
Pancreatic Cancer in Chronic Pancreatitis: Pathogenesis and Diagnostic Approach

 , , , ,
, , , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. How Does Pancreatic Adenocarcinoma Develop from Chronic Pancreatitis?
2.1. Genetics of Pancreatic Carcinogenesis
2.2. Pancreatic Carcinogenesis during Chronic Pancreatitis: Role of Acinar Ductal Metaplasia
2.3. Mouse Models of PDAC
3. Incidence of Pancreatic Cancer in Chronic Pancreatitis
4. Which Clinical and Paraclinical Signs in Chronic Pancreatitis Are an Indication of Cancer?
4.1. Clinical Signs
4.2. Standard Biology
4.3. Imaging
4.3.1. Ultrasonography, Tomodensitometry (CT Scan), and MRI
4.3.2. Endoscopic Ultrasound
4.4. Role of Liquid Biopsy
4.5. Role of Surgery
5. Can Pancreatic Cancer Developed in Chronic Pancreatitis Be Detected and Prevented?
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hegyi, P.; Párniczky, A.; Lerch, M.M.; Sheel, A.R.G.; Rebours, V.; Forsmark, C.E.; Del Chiaro, M.; Rosendahl, J.; de-Madaria, E.; Szücs, Á.; et al. International Consensus Guidelines for Risk Factors in Chronic Pancreatitis. Recommendations from the Working Group for the International Consensus Guidelines for Chronic Pancreatitis in Collaboration with the International Association of Pancreatology, the American Pancreatic Association, the Japan Pancreas Society, and European Pancreatic Club. Pancreatology 2020, 20, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Yadav, D.; Garg, P.K. Diagnosis and Management of Chronic Pancreatitis: A Review. JAMA 2019, 322, 2422–2434. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Muscari, F.; Carrère, N.; Otal, P. Traité de pancréatologie; Elsevier Masson: Paris, France, 2021; ISBN 978-2-294-77623-6. [Google Scholar]
- Lévy, P.; Ruszniewski, P.; Bernades, P. Natural history of chronic alcoholic pancreatitis. Gastroenterol. Clin. Biol. 2000, 24, 725–741. [Google Scholar] [PubMed]
- Molina-Montes, E.; Van Hoogstraten, L.; Gomez-Rubio, P.; Löhr, M.; Sharp, L.; Molero, X.; Márquez, M.; Michalski, C.W.; Farré, A.; Perea, J.; et al. Pancreatic Cancer Risk in Relation to Lifetime Smoking Patterns, Tobacco Type, and Dose-Response Relationships. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1009–1018. [Google Scholar] [CrossRef]
- Rebours, V.; Vullierme, M.-P.; Hentic, O.; Maire, F.; Hammel, P.; Ruszniewski, P.; Lévy, P. Smoking and the Course of Recurrent Acute and Chronic Alcoholic Pancreatitis: A Dose-Dependent Relationship. Pancreas 2012, 41, 1219–1224. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P. Defining the Role of Smoking in Chronic Pancreatitis. Clin. Gastroenterol. Hepatol. 2011, 9, 196–197. [Google Scholar] [CrossRef]
- Delpu, Y.; Hanoun, N.; Lulka, H.; Sicard, F.; Selves, J.; Buscail, L.; Torrisani, J.; Cordelier, P. Genetic and Epigenetic Alterations in Pancreatic Carcinogenesis. Curr. Genom. 2011, 12, 15. [Google Scholar] [CrossRef]
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting KRAS for Diagnosis, Prognosis, and Treatment of Pancreatic Cancer: Hopes and Realities. Eur. J. Cancer 2016, 54, 75–83. [Google Scholar] [CrossRef]
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in Pancreatic Tumor Development and Progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of Oncogenic KRAS in the Diagnosis, Prognosis and Treatment of Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Saiki, Y.; Jiang, C.; Ohmuraya, M.; Furukawa, T. Genetic Mutations of Pancreatic Cancer and Genetically Engineered Mouse Models. Cancers 2021, 14, 71. [Google Scholar] [CrossRef]
- Bijlsma, M.F.; Sadanandam, A.; Tan, P.; Vermeulen, L. Molecular Subtypes in Cancers of the Gastrointestinal Tract. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 333–342. [Google Scholar] [CrossRef]
- Jonckheere, N.; Vasseur, R.; Van Seuningen, I. The Cornerstone K-RAS Mutation in Pancreatic Adenocarcinoma: From Cell Signaling Network, Target Genes, Biological Processes to Therapeutic Targeting. Crit. Rev. Oncol. Hematol. 2017, 111, 7–19. [Google Scholar] [CrossRef]
- Ebert, M.; Schandl, L.; Schmid, R.M. Differentiation of Chronic Pancreatitis from Pancreatic Cancer: Recent Advances in Molecular Diagnosis. Dig. Dis. 2001, 19, 32–36. [Google Scholar] [CrossRef]
- Pinho, A.V.; Chantrill, L.; Rooman, I. Chronic Pancreatitis: A Path to Pancreatic Cancer. Cancer Lett. 2014, 345, 203–209. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- De La O, J.-P.; Emerson, L.L.; Goodman, J.L.; Froebe, S.C.; Illum, B.E.; Curtis, A.B.; Murtaugh, L.C. Notch and Kras Reprogram Pancreatic Acinar Cells to Ductal Intraepithelial Neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 18907–18912. [Google Scholar] [CrossRef]
- Gidekel Friedlander, S.Y.; Chu, G.C.; Snyder, E.L.; Girnius, N.; Dibelius, G.; Crowley, D.; Vasile, E.; DePinho, R.A.; Jacks, T. Context-Dependent Transformation of Adult Pancreatic Cells by Oncogenic K-Ras. Cancer Cell 2009, 16, 379–389. [Google Scholar] [CrossRef]
- Pérez-Mancera, P.A.; Guerra, C.; Barbacid, M.; Tuveson, D.A. What We Have Learned about Pancreatic Cancer from Mouse Models. Gastroenterology 2012, 142, 1079–1092. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Wang, Q.; Song, Y.; Chen, S.; Cheng, B.; Zhang, Y.; Cui, Z.; Wu, Z.; Zhu, C. MIF Inhibitor ISO-1 Alleviates Severe Acute Pancreatitis-Associated Acute Kidney Injury by Suppressing the NLRP3 Inflammasome Signaling Pathway. Int. Immunopharmacol. 2021, 96, 107555. [Google Scholar] [CrossRef]
- Al Mamun, A.; Suchi, S.A.; Aziz, M.A.; Zaeem, M.; Munir, F.; Wu, Y.; Xiao, J. Pyroptosis in Acute Pancreatitis and Its Therapeutic Regulation. Apoptosis 2022, 27, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic Kras Is Required for Both the Initiation and Maintenance of Pancreatic Cancer in Mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Müller-Decker, K.; Fürstenberger, G.; Annan, N.; Kucher, D.; Pohl-Arnold, A.; Steinbauer, B.; Esposito, I.; Chiblak, S.; Friess, H.; Schirmacher, P.; et al. Preinvasive Duct-Derived Neoplasms in Pancreas of Keratin 5-Promoter Cyclooxygenase-2 Transgenic Mice. Gastroenterology 2006, 130, 2165–2178. [Google Scholar] [CrossRef]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.-F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-Induced IKK2/β/NF-ΚB Activation by IL-1α and P62 Feedforward Loops Is Required for Development of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Maniati, E.; Bossard, M.; Cook, N.; Candido, J.B.; Emami-Shahri, N.; Nedospasov, S.A.; Balkwill, F.R.; Tuveson, D.A.; Hagemann, T. Crosstalk between the Canonical NF-ΚB and Notch Signaling Pathways Inhibits Pparγ Expression and Promotes Pancreatic Cancer Progression in Mice. J. Clin. Investig. 2011, 121, 4685–4699. [Google Scholar] [CrossRef]
- Kandikattu, H.K.; Venkateshaiah, S.U.; Yadavalli, C.S.; Oruganti, L.; Mishra, A. Development and Characterization of Novel Chronic Eosinophilic Inflammation-Mediated Murine Model of Malignant Pancreatitis. Endocr. Metab. Immune Disord. Drug Targets 2022. [Google Scholar] [CrossRef]
- Kandikattu, H.K.; Manohar, M.; Upparahalli Venkateshaiah, S.; Yadavalli, C.; Mishra, A. Chronic Inflammation Promotes Epithelial-Mesenchymal Transition-Mediated Malignant Phenotypes and Lung Injury in Experimentally-Induced Pancreatitis. Life Sci. 2021, 278, 119640. [Google Scholar] [CrossRef]
- Al Saati, T.; Clerc, P.; Hanoun, N.; Peuget, S.; Lulka, H.; Gigoux, V.; Capilla, F.; Béluchon, B.; Couvelard, A.; Selves, J.; et al. Oxidative Stress Induced by Inactivation of TP53INP1 Cooperates with KrasG12D to Initiate and Promote Pancreatic Carcinogenesis in the Murine Pancreas. Am. J. Pathol. 2013, 182, 1996–2004. [Google Scholar] [CrossRef]
- Daniluk, J.; Liu, Y.; Deng, D.; Chu, J.; Huang, H.; Gaiser, S.; Cruz-Monserrate, Z.; Wang, H.; Ji, B.; Logsdon, C.D. An NF-ΚB Pathway-Mediated Positive Feedback Loop Amplifies Ras Activity to Pathological Levels in Mice. J. Clin. Investig. 2012, 122, 1519–1528. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-Induced Inflammation Contributes to Pancreatic Cancer by Inhibiting Oncogene-Induced Senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef]
- Karlson, B.M.; Ekbom, A.; Josefsson, S.; McLaughlin, J.K.; Fraumeni, J.F.; Nyrén, O. The Risk of Pancreatic Cancer Following Pancreatitis: An Association Due to Confounding? Gastroenterology 1997, 113, 587–592. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andrén-Sandberg, A.; Domellöf, L. Pancreatitis and the Risk of Pancreatic Cancer. International Pancreatitis Study Group. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef]
- Rebours, V.; Lévy, P.; Mosnier, J.-F.; Scoazec, J.-Y.; Soubeyrand, M.-S.; Fléjou, J.-F.; Turlin, B.; Hammel, P.; Ruszniewski, P.; Bedossa, P.; et al. Pathology analysis reveals that dysplastic pancreatic ductal lesions are frequent in patients with hereditary pancreatitis. Clin. Gastroenterol. Hepatol. 2010, 8, 206–212. [Google Scholar] [CrossRef]
- Rebours, V.; Boutron-Ruault, M.-C.; Schnee, M.; Férec, C.; Le Maréchal, C.; Hentic, O.; Maire, F.; Hammel, P.; Ruszniewski, P.; Lévy, P. The Natural History of Hereditary Pancreatitis: A National Series. Gut 2009, 58, 97–103. [Google Scholar] [CrossRef]
- Howes, N.; Lerch, M.M.; Greenhalf, W.; Stocken, D.D.; Ellis, I.; Simon, P.; Truninger, K.; Ammann, R.; Cavallini, G.; Charnley, R.M.; et al. Clinical and Genetic Characteristics of Hereditary Pancreatitis in Europe. Clin. Gastroenterol. Hepatol. 2004, 2, 252–261. [Google Scholar] [CrossRef]
- Muller, N.; Sarantitis, I.; Rouanet, M.; de Mestier, L.; Halloran, C.; Greenhalf, W.; Férec, C.; Masson, E.; Ruszniewski, P.; Lévy, P.; et al. Natural History of SPINK1 Germline Mutation Related-Pancreatitis. EBioMedicine 2019, 48, 581–591. [Google Scholar] [CrossRef]
- Cazacu, I.M.; Farkas, N.; Garami, A.; Balaskó, M.; Mosdósi, B.; Alizadeh, H.; Gyöngyi, Z.; Rakonczay, Z.; Vigh, É.; Habon, T.; et al. Pancreatitis-Associated Genes and Pancreatic Cancer Risk: A Systematic Review and Meta-Analysis. Pancreas 2018, 47, 1078–1086. [Google Scholar] [CrossRef]
- Hamoir, C.; Pepermans, X.; Piessevaux, H.; Jouret-Mourin, A.; Weynand, B.; Habyalimana, J.-B.; Leal, T.; Geubel, A.; Gigot, J.-F.; Deprez, P.H. Clinical and Morphological Characteristics of Sporadic Genetically Determined Pancreatitis as Compared to Idiopathic Pancreatitis: Higher Risk of Pancreatic Cancer in CFTR Variants. Digestion 2013, 87, 229–239. [Google Scholar] [CrossRef]
- Gandhi, S.; de la Fuente, J.; Murad, M.H.; Majumder, S. Chronic Pancreatitis Is a Risk Factor for Pancreatic Cancer, and Incidence Increases With Duration of Disease: A Systematic Review and Meta-Analysis. Clin. Transl. Gastroenterol. 2022, 13, e00463. [Google Scholar] [CrossRef] [PubMed]
- Poddighe, D. Autoimmune Pancreatitis and Pancreatic Cancer: Epidemiological Aspects and Immunological Considerations. World J. Gastroenterol. WJG 2021, 27, 3825–3836. [Google Scholar] [CrossRef] [PubMed]
- Bang, U.C.; Benfield, T.; Hyldstrup, L.; Bendtsen, F.; Beck Jensen, J. Mortality, Cancer, and Comorbidities Associated with Chronic Pancreatitis: A Danish Nationwide Matched-Cohort Study. Gastroenterology 2014, 146, 989–994.e1. [Google Scholar] [CrossRef] [PubMed]
- Kirkegård, J.; Mortensen, F.V.; Cronin-Fenton, D. Chronic Pancreatitis and Pancreatic Cancer Risk: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2017, 112, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Lowenfels, A.B.; Morselli-Labate, A.M.; Maisonneuve, P.; Pezzilli, R. Pancreatic Cancer in Chronic Pancreatitis; Aetiology, Incidence, and Early Detection. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 349–358. [Google Scholar] [CrossRef]
- Duell, E.J.; Lujan-Barroso, L.; Sala, N.; Deitz McElyea, S.; Overvad, K.; Tjonneland, A.; Olsen, A.; Weiderpass, E.; Busund, L.-T.; Moi, L.; et al. Plasma MicroRNAs as Biomarkers of Pancreatic Cancer Risk in a Prospective Cohort Study. Int. J. Cancer 2017, 141, 905–915. [Google Scholar] [CrossRef]
- Molina-Montes, E.; Coscia, C.; Gómez-Rubio, P.; Fernández, A.; Boenink, R.; Rava, M.; Márquez, M.; Molero, X.; Löhr, M.; Sharp, L.; et al. Deciphering the Complex Interplay between Pancreatic Cancer, Diabetes Mellitus Subtypes and Obesity/BMI through Causal Inference and Mediation Analyses. Gut 2021, 70, 319–329. [Google Scholar] [CrossRef]
- Téllez-Ávila, F.I.; Villalobos-Garita, A.; Giovannini, M.; Chan, C.; Hernández-Calleros, J.; Uscanga, L.; Ramírez-Luna, M.Á. Follow-up of Patients with Pseudotumoral Chronic Pancreatitis: Outcome and Surveillance. World J. Gastroenterol. 2014, 20, 8612–8616. [Google Scholar] [CrossRef]
- Goggins, M.; Overbeek, K.A.; Brand, R.; Syngal, S.; Del Chiaro, M.; Bartsch, D.K.; Bassi, C.; Carrato, A.; Farrell, J.; Fishman, E.K.; et al. Management of Patients with Increased Risk for Familial Pancreatic Cancer: Updated Recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 2020, 69, 7–17. [Google Scholar] [CrossRef]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef]
- Boyd, L.N.C.; Ali, M.; Leeflang, M.M.G.; Treglia, G.; de Vries, R.; Le Large, T.Y.S.; Besselink, M.G.; Giovannetti, E.; van Laarhoven, H.W.M.; Kazemier, G. Diagnostic Accuracy and Added Value of Blood-Based Protein Biomarkers for Pancreatic Cancer: A Meta-Analysis of Aggregate and Individual Participant Data. eClinicalMedicine 2023, 55, 101747. [Google Scholar] [CrossRef] [PubMed]
- Fahrmann, J.F.; Schmidt, C.M.; Mao, X.; Irajizad, E.; Loftus, M.; Zhang, J.; Patel, N.; Vykoukal, J.; Dennison, J.B.; Long, J.P.; et al. Lead-Time Trajectory of CA19-9 as an Anchor Marker for Pancreatic Cancer Early Detection. Gastroenterology 2021, 160, 1373–1383.e6. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Z.-X.; Chen, X.-Y.; Xu, Y.-L.; Yin, N.; Yang, J.; Zhu, D.-M.; Li, D.-C.; Zhou, J. A Panel of Three Biomarkers Identified by ITRAQ for the Early Diagnosis of Pancreatic Cancer. Proteom. Clin. Appl. 2019, 13, e1800195. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bamlet, W.R.; Oberg, A.L.; Chaffee, K.G.; Donahue, G.; Cao, X.-J.; Chari, S.; Garcia, B.A.; Petersen, G.M.; Zaret, K.S. Detection of Early Pancreatic Ductal Adenocarcinoma with Thrombospondin-2 and CA19-9 Blood Markers. Sci. Transl. Med. 2017, 9, eaah5583. [Google Scholar] [CrossRef]
- Staal, B.; Liu, Y.; Barnett, D.; Hsueh, P.; He, Z.; Gao, C.; Partyka, K.; Hurd, M.W.; Singhi, A.D.; Drake, R.R.; et al. The STRA Plasma Biomarker: Blinded Validation of Improved Accuracy Over CA19-9 in Pancreatic Cancer Diagnosis. Clin. Cancer Res. 2019, 25, 2745–2754. [Google Scholar] [CrossRef]
- Brand, R.E.; Persson, J.; Bratlie, S.O.; Chung, D.C.; Katona, B.W.; Carrato, A.; Castillo, M.; Earl, J.; Kokkola, A.; Lucas, A.L.; et al. Detection of Early-Stage Pancreatic Ductal Adenocarcinoma From Blood Samples: Results of a Multiplex Biomarker Signature Validation Study. Clin. Transl. Gastroenterol. 2022, 13, e00468. [Google Scholar] [CrossRef]
- Mahajan, U.M.; Oehrle, B.; Sirtl, S.; Alnatsha, A.; Goni, E.; Regel, I.; Beyer, G.; Vornhülz, M.; Vielhauer, J.; Chromik, A.; et al. Independent Validation and Assay Standardization of Improved Metabolic Biomarker Signature to Differentiate Pancreatic Ductal Adenocarcinoma from Chronic Pancreatitis. Gastroenterology 2022, 163, 1407–1422. [Google Scholar] [CrossRef]
- Sogawa, K.; Takano, S.; Iida, F.; Satoh, M.; Tsuchida, S.; Kawashima, Y.; Yoshitomi, H.; Sanda, A.; Kodera, Y.; Takizawa, H.; et al. Identification of a Novel Serum Biomarker for Pancreatic Cancer, C4b-Binding Protein α-Chain (C4BPA) by Quantitative Proteomic Analysis Using Tandem Mass Tags. Br. J. Cancer 2016, 115, 949–956. [Google Scholar] [CrossRef]
- Capello, M.; Vykoukal, J.V.; Katayama, H.; Bantis, L.E.; Wang, H.; Kundnani, D.L.; Aguilar-Bonavides, C.; Aguilar, M.; Tripathi, S.C.; Dhillon, D.S.; et al. Exosomes Harbor B Cell Targets in Pancreatic Adenocarcinoma and Exert Decoy Function against Complement-Mediated Cytotoxicity. Nat. Commun. 2019, 10, 254. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and Localization of Surgically Resectable Cancers with a Multi-Analyte Blood Test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 Identifies Cancer Exosomes and Detects Early Pancreatic Cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef]
- Alizadeh Savareh, B.; Asadzadeh Aghdaie, H.; Behmanesh, A.; Bashiri, A.; Sadeghi, A.; Zali, M.; Shams, R. A Machine Learning Approach Identified a Diagnostic Model for Pancreatic Cancer through Using Circulating MicroRNA Signatures. Pancreatology 2020, 20, 1195–1204. [Google Scholar] [CrossRef]
- Schultz, N.A.; Dehlendorff, C.; Jensen, B.V.; Bjerregaard, J.K.; Nielsen, K.R.; Bojesen, S.E.; Calatayud, D.; Nielsen, S.E.; Yilmaz, M.; Holländer, N.H.; et al. MicroRNA Biomarkers in Whole Blood for Detection of Pancreatic Cancer. JAMA 2014, 311, 392–404. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Sun, Y.-W.; Liu, D.-J.; Zhang, J.-F.; Li, J.; Hua, R. MicroRNAs in Stool Samples as Potential Screening Biomarkers for Pancreatic Ductal Adenocarcinoma Cancer. Am. J. Cancer Res. 2014, 4, 663–673. [Google Scholar]
- Kisiel, J.B.; Yab, T.C.; Taylor, W.R.; Chari, S.T.; Petersen, G.M.; Mahoney, D.W.; Ahlquist, D.A. Stool DNA Testing for the Detection of Pancreatic Cancer: Assessment of Methylation Marker Candidates. Cancer 2012, 118, 2623–2631. [Google Scholar] [CrossRef]
- Yi, J.M.; Guzzetta, A.A.; Bailey, V.J.; Downing, S.R.; Van Neste, L.; Chiappinelli, K.B.; Keeley, B.P.; Stark, A.; Herrera, A.; Wolfgang, C.; et al. Novel Methylation Biomarker Panel for the Early Detection of Pancreatic Cancer. Clin. Cancer Res. 2013, 19, 6544–6555. [Google Scholar] [CrossRef]
- Arasaradnam, R.P.; Wicaksono, A.; O’Brien, H.; Kocher, H.M.; Covington, J.A.; Crnogorac-Jurcevic, T. Noninvasive Diagnosis of Pancreatic Cancer Through Detection of Volatile Organic Compounds in Urine. Gastroenterology 2018, 154, 485–487.e1. [Google Scholar] [CrossRef]
- Mayerle, J.; Kalthoff, H.; Reszka, R.; Kamlage, B.; Peter, E.; Schniewind, B.; González Maldonado, S.; Pilarsky, C.; Heidecke, C.-D.; Schatz, P.; et al. Metabolic Biomarker Signature to Differentiate Pancreatic Ductal Adenocarcinoma from Chronic Pancreatitis. Gut 2018, 67, 128–137. [Google Scholar] [CrossRef]
- Liu, H.-J.; Guo, Y.-Y.; Li, D.-J. Predicting Novel Salivary Biomarkers for the Detection of Pancreatic Cancer Using Biological Feature-Based Classification. Pathol. Res. Pract. 2017, 213, 394–399. [Google Scholar] [CrossRef]
- Pereira, S.P.; Oldfield, L.; Ney, A.; Hart, P.A.; Keane, M.G.; Pandol, S.J.; Li, D.; Greenhalf, W.; Jeon, C.Y.; Koay, E.J.; et al. Early Detection of Pancreatic Cancer. Lancet Gastroenterol. Hepatol. 2020, 5, 698–710. [Google Scholar] [CrossRef]
- Tong, T.; Gu, J.; Xu, D.; Song, L.; Zhao, Q.; Cheng, F.; Yuan, Z.; Tian, S.; Yang, X.; Tian, J.; et al. Deep Learning Radiomics Based on Contrast-Enhanced Ultrasound Images for Assisted Diagnosis of Pancreatic Ductal Adenocarcinoma and Chronic Pancreatitis. BMC Med. 2022, 20, 74. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-L.; Wu, T.; Chen, P.-T.; Tsai, Y.M.; Roth, H.; Wu, M.-S.; Liao, W.-C.; Wang, W. Deep Learning to Distinguish Pancreatic Cancer Tissue from Non-Cancerous Pancreatic Tissue: A Retrospective Study with Cross-Racial External Validation. Lancet Digit. Health 2020, 2, e303–e313. [Google Scholar] [CrossRef] [PubMed]
- Aslan, S.; Nural, M.S.; Camlidag, I.; Danaci, M. Efficacy of Perfusion CT in Differentiating of Pancreatic Ductal Adenocarcinoma from Mass-Forming Chronic Pancreatitis and Characterization of Isoattenuating Pancreatic Lesions. Abdom. Radiol. 2019, 44, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.K.; Sharma, R.; Kandasamy, D.; Pradhan, R.K.; Garg, P.K.; Bhalla, A.S.; Gamanagatti, S.; Srivastava, D.N.; Sahni, P.; Upadhyay, A.D. Perfusion CT—Can It Resolve the Pancreatic Carcinoma versus Mass Forming Chronic Pancreatitis Conundrum? Pancreatology 2016, 16, 979–987. [Google Scholar] [CrossRef]
- Conwell, D.L.; Lee, L.S.; Yadav, D.; Longnecker, D.S.; Miller, F.H.; Mortele, K.J.; Levy, M.J.; Kwon, R.; Lieb, J.G.; Stevens, T.; et al. American Pancreatic Association Practice Guidelines in Chronic Pancreatitis: Evidence-Based Report on Diagnostic Guidelines. Pancreas 2014, 43, 1143–1162. [Google Scholar] [CrossRef]
- Boninsegna, E.; Manfredi, R.; Negrelli, R.; Avesani, G.; Mehrabi, S.; Pozzi Mucelli, R. Pancreatic Duct Stenosis: Differential Diagnosis between Malignant and Benign Conditions at Secretin-Enhanced MRCP. Clin. Imaging 2017, 41, 137–143. [Google Scholar] [CrossRef]
- Deng, Y.; Ming, B.; Zhou, T.; Wu, J.-L.; Chen, Y.; Liu, P.; Zhang, J.; Zhang, S.-Y.; Chen, T.-W.; Zhang, X.-M. Radiomics Model Based on MR Images to Discriminate Pancreatic Ductal Adenocarcinoma and Mass-Forming Chronic Pancreatitis Lesions. Front. Oncol. 2021, 11, 620981. [Google Scholar] [CrossRef]
- Wolske, K.M.; Ponnatapura, J.; Kolokythas, O.; Burke, L.M.B.; Tappouni, R.; Lalwani, N. Chronic Pancreatitis or Pancreatic Tumor? A Problem-Solving Approach. RadioGraphics 2019, 39, 1965–1982. [Google Scholar] [CrossRef]
- Uchida, K.; Okazaki, K. Clinical and Pathophysiological Aspects of Type 1 Autoimmune Pancreatitis. J. Gastroenterol. 2018, 53, 475–483. [Google Scholar] [CrossRef]
- Sandrasegaran, K.; Menias, C.O. Imaging in Autoimmune Pancreatitis and Immunoglobulin G4-Related Disease of the Abdomen. Gastroenterol. Clin. N. Am. 2018, 47, 603–619. [Google Scholar] [CrossRef]
- Schima, W.; Böhm, G.; Rösch, C.S.; Klaus, A.; Függer, R.; Kopf, H. Mass-Forming Pancreatitis versus Pancreatic Ductal Adenocarcinoma: CT and MR Imaging for Differentiation. Cancer Imaging 2020, 20, 52. [Google Scholar] [CrossRef]
- Elsherif, S.B.; Virarkar, M.; Javadi, S.; Ibarra-Rovira, J.J.; Tamm, E.P.; Bhosale, P.R. Pancreatitis and PDAC: Association and Differentiation. Abdom. Radiol. 2020, 45, 1324–1337. [Google Scholar] [CrossRef]
- Srisajjakul, S.; Prapaisilp, P.; Bangchokdee, S. CT and MR Features That Can Help to Differentiate between Focal Chronic Pancreatitis and Pancreatic Cancer. Radiol. Med. 2020, 125, 356–364. [Google Scholar] [CrossRef]
- Overbeek, K.A.; Goggins, M.G.; Dbouk, M.; Levink, I.J.M.; Koopmann, B.D.M.; Chuidian, M.; Konings, I.C.A.W.; Paiella, S.; Earl, J.; Fockens, P.; et al. Timeline of Development of Pancreatic Cancer and Implications for Successful Early Detection in High-Risk Individuals. Gastroenterology 2022, 162, 772–785.e4. [Google Scholar] [CrossRef]
- Barthet, M.; Portal, I.; Boujaoude, J.; Bernard, J.-P.; Sahel, J. Endoscopic Ultrasonographic Diagnosis of Pancreatic Cancer Complicating Chronic Pancreatitis. Endoscopy 1996, 28, 487–491. [Google Scholar] [CrossRef]
- Tonozuka, R.; Itoi, T.; Nagata, N.; Kojima, H.; Sofuni, A.; Tsuchiya, T.; Ishii, K.; Tanaka, R.; Nagakawa, Y.; Mukai, S. Deep Learning Analysis for the Detection of Pancreatic Cancer on Endosonographic Images: A Pilot Study. J. Hepatobiliary Pancreat. Sci. 2021, 28, 95–104. [Google Scholar] [CrossRef]
- Grassia, R.; Imperatore, N.; Capone, P.; Cereatti, F.; Forti, E.; Antonini, F.; Tanzi, G.P.; Martinotti, M.; Buffoli, F.; Mutignani, M.; et al. EUS-Guided Tissue Acquisition in Chronic Pancreatitis: Differential Diagnosis between Pancreatic Cancer and Pseudotumoral Masses Using EUS-FNA or Core Biopsy. Endosc. Ultrasound 2020, 9, 122–129. [Google Scholar] [CrossRef]
- Ardengh, J.C.; Lopes, C.V.; Campos, A.D.; Pereira de Lima, L.F.; Venco, F.; Módena, J.L.P. Endoscopic Ultrasound and Fine Needle Aspiration in Chronic Pancreatitis: Differential Diagnosis between Pseudotumoral Masses and Pancreatic Cancer. JOP 2007, 8, 413–421. [Google Scholar]
- Bournet, B.; Souque, A.; Senesse, P.; Assenat, E.; Barthet, M.; Lesavre, N.; Aubert, A.; O’Toole, D.; Hammel, P.; Levy, P.; et al. Endoscopic Ultrasound-Guided Fine-Needle Aspiration Biopsy Coupled with KRAS Mutation Assay to Distinguish Pancreatic Cancer from Pseudotumoral Chronic Pancreatitis. Endoscopy 2009, 41, 552–557. [Google Scholar] [CrossRef]
- Gheonea, D.I.; Streba, C.T.; Ciurea, T.; Săftoiu, A. Quantitative Low Mechanical Index Contrast-Enhanced Endoscopic Ultrasound for the Differential Diagnosis of Chronic Pseudotumoral Pancreatitis and Pancreatic Cancer. BMC Gastroenterol. 2013, 13, 2. [Google Scholar] [CrossRef]
- Săftoiu, A.; Iordache, S.A.; Gheonea, D.I.; Popescu, C.; Maloş, A.; Gorunescu, F.; Ciurea, T.; Iordache, A.; Popescu, G.L.; Manea, C.T.L. Combined Contrast-Enhanced Power Doppler and Real-Time Sonoelastography Performed during EUS, Used in the Differential Diagnosis of Focal Pancreatic Masses (with Videos). Gastrointest. Endosc. 2010, 72, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Garcia, J.; Domínguez-Muñoz, J.E.; Castiñeira-Alvariño, M.; Luaces-Regueira, M.; Lariño-Noia, J. Quantitative Elastography Associated with Endoscopic Ultrasound for the Diagnosis of Chronic Pancreatitis. Endoscopy 2013, 45, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Shimokawa, T.; Napoléon, B.; Fusaroli, P.; Gincul, R.; Kudo, M.; Kitano, M. Value of Contrast-Enhanced Harmonic Endoscopic Ultrasonography with Enhancement Pattern for Diagnosis of Pancreatic Cancer: A Meta-Analysis. Dig. Endosc. 2019, 31, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Selves, J.; Grand, D.; Danjoux, M.; Hanoun, N.; Cordelier, P.; Buscail, L. Endoscopic Ultrasound-Guided Fine-Needle Aspiration Biopsy Coupled with a KRAS Mutation Assay Using Allelic Discrimination Improves the Diagnosis of Pancreatic Cancer. J. Clin. Gastroenterol. 2015, 49, 50–56. [Google Scholar] [CrossRef]
- Bournet, B.; Gayral, M.; Torrisani, J.; Selves, J.; Cordelier, P.; Buscail, L. Role of Endoscopic Ultrasound in the Molecular Diagnosis of Pancreatic Cancer. World J. Gastroenterol. 2014, 20, 10758–10768. [Google Scholar] [CrossRef]
- Bournet, B.; Pointreau, A.; Souque, A.; Oumouhou, N.; Muscari, F.; Lepage, B.; Senesse, P.; Barthet, M.; Lesavre, N.; Hammel, P.; et al. Gene Expression Signature of Advanced Pancreatic Ductal Adenocarcinoma Using Low Density Array on Endoscopic Ultrasound-Guided Fine Needle Aspiration Samples. Pancreatology 2012, 12, 27–34. [Google Scholar] [CrossRef]
- Bournet, B.; Muscari, F.; Buscail, C.; Assenat, E.; Barthet, M.; Hammel, P.; Selves, J.; Guimbaud, R.; Cordelier, P.; Buscail, L. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin. Transl. Gastroenterol. 2016, 7, e157. [Google Scholar] [CrossRef]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical Implications of Genomic Alterations in the Tumour and Circulation of Pancreatic Cancer Patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef]
- Brychta, N.; Krahn, T.; von Ahsen, O. Detection of KRAS Mutations in Circulating Tumor DNA by Digital PCR in Early Stages of Pancreatic Cancer. Clin. Chem. 2016, 62, 1482–1491. [Google Scholar] [CrossRef]
- Park, G.; Park, J.K.; Son, D.-S.; Shin, S.-H.; Kim, Y.J.; Jeon, H.-J.; Lee, J.; Park, W.-Y.; Lee, K.H.; Park, D. Utility of Targeted Deep Sequencing for Detecting Circulating Tumor DNA in Pancreatic Cancer Patients. Sci. Rep. 2018, 8, 11631. [Google Scholar] [CrossRef]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined Circulating Tumor DNA and Protein Biomarker-Based Liquid Biopsy for the Earlier Detection of Pancreatic Cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef]
- Lee, B.; Lipton, L.; Cohen, J.; Tie, J.; Javed, A.A.; Li, L.; Goldstein, D.; Burge, M.; Cooray, P.; Nagrial, A.; et al. Circulating Tumor DNA as a Potential Marker of Adjuvant Chemotherapy Benefit Following Surgery for Localized Pancreatic Cancer. Ann. Oncol. 2019, 30, 1472–1478. [Google Scholar] [CrossRef]
- Buscail, E.; Maulat, C.; Muscari, F.; Chiche, L.; Cordelier, P.; Dabernat, S.; Alix-Panabières, C.; Buscail, L. Liquid Biopsy Approach for Pancreatic Ductal Adenocarcinoma. Cancers 2019, 11, 852. [Google Scholar] [CrossRef]
- Amintas, S.; Bedel, A.; Moreau-Gaudry, F.; Boutin, J.; Buscail, L.; Merlio, J.-P.; Vendrely, V.; Dabernat, S.; Buscail, E. Circulating Tumor Cell Clusters: United We Stand Divided We Fall. Int. J. Mol. Sci. 2020, 21, 2653. [Google Scholar] [CrossRef]
- Amintas, S.; Vendrely, V.; Dupin, C.; Buscail, L.; Laurent, C.; Bournet, B.; Merlio, J.-P.; Bedel, A.; Moreau-Gaudry, F.; Boutin, J.; et al. Next-Generation Cancer Biomarkers: Extracellular Vesicle DNA as a Circulating Surrogate of Tumor DNA. Front. Cell Dev. Biol. 2020, 8, 622048. [Google Scholar] [CrossRef]
- du Rieu, M.C.; Torrisani, J.; Selves, J.; Al Saati, T.; Souque, A.; Dufresne, M.; Tsongalis, G.J.; Suriawinata, A.A.; Carrère, N.; Buscail, L.; et al. MicroRNA-21 Is Induced Early in Pancreatic Ductal Adenocarcinoma Precursor Lesions. Clin. Chem. 2010, 56, 603–612. [Google Scholar] [CrossRef]
- Hanoun, N.; Delpu, Y.; Suriawinata, A.A.; Bournet, B.; Bureau, C.; Selves, J.; Tsongalis, G.J.; Dufresne, M.; Buscail, L.; Cordelier, P.; et al. The Silencing of MicroRNA 148a Production by DNA Hypermethylation Is an Early Event in Pancreatic Carcinogenesis. Clin. Chem. 2010, 56, 1107–1118. [Google Scholar] [CrossRef]
- Sicard, F.; Gayral, M.; Lulka, H.; Buscail, L.; Cordelier, P. Targeting MiR-21 for the Therapy of Pancreatic Cancer. Mol. Ther. 2013, 21, 986–994. [Google Scholar] [CrossRef]
- Delpu, Y.; Lulka, H.; Sicard, F.; Saint-Laurent, N.; Lopez, F.; Hanoun, N.; Buscail, L.; Cordelier, P.; Torrisani, J. The Rescue of MiR-148a Expression in Pancreatic Cancer: An Inappropriate Therapeutic Tool. PLoS ONE 2013, 8, e55513. [Google Scholar] [CrossRef]
- Humeau, M.; Vignolle-Vidoni, A.; Sicard, F.; Martins, F.; Bournet, B.; Buscail, L.; Torrisani, J.; Cordelier, P. Salivary MicroRNA in Pancreatic Cancer Patients. PLoS ONE 2015, 10, e0130996. [Google Scholar] [CrossRef]
- Diaz-Riascos, Z.V.; Ginesta, M.M.; Fabregat, J.; Serrano, T.; Busquets, J.; Buscail, L.; Cordelier, P.; Capellá, G. Expression and Role of MicroRNAs from the MiR-200 Family in the Tumor Formation and Metastatic Propensity of Pancreatic Cancer. Mol. Nucleic Acids 2019, 17, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Tijunelyte, I.; Malbec, R.; Chami, B.; Cacheux, J.; Dez, C.; Leichlé, T.; Cordelier, P.; Bancaud, A. Micro-RNA 21 Detection with a Limit of 2 PM in 1 Min Using a Size-Accordable Concentration Module Operated by Electrohydrodynamic Actuation. Biosens. Bioelectron. 2021, 178, 112992. [Google Scholar] [CrossRef] [PubMed]
- Vila-Navarro, E.; Duran-Sanchon, S.; Vila-Casadesús, M.; Moreira, L.; Ginès, À.; Cuatrecasas, M.; Lozano, J.J.; Bujanda, L.; Castells, A.; Gironella, M. Novel Circulating MiRNA Signatures for Early Detection of Pancreatic Neoplasia. Clin. Transl. Gastroenterol. 2019, 10, e00029. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.A.; Rashid, S.; Singh, N.; Rashid, S.; Singh, V.; Gunjan, D.; Das, P.; Dash, N.R.; Pandey, R.M.; Chauhan, S.S.; et al. Panel of Serum MiRNAs as Potential Non-Invasive Biomarkers for Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2021, 11, 2824. [Google Scholar] [CrossRef]
- Reese, M.; Flammang, I.; Yang, Z.; Dhayat, S.A. Potential of Exosomal MicroRNA-200b as Liquid Biopsy Marker in Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 197. [Google Scholar] [CrossRef]
- Wang, L.; Wu, J.; Ye, N.; Li, F.; Zhan, H.; Chen, S.; Xu, J. Plasma-Derived Exosome MiR-19b Acts as a Diagnostic Marker for Pancreatic Cancer. Front. Oncol. 2021, 11, 739111. [Google Scholar] [CrossRef]
- Greenhalf, W.; Lévy, P.; Gress, T.; Rebours, V.; Brand, R.E.; Pandol, S.; Chari, S.; Jørgensen, M.T.; Mayerle, J.; Lerch, M.M.; et al. International Consensus Guidelines on Surveillance for Pancreatic Cancer in Chronic Pancreatitis. Recommendations from the Working Group for the International Consensus Guidelines for Chronic Pancreatitis in Collaboration with the International Association of Pancreatology, the American Pancreatic Association, the Japan Pancreas Society, and European Pancreatic Club. Pancreatology 2020, 20, 910–918. [Google Scholar] [CrossRef]
- Culetto, A.; Bournet, B.; Buscail, L. Clinical Profile of Cannabis-Associated Acute Pancreatitis. Dig. Liver Dis. 2017, 49, 1284–1285. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors, Year, [Ref.] | Genetically Engineered Model | Pathway Affected and Phenotype |
|---|---|---|
| Muller-Decker et al., 2006 [24] | Krt5-Cox2Tg | Cox2 overexpression: chronic pancreatitis and ductal neoplastic lesions |
| Al Saati et al., 2013 [29] | Pdx1-Cre;KrasG12D;TP53INP1-/- | Oncogenic RAS + oxidative status dysregulation: accelerated PanINs formation |
| Daniluk et al., 2012 [30] | Ela-CreERT;KrasG12D;CoxTg | Oncogenic RAS + Cox2 overexpression: rapid development of chronic inflammation and PanINs |
| Maniati et al., 2011 [26] | Pdx1-Cre;KrasG12D;IKK2 FI/FI | Oncogenic RAS + NfκB pathway inhibition: impaired PanIN formation and decreased PDAC development |
| Daniluk et al., 2012 [30] | Ela-CreERT;KrasG12D;IKK2Tg | Oncogenic RAS + NfκB pathway activation: increased fibrosis and rapid development of PanINs |
| Lesina et al., 2011 [31] | Ptf1a-CreEx1;KrasG12D;Socs3 FI/FI | Oncogenic RAS + Stat3 activation: accelerated PanIN progression and increased PDAC formation |
| Guerra et al., 2011 [32] | KrasG12V;p16Ink4a/p19arf Iox/lox;Ela-tTA/tetO-Cre KrasG12V;Trp53 Iox/lox;Elas-tTA/tetO-Cre | Oncogenic RAS + loss of p16Ink4a/p19arf or Trp53 + cerulein injections: increased PDAC formation and progression |
| Liou et al., 2016 [33] | Ptf1a/p48Cre/+ LSL-KrasG12D/+ PKD1fl/fl | Treatment of mice by mitochondria-targeted antioxidant MitoQ: reduced KRAS-induced formation of ROS with reduced formation of PanINs |
| Etiology | Pancreatic Cancer Estimated Risk |
|---|---|
| Alcoholic CP | Incidence of 2 and 4% after 5 and 20 years of evolution, respectively. |
| Hereditary pancreatitis (PRSS1) | Incidence of 10, 19, and 53.5% at 50, 60, and 75 years, respectively. |
| SPINK1 mutations | Incidence of 12, 28, and 52% at 60, 70, and 80 years. |
| CFTR mutations | Increased risk of PDAC of 1.41 compared to control patients. |
| CTRC, CASR, CLDN2, CPA1, TRPV6, CEL-HYB mutations | No available data due to very low incidence of these mutations. |
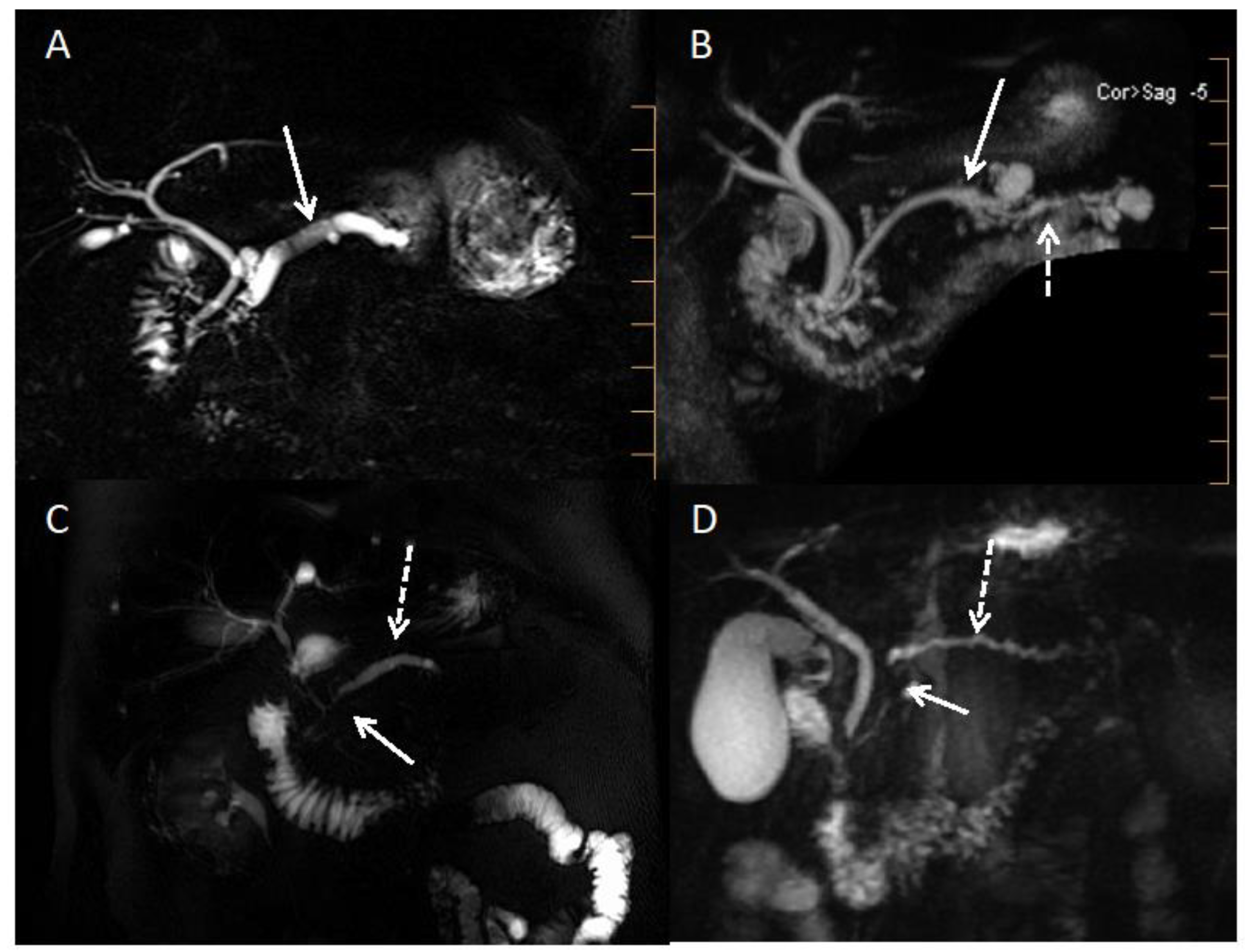
| Signs Evocative of Benign Disease | Signs Evocative of Malignant Disease | |
|---|---|---|
| Parenchymal signs | Pseudotumorous CP and IPMN:
|
|
Paraduodenal pancreatitis:
| ||
AIP:
| ||
| Duct signs | Obstructive CP and IPMN:
|
|
| Vessels signs | Pseudotumorous CP:
|
|
| Other signs | AIP:
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Cosquer, G.; Maulat, C.; Bournet, B.; Cordelier, P.; Buscail, E.; Buscail, L. Pancreatic Cancer in Chronic Pancreatitis: Pathogenesis and Diagnostic Approach. Cancers 2023, 15, 761. https://doi.org/10.3390/cancers15030761
Le Cosquer G, Maulat C, Bournet B, Cordelier P, Buscail E, Buscail L. Pancreatic Cancer in Chronic Pancreatitis: Pathogenesis and Diagnostic Approach. Cancers. 2023; 15(3):761. https://doi.org/10.3390/cancers15030761
Chicago/Turabian StyleLe Cosquer, Guillaume, Charlotte Maulat, Barbara Bournet, Pierre Cordelier, Etienne Buscail, and Louis Buscail. 2023. "Pancreatic Cancer in Chronic Pancreatitis: Pathogenesis and Diagnostic Approach" Cancers 15, no. 3: 761. https://doi.org/10.3390/cancers15030761
APA StyleLe Cosquer, G., Maulat, C., Bournet, B., Cordelier, P., Buscail, E., & Buscail, L. (2023). Pancreatic Cancer in Chronic Pancreatitis: Pathogenesis and Diagnostic Approach. Cancers, 15(3), 761. https://doi.org/10.3390/cancers15030761