 (3.9%) | 0.032 |
| Preterm delivery | 212 | 9 (17.3%) | 105 (32.6%) | 21 (15.2%) | 7 (58.3%) | 70 (22.8%) | <0.001 |
| Gestational age (weeks) | 37.0 ± 2.9 | 37.3 ± 2.7 | 37.0 ± 2.6 | 37.9 ± 2.4 | 33.2 ± 4.1 * | 36.4 ± 3.4 | <0.001 |
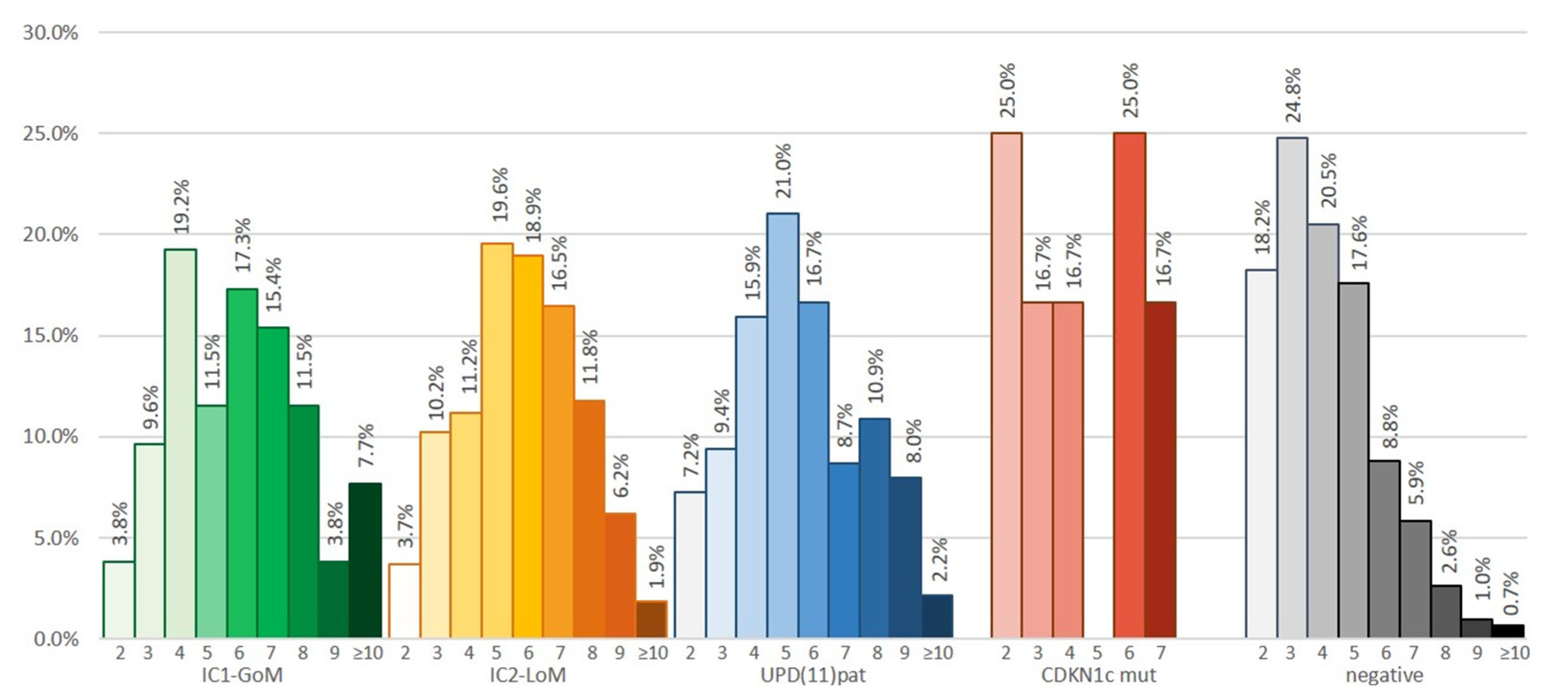
| Consensus score (mean ± SD) | 5.1 ± 2.1 | 5.9 ± 2.2 | 5.8 ± 1.9 | 5.5 ± 2.1 | 5.3 ± 2.0 | 4.1 ± 1.7 | <0.001 |
| Cardinal features (mean ± SD) | 1.5 ± 1.7 | 1.6 ± 0.8 ⁑ | 1.7 ± 0.7 | 1.8 ± 0.7 | 1.6 ± 0.7 | 1.3 ± 0.6 | <0.001 |
| Suggestive features (mean ± SD) | 2.2 ± 0.7 | 3.1 ± 1.7 • | 2.5 ± 1.4 | 2.3 ± 1.5 | 2.5 ± 1.6 | 2.5 ± 1.6 | <0.001 |
| ConsensusScore a | n (%) | IC1-GoM | IC2-LoM | UPD(11)pat | CDKN1C Mutation | Negative |
|---|---|---|---|---|---|---|
| Any scoresScores | 48/831 (5.8%) | 10 (19.2%) | 5 (1.6%) | 17 (12.3%) | 1 (8.3%) | 15 (4.9%) |
| 2 | 5/85 (5.9%) | - | - | 1 AK, 2 HB | - | 2 WT |
| 3 | 12/137 (8.8%) | 1 WT | 1 RMS | 1 AK, 1 WT, 1 HB, 1 leu | - | 2 WT + 1 bWT, 1 AK, 1 LH, 1 PCC |
| 4 | 2/124 (1.6%) | - | - | 1 WT, 1 NB | - | - |
| 5 | 5/156 (3.2%) | - | 1 AK | 1 PB * 1 NB * | - | 2 WT + 1 bWT |
| 6 | 7/123 (5.7%) | 2 WT + 1 bWT | 1 AK ′, 1 SCT ′ | - | 1 NB | 1 HCC, 1 WT |
| 7 | 8/95 (8.4%) | 1 PB 1 WT | 1 HB, 1 NB | 2 WT, 1 HB | - | 1 WT |
| 8 | 4/64 (6.2%) | 1 WT | - | 2 WT | - | 1 bWT |
| 9 | 2/35 (5.7%%) | 1 WT | - | 1 HB | - | - |
| ≥10 | 3/12 (25%%) | 2 WT | - | 1 HB | - | - |
| Elliott et al., 1994 [14] | DeBaun and Tucker 1998 [15] | Weksberg et al., 2001 [3] | Gaston et al., 2001 [17] | Zarate et al., 2009 [16] | Ibrahim et al., 2014 [2] | Brioude et al., 2018 [10] | |
|---|---|---|---|---|---|---|---|
| Performance against a Positive/Negative Molecular Test | |||||||
| Patients with/without clinical criteria | 253/578 | 598/233 | 565/266 | 684/147 | 455/376 | 535/296 | 621/210 |
| Positive test | 210/314 | 440/84 | 412/112 | 462/62 | 344/180 | 414/110 | 446/78 |
| IC1-GoM | 27/25 | 43/9 | 44/8 | 45/7 | 38/14 | 42/10 | 45/7 |
| IC2-LoM | 136/186 | 282/40 | 258/64 | 290/32 | 222/100 | 268/54 | 277/45 |
| UPD(11)pat | 42/96 | 103/35 | 102/36 | 118/20 | 76/62 | 95/43 | 115/23 |
| CDKN1C mutation | 5/7 | 12/0 | 8/4 | 9/3 | 8/4 | 9/3 | 9/3 |
| Negative test | 43/264 | 158/149 | 153/154 | 222/85 | 111/196 | 121/186 | 175/132 |
| ROC Analysis | |||||||
| Area under curve (AUC) | 0.706 | 0.706 | 0.692 | 0.704 | 0.724 | 0.761 | 0.731 |
| st.Err | 0.018 | 0.019 | 0.019 | 0.019 | 0.018 | 0.017 | 0.018 |
| 95% Confidence Interval | 0.67–0.74 | 0.69–0.74 | 0.65–0.73 | 0.69–0.74 | 0.68–0.76 | 0.73–0.79 | 0.70–0.77 |
| p value | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| Sensitivity | 0.40 | 0.84 | 0.79 | 0.88 | 0.66 | 0.79 | 0.85 |
| Specificity | 0.86 | 0.49 | 0.5 | 0.28 | 0.64 | 0.61 | 0.43 |
| Positive Predictive Value (PPV) | 0.83 | 0.74 | 0.73 | 0.68 | 0.76 | 0.77 | 0.72 |
| Negative Predictive Value (NPV) | 0.46 | 0.64 | 0.58 | 0.58 | 0.52 | 0.63 | 0.63 |
| Diagnostic Accuracy (ACC) | 0.57 | 0.71 | 0.68 | 0.66 | 0.65 | 0.72 | 0.69 |
| Performance against Tumor Development (n = 48) * | |||||||
| Patients with tumor diagnosed | 16 (33.3%) | 30 (62.5%) | 18 (37.5%) | 40 (83.3%) | 24 (50.0%) | 27 (56.8%) | 31 + 9 ⁑ (83.3%) |
| With positive molecular tests | 13 | 25 | 15 | 28 | 20 | 22 | 24 + 9 ⁑ |
| With negative molecular tests | 3 | 5 | 3 | 12 | 4 | 5 | 7 |
| Patients with tumor missed | 32 (66.7%) | 18 (37.5%) | 30 (62.5%) | 8 (16.7%) | 24 (50.0%) | 21 (43.2%) | 8 (16.7%) |
| With positive molecular tests | 20 | 8 | 18 | 5 | 13 | 11 | 0 |
| With negative molecular tests | 12 | 10 | 12 | 3 | 11 | 10 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luca, M.; Carli, D.; Cardaropoli, S.; Milani, D.; Cocchi, G.; Leoni, C.; Macchiaiolo, M.; Bartuli, A.; Tarani, L.; Melis, D.; et al. Performance Metrics of the Scoring System for the Diagnosis of the Beckwith–Wiedemann Spectrum (BWSp) and Its Correlation with Cancer Development. Cancers 2023, 15, 773. https://doi.org/10.3390/cancers15030773
Luca M, Carli D, Cardaropoli S, Milani D, Cocchi G, Leoni C, Macchiaiolo M, Bartuli A, Tarani L, Melis D, et al. Performance Metrics of the Scoring System for the Diagnosis of the Beckwith–Wiedemann Spectrum (BWSp) and Its Correlation with Cancer Development. Cancers. 2023; 15(3):773. https://doi.org/10.3390/cancers15030773
Chicago/Turabian StyleLuca, Maria, Diana Carli, Simona Cardaropoli, Donatella Milani, Guido Cocchi, Chiara Leoni, Marina Macchiaiolo, Andrea Bartuli, Luigi Tarani, Daniela Melis, and et al. 2023. "Performance Metrics of the Scoring System for the Diagnosis of the Beckwith–Wiedemann Spectrum (BWSp) and Its Correlation with Cancer Development" Cancers 15, no. 3: 773. https://doi.org/10.3390/cancers15030773
APA StyleLuca, M., Carli, D., Cardaropoli, S., Milani, D., Cocchi, G., Leoni, C., Macchiaiolo, M., Bartuli, A., Tarani, L., Melis, D., Bontempo, P., D’Elia, G., Prada, E., Vitale, R., Grammegna, A., Tannorella, P., Sparago, A., Pignata, L., Riccio, A., ... Mussa, A. (2023). Performance Metrics of the Scoring System for the Diagnosis of the Beckwith–Wiedemann Spectrum (BWSp) and Its Correlation with Cancer Development. Cancers, 15(3), 773. https://doi.org/10.3390/cancers15030773