
CAR-T: What Is Next?
Abstract
Simple Summary
Abstract
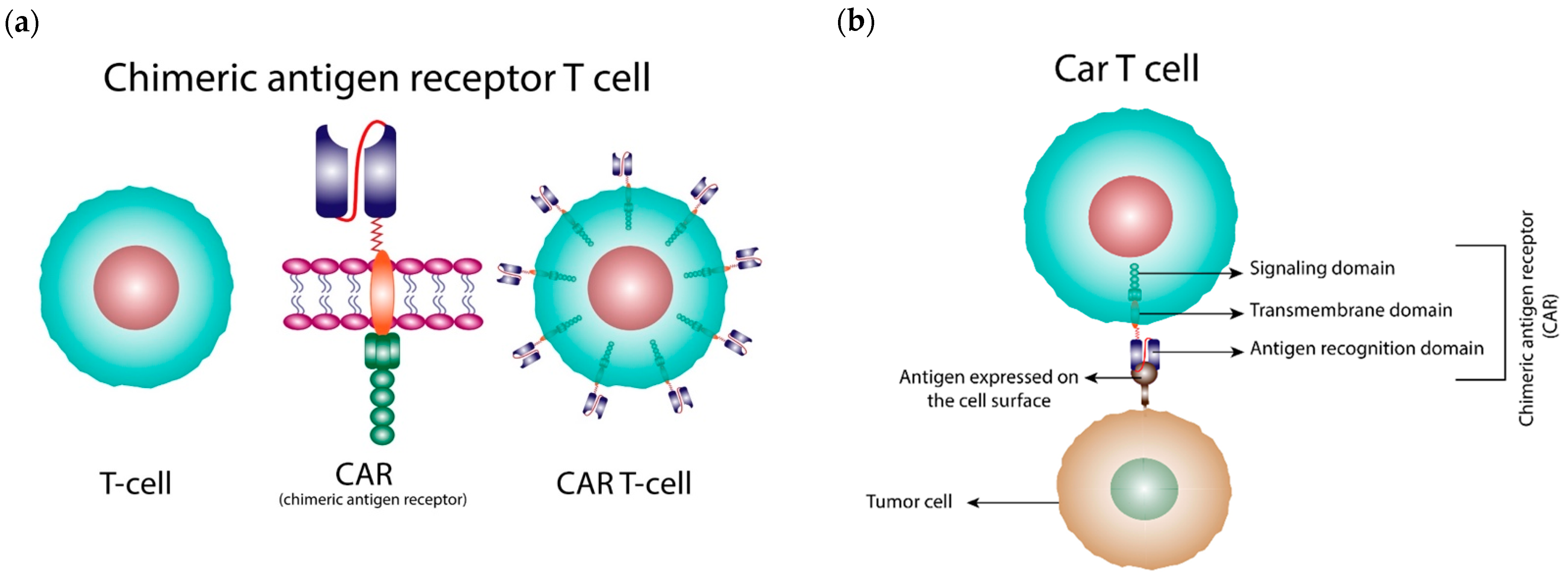
1. Introduction
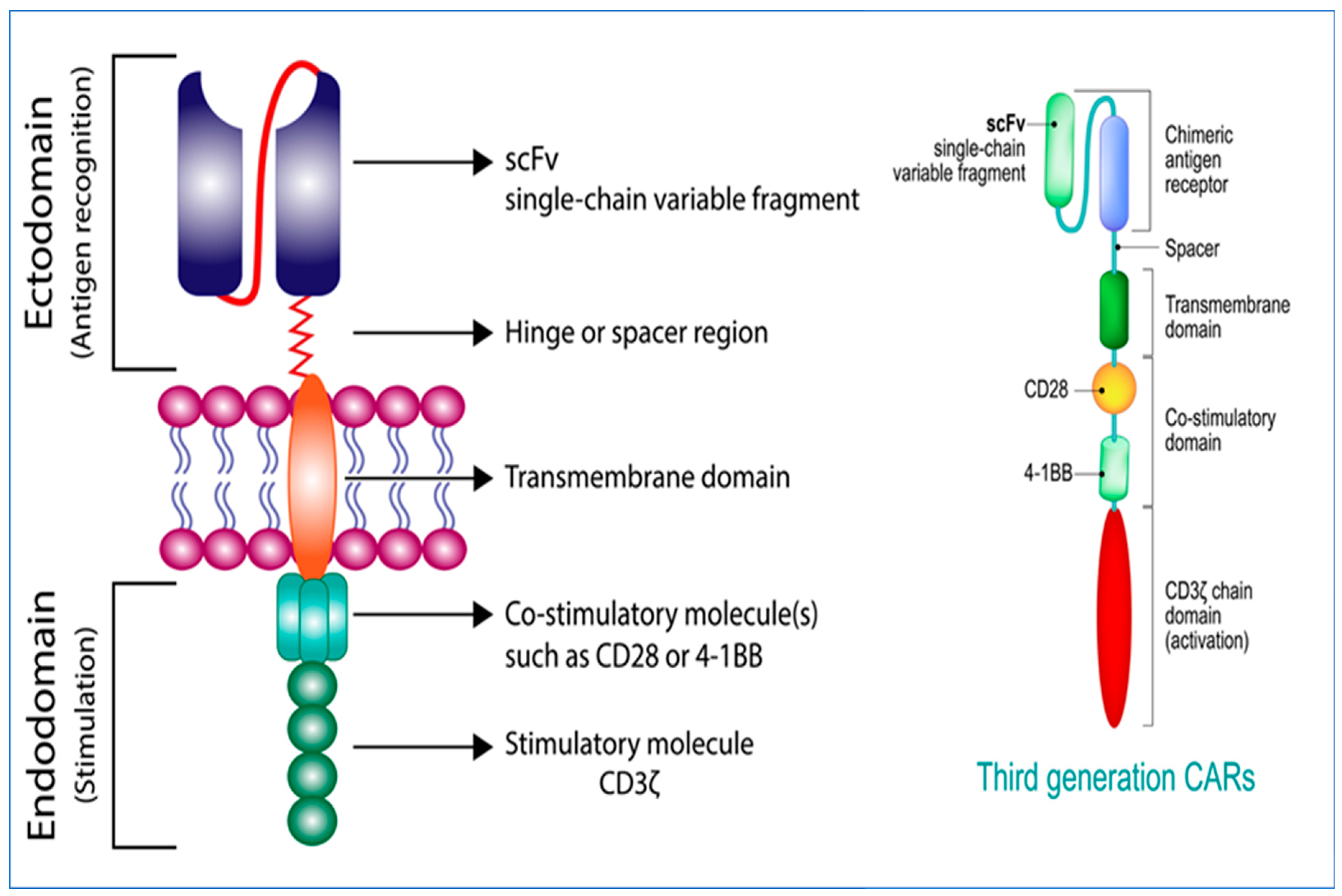
2. Generations of CARs
3. Efficacy Issues and Strategies to Overcome
3.1. Mechanisms of Relapse to CAR-T Cell Therapy
3.2. Strategies to Overcome CAR-T Relapse
3.3. Strategies to Overcome Antigen-Positive Relapse
3.3.1. Combination with Other Therapy
3.3.2. Novel CAR Designs
3.4. Strategies to Overcome Antigen-Negative Relapse
3.4.1. Combination with Other Therapy
3.4.2. Novel CAR Designs
4. CAR-T Therapy Side Effects and Strategies to Overcome Them
4.1. On-Target, On-Tumor Activity: Cytokine-Related Toxicity
4.2. Strategies to Minimize Cytokine-Related Toxicity
4.2.1. Combination with Other Therapy
4.2.2. Novel CAR Designs
ON-Switch CARs and OFF-Switch CARs
Suicide Gene/Receptors
4.3. On-Target, Off-Tumor Activity
4.4. Novel CAR Designs to Avoid On-Target, Off-Tumor Activity
4.4.1. Dual CARs with a “NOT” Logic Gate
4.4.2. Dual CARs with an “AND” Logic Gate
5. Challenges of CAR-T Cells in Solid Tumors and Strategies to Overcome Them
5.1. Antigen Specificity/Heterogeneity
5.2. Cell Trafficking and Infiltration
5.3. The Tumor Microenvironment
5.4. CAR-T Studies on Solid Tumors
6. CAR-Based Cellular Therapies in the Future: Off-the-Shelf CARs and Next-Generation CARs
6.1. Off-the-Shelf CAR-T Cells
6.2. Next-Generation CAR Cells
7. Discussion: Current Challenges of CAR-T Cell Therapy and Future Development Activity
7.1. Target Antigen Selection
7.2. Efficacy, Safety, and Clinical Application Extensions
7.3. Treatment Costs
7.4. Future Perspectives
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- FDA. Package Insert-KYMRIAH. 2022. Available online: https://www.fda.gov/media/107296/download (accessed on 13 December 2022).
- FDA. Package Insert-YESCARTA. 2022. Available online: https://www.fda.gov/media/108377/download (accessed on 13 December 2022).
- FDA. Package Insert-TECARTUS. 2022. Available online: https://www.fda.gov/media/140409/download (accessed on 13 December 2022).
- FDA. Package Insert-BREYANZI. 2022. Available online: https://www.fda.gov/media/145711/download (accessed on 13 December 2022).
- FDA. Package Insert-ABECMA. 2021. Available online: https://www.fda.gov/media/147055/download (accessed on 13 December 2022).
- FDA. Package Insert-CARVYKTI. 2022. Available online: https://www.fda.gov/media/156560/download (accessed on 13 December 2022).
- Srour, S.A.; Singh, H.; McCarty, J.; De Groot, E.; Huls, H.; Rondon, G.; Qazilbash, M.; Ciurea, S.; Bardelli, G.; Buck, J.; et al. Long-term outcomes of Sleeping Beauty-generated CD19-specific CAR T-cell therapy for relapsed-refractory B-cell lymphomas. Blood 2020, 135, 862–865. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V.; Porter, D.L. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Locke, F.L.; Lin, Y.; Jain, N.; Daver, N.; Gulbis, A.M.; Adkins, S.; et al. Toxicity management after chimeric antigen receptor T cell therapy: One size does not fit ‘ALL’. Nat. Rev. Clin. Oncol. 2018, 15, 218. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Akhoundi, M.; Mohammadi, M.; Sahraei, S.S.; Sheykhhasan, M.; Fayazi, N. CAR T cell therapy as a promising approach in cancer immunotherapy: Challenges and opportunities. Cell Oncol. 2021, 44, 495–523. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Hill, T.; Shamah, S.M.; Salter, A.I.; Chen, Y.; Mohler, K.M.; Riddell, S.R. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia 2017, 31, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J. Immunother. 2017, 40, 187–195. [Google Scholar] [CrossRef]
- Inoue, D.; Bradley, R.K.; Abdel-Wahab, O. Spliceosomal gene mutations in myelodysplasia: Molecular links to clonal abnormalities of hematopoiesis. Genes Dev. 2016, 30, 989–1001. [Google Scholar] [CrossRef]
- Butler, M.O.; Hirano, N. Human cell-based artificial antigen-presenting cells for cancer immunotherapy. Immunol. Rev. 2014, 257, 191–209. [Google Scholar] [CrossRef]
- Hasan, A.N.; Selvakumar, A.; O’Reilly, R.J. Artificial Antigen Presenting Cells: An Off the Shelf Approach for Generation of Desirable T-Cell Populations for Broad Application of Adoptive Immunotherapy. Adv. Genet. Eng. 2015, 4, 130. [Google Scholar]
- ClinicalTrials.gov. Pilot Study of T-APCs Following CAR T Cell Immunotherapy for CD19+ Leukemia. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03186118 (accessed on 17 July 2022).
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Zheng, W.; O’Hear, C.E.; Alli, R.; Basham, J.H.; Abdelsamed, H.A.; Palmer, L.E.; Jones, L.L.; Youngblood, B.; Geiger, T.L. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia 2018, 32, 1157–1167. [Google Scholar] [CrossRef]
- Edeline, J.; Houot, R.; Marabelle, A.; Alcantara, M. CAR-T cells and BiTEs in solid tumors: Challenges and perspectives. J. Hematol. Oncol. 2021, 14, 65. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Zah, E.; Lin, M.Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef]
- Hanada, K.; Restifo, N.P. Double or nothing on cancer immunotherapy. Nat. Biotechnol. 2013, 31, 33–34. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 125. [Google Scholar] [CrossRef] [PubMed]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [PubMed]
- de Lima, S.C.G.; Fantacini, D.M.C.; de Castro Batista, L.; Silveira, R.M.; Furtado, I.P.; Rossetti, R.; Brand, H.; Covas, D.T.; de Souza, L.E.B. Strategies to Enhance the Therapeutic Efficacy, Applicability, and Safety of Genetically Engineered Immune Cells. Crit. Rev. Immunol. 2021, 41, 41–67. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Dietrich, J.; Frigault, M. Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS) UpToDate. 2021. Available online: https://www.uptodate.com/contents/immune-effector-cell-associated-neurotoxicity-syndrome-icans?search=Immune%20%20Effector%20%20Cell-Associated%20%20Neurotoxicity%20%20Syndrome%20%20(ICANS)&source=search_result&selectedTitle=1~14&usage_type=default&display_rank=1 (accessed on 17 July 2022).
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Tkach, D.; Busser, B.W.; Temburni, S.; Valton, J.; Duclert, A.; Poirot, L.; Depil, S.; Duchateau, P. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. 2019, 19, 44. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.S.; Lamb, L.S.; Goldman, F.; Di Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5, 254. [Google Scholar] [CrossRef] [PubMed]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.Y.; Chung, W.H.; Chu, M.T.; Chen, S.J.; Chen, H.C.; Zheng, L.; Hung, S.I. Recent Development and Clinical Application of Cancer Vaccine: Targeting Neoantigens. J. Immunol.Res. 2018, 2018, 4325874. [Google Scholar] [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef]
- Wing, A.; Fajardo, C.A.; Posey, A.D.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol. Res. 2018, 6, 605–616. [Google Scholar] [CrossRef]
- Lohmueller, J.J.; Ham, J.D.; Kvorjak, M.; Finn, O.J. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology 2017, 7, e1368604. [Google Scholar] [CrossRef] [PubMed]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef]
- Wang, L.C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; Moon, E.; Albelda, S.M. Chimeric antigen receptor T-cell therapy for solid tumors. Mol. Ther. Oncolytics 2016, 3, 16006. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Sun, C.; Bernatchez, C.; Xia, X.; Hwu, P.; Dotti, G.; Li, S. T-cell Homing Therapy for Reducing Regulatory T Cells and Preserving Effector T-cell Function in Large Solid Tumors. Clin. Cancer Res. 2018, 24, 2920–2934. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132 (Suppl. 1), 556. [Google Scholar] [CrossRef]
- Kueberuwa, G.; Kalaitsidou, M.; Cheadle, E.; Hawkins, R.E.; Gilham, D.E. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol. Ther. Oncolytics 2018, 8, 41–51. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- Patel, U.; Abernathy, J.; Savani, B.N.; Oluwole, O.; Sengsayadeth, S.; Dholaria, B. CAR T cell therapy in solid tumors: A review of current clinical trials. EJHaem 2022, 3 (Suppl. 1), 24–31. [Google Scholar] [CrossRef]
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tang, Q. Recent updates on chimeric antigen receptor T cell therapy for hepatocellular carcinoma. Cancer Gene Ther. 2021, 28, 1075–1087. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [PubMed]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug. Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Liu, P.Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovitch, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef]
- Leen, A.M.; Bollard, C.M.; Mendizabal, A.M.; Shpall, E.J.; Szabolcs, P.; Antin, J.H.; Kapoor, N.; Pai, S.Y.; Rowley, S.D.; Kebriaei, P.; et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood 2013, 121, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, K.J.; Gottschalk, S.; Talleur, A.C. Allogeneic CAR Cell Therapy-More Than a Pipe Dream. Front. Immunol. 2020, 11, 618427. [Google Scholar] [CrossRef] [PubMed]
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K.; et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef]
- Mehta, R.S.; Rezvani, K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Z.; Tan, X.; Jiang, H.; Xu, Z.; Fang, Y.; Han, D.; Hong, W.; Wei, W.; Tu, J. CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 2021, 139, 111605. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Studies Found for: CAR-NK|Recruiting, Active, Not Recruiting Studies. 2022. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=CAR-NK&cntry=&state=&city=&dist= (accessed on 29 September 2022).
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. CAR-Macrophages for the Treatment of HER2 Overexpressing Solid Tumors. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04660929?term=CAR-macrophages&draw=2&rank=2 (accessed on 1 October 2022).
- Wei, J.; Han, X.; Bo, J.; Han, W. Target selection for CAR-T therapy. J. Hematol. Oncol. 2019, 12, 62. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow. Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- de Lima Lopes, G.; Nahas, G.R. Chimeric antigen receptor T cells, a savior with a high price. Chin. Clin. Oncol. 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| CAR-T Cell Product Name and FDA Approved Date | Indication(s) | Target Antigen |
|---|---|---|
| Kymriah® (tisagenlecleucel) - Approved by FDA in 2017 |
| CD19 |
| Yescarta® (axicabtagene ciloleucel) - Approved by FDA in 2017 |
| CD19 |
| Tecartus® (brexucabtagene autoleucal) - Approved by FDA in 2020 |
| CD19 |
| Breyanzi® (lisocabtagene maraleucel) - Approved by FDA in 2021 | Adult patients with large B-cell lymphoma (LBCL), including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (including DLBCL arising from indolent lymphoma), highgrade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B, who have:
| CD19 |
| Abecma® (idecabtagene vicleucel) - Approved by FDA in 2021 | Adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. | BCMA |
| Carvykti® (ciltacabtagene autoleucel) - Approved by FDA in 2022 | Adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody. | BCMA |
| Efficacy | Safety | Solid Tumours | |||||
|---|---|---|---|---|---|---|---|
| Antigen-Positive Relapse | Antigen-Negative Relapse | Cytokine-Related Toxicity | On-Target Off-Tumor Toxicity | Antigen Specificity/Heterogeneity | Cell Trafficking | Tumor Microenvironment | |
| Novel CAR designs | |||||||
| The fifth-generation CARs | ● | ||||||
| Multiantigen CARs with “OR” logic gate | ● | ● | |||||
| Pooled CAR-T cells | ● | ● | |||||
| Universal CARs | ● | ● | |||||
| On-switch CARs | ● | ||||||
| Off-switch CARs | ● | ||||||
| Suicide gene | ● | ||||||
| Suicide receptor (Antibody-mediated depletion) | ● | ||||||
| CAR-T cells with tumour-associated chemokine receptors | ● | ||||||
| FAP-specific CARs | ● | ||||||
| Modify CAR-T cells to express heparinase by gene editing | ● | ||||||
| Dual CARs with “NOT” logic gate: inhibitory CARs (iCARs) | ● | ||||||
| Dual CARs with “AND” logic gate: SynNotch receptor system | ● | ● | |||||
| Dual CARs with “AND” logic gate: Split CARs (Combination CARs) | ● | ● | |||||
| Armoured CAR-T cells | ● | ||||||
| The fourth-generation CARs (TRUCKs) | ● | ||||||
| Combination with other therapeutic agents | |||||||
| Artificial antigen-presenting cells (AAPCs) | ● | ||||||
| Bi-specific T cell engagers (BiTEs) | ● | ● | |||||
| Haematopoietic stem cell transplantation after remission | ● | ||||||
| Cytokine inhibitors | ● | ||||||
| Dasatinib to inhibit CD3ζ downstream signal | ● | ||||||
| Antibodies for depleting suppressive immune cells/cytokines | ● | ||||||
| Immune checkpoint inhibitors (ICIs) | ● | ||||||
| Other strategy | |||||||
| Regional delivery | ● | ● | |||||
| NCT Number | Stage | Status | Study Title | Cell Target |
|---|---|---|---|---|
| NCT05215015 | Early Phase 1 | Recruiting | Study of Anti-CD33/CLL1 CAR-NK in Acute Myeloid Leukemia | CD33/CLL1 |
| NCT05194709 | Early Phase 1 | Recruiting | Study of Anti-5T4 CAR-NK Cell Therapy in Advanced Solid Tumors | 5T4 |
| NCT05008536 | Early Phase 1 | Recruiting | Anti-BCMA CAR-NK Cell Therapy for the Relapsed or Refractory Multiple Myeloma | BCMA |
| NCT03692663 | Early Phase 1 | Recruiting | Study of Anti-PSMA CAR NK Cell (TABP EIC) in Metastatic Castration-Resistant Prostate Cancer | PSMA |
| NCT05248048 | Early Phase 1 | Recruiting | NKG2D CAR-T Cells to Treat Patients With Previously Treated Liver Metastatic Colorectal Cancer | NKG2D |
| NCT05247957 | Phase 1 | Recruiting | NKG2D CAR-NK Cell Therapy in Patients With Relapsed or Refractory Acute Myeloid Leukemia | NKG2DL |
| NCT05472558 | Phase 1 | Recruiting | Clinical Study of Cord Blood-derived CAR-NK Cells Targeting CD19 in the Treatment of Refractory/Relapsed B-cell NHL | CD19 |
| NCT04887012 | Phase 1 | Recruiting | Clinical Study of HLA Haploidentical CAR-NK Cells Targeting CD19 in the Treatment of Refractory/Relapsed B-cell NHL | CD19 |
| NCT05213195 | Phase 1 | Recruiting | NKG2D CAR-NK Cell Therapy in Patients With Refractory Metastatic Colorectal Cancer | NKG2D |
| NCT05008575 | Phase 1 | Recruiting | Anti-CD33 CAR NK Cells in the Treatment of Relapsed/Refractory Acute Myeloid Leukemia | CD33 |
| NCT05507593 | Phase 1 | Recruiting | Study of DLL3-CAR-NK Cells in the Treatment of Extensive Stage Small Cell Lung Cancer | DLL3 |
| NCT05410041 | Phase 1 | Recruiting | Anti-CD19 CAR-Engineered NK Cells in the Treatment of Relapsed/Refractory B-cell Malignancies | CD19 |
| NCT04796675 | Phase 1 | Recruiting | Cord Blood Derived Anti-CD19 CAR-Engineered NK Cells for B Lymphoid Malignancies | CD19 |
| NCT04623944 | Phase 1 | Recruiting | NKX101, Intravenous Allogeneic CAR NK Cells, in Adults With AML or MDS | NKG2D |
| NCT05020678 | Phase 1 | Recruiting | NKX019, Intravenous Allogeneic Chimeric Antigen Receptor Natural Killer Cells (CAR NK), in Adults With B-cell Cancers | CD19 |
| NCT05563545 | Phase 1 | Recruiting | Anti-CD19 CAR-Engineered NK Cells in the Treatment of Relapsed/Refractory Acute Lymphoblastic Leukemia | CD19 |
| NCT04796688 | Phase 1 | Recruiting | Universal Chimeric Antigen Receptor-modified AT19 Cells for CD19+ Relapsed/Refractory Hematological Malignancies | CD19 |
| NCT05379647 | Phase 1 | Recruiting | Natural Killer (NK) Cell Therapy for B-Cell Malignancies | CD19 |
| NCT05182073 | Phase 1 | Recruiting | FT576 in Subjects With Multiple Myeloma | - |
| NCT05410717 | Phase 1/Phase 2 | Recruiting | CLDN6-CAR-NK Cell Therapy for Advanced Solid Tumors | Claudin6 |
| NCT05528341 | Phase 1/Phase 2 | Recruiting | NKG2D-CAR-NK92 Cells Immunotherapy for Solid Tumors | NKG2D |
| NCT03056339 | Phase 1/Phase 2 | Active, not recruiting | Umbilical & Cord Blood (CB) Derived CAR-Engineered NK Cells for B Lymphoid Malignancies | CD19 |
| NCT04847466 | Phase 2 | Recruiting | Immunotherapy Combination: Irradiated PD-L1 CAR-NK Cells Plus Pembrolizumab Plus N-803 for Subjects With Recurrent/Metastatic Gastric or Head and Neck Cancer | - |
| NCT Number | Stage | Status | Study Title | Cell Target |
|---|---|---|---|---|
| NCT04660929 | Phase I | Recruiting | CAR-macrophages for the Treatment of HER2 Overexpressing Solid Tumors | HER2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-J.; Abila, B.; Mostafa Kamel, Y. CAR-T: What Is Next? Cancers 2023, 15, 663. https://doi.org/10.3390/cancers15030663
Chen Y-J, Abila B, Mostafa Kamel Y. CAR-T: What Is Next? Cancers. 2023; 15(3):663. https://doi.org/10.3390/cancers15030663
Chicago/Turabian StyleChen, Yi-Ju, Bams Abila, and Yasser Mostafa Kamel. 2023. "CAR-T: What Is Next?" Cancers 15, no. 3: 663. https://doi.org/10.3390/cancers15030663
APA StyleChen, Y.-J., Abila, B., & Mostafa Kamel, Y. (2023). CAR-T: What Is Next? Cancers, 15(3), 663. https://doi.org/10.3390/cancers15030663