
Promises and Pitfalls of Next-Generation Treg Adoptive Immunotherapy

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction

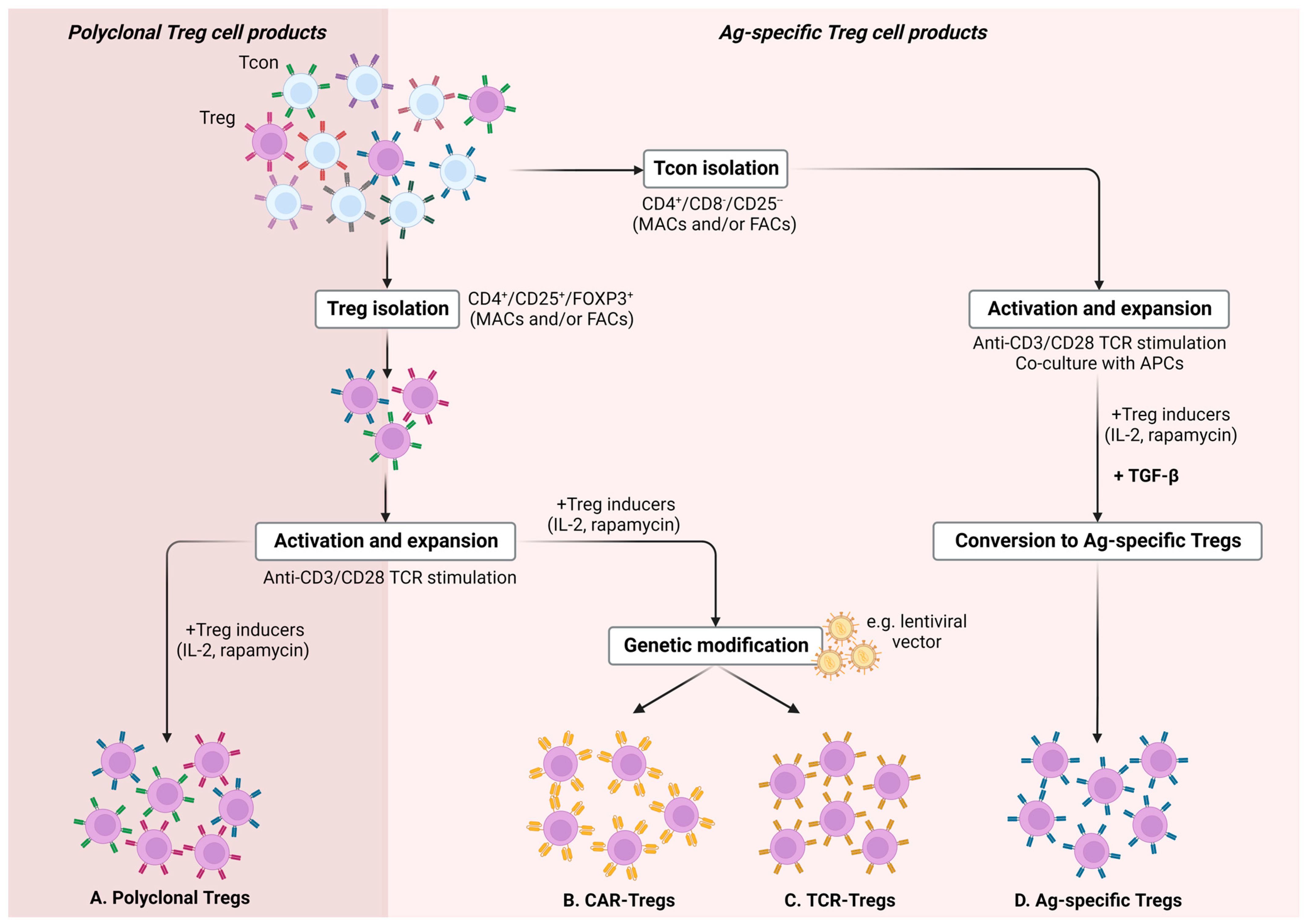{kind=link}
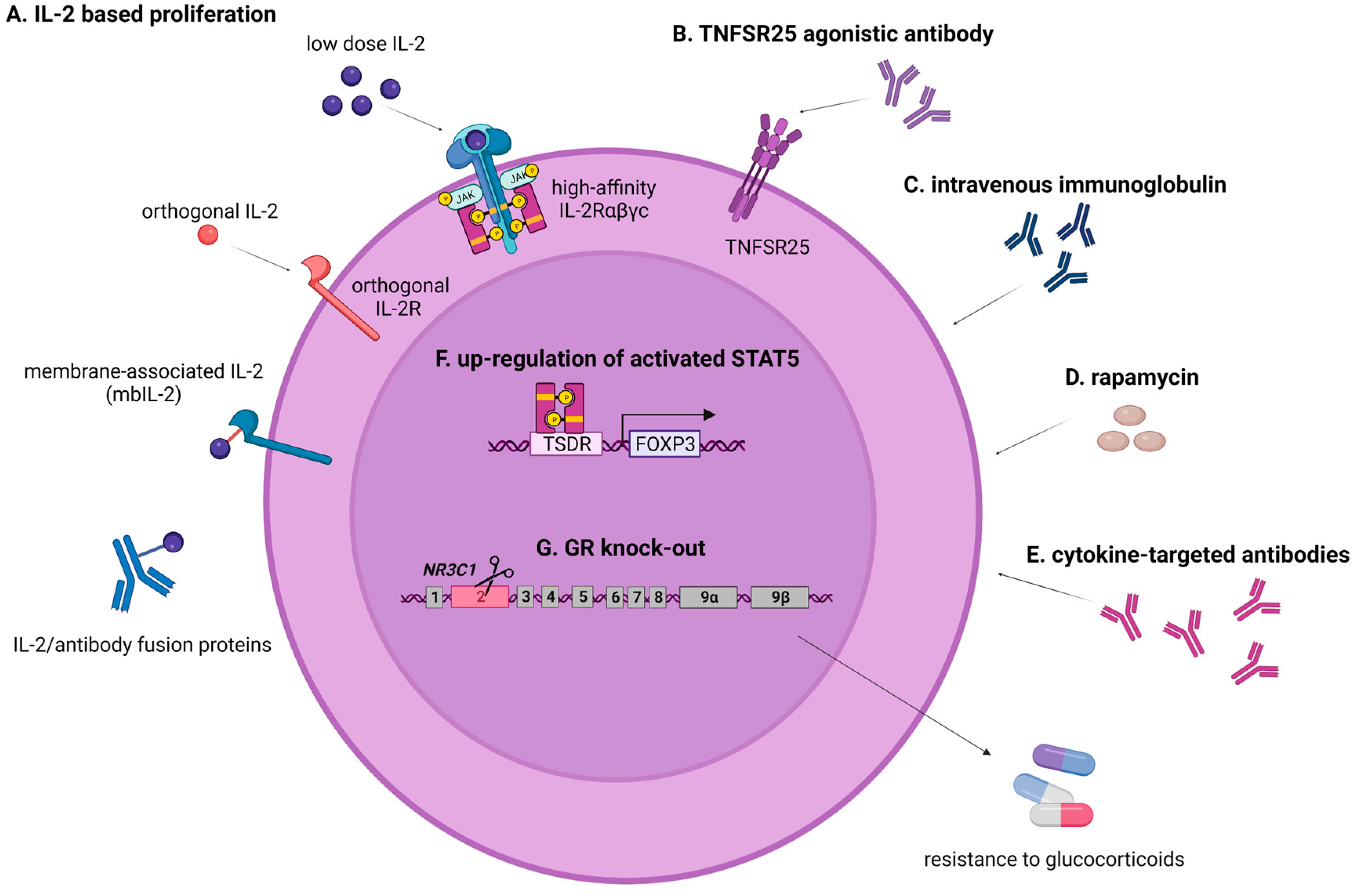{kind=link}
{kind=link}
| Treg Subset | Origin | Markers | Transcription Factors | Suppressive Mechanism | References |
|---|---|---|---|---|---|
| tTregs | Generating in the thymus | CD4+, CD25hi, CD27lo, CTLA-4+, LAG-3+, TIGIT+, TIM-3+, PD-1+ | FOXP3pos | Cell-contact-dependent immunosuppression via receptors like CTLA-4 and PD-1 | [33] |
| pTregs | Differentiating from peripheral naive CD4+ T cells | CD4+, CD25hi, CD27lo, CTLA-4+, LAG-3+, TIGIT+, Tim-3+, PD-1+ | FOXP3pos | Inhibitory function via soluble factors such as TGF-β1 and IL-10 | [33] |
| Tr1 Tregs | Differentiating from peripheral naive CD4+ T cells | CD4+, CD25, CD49b+, LAG-3+ [3] | Tbet, Blimp-1, FOXP3neg [1] | Inhibitory function via IL-10 production | [34,35] |
| Th3 Tregs | Differentiating from peripheral naive CD4+ T cells | CD4+, CD25+, CD69+, LAP+ | TGF-β, FOXP3neg | Inhibitory function via TGF-β production | [36] |
| CD8+ Tregs | Differentiating from peripheral naive CD8+ T cells | CD8+, CD25+, CD122+, CD49d+ | FOXP3pos, Eomes, Helios, TGF-β | TGF-dependent control of Helios and homeostatic cytokine IL-15 [4] | [37,38] |
| Cell Product | Source | Disease | Treg Manufacturing | Study Phase | Patients | Safety | Efficacy | Trial ID | References |
|---|---|---|---|---|---|---|---|---|---|
| Tregs for autoimmune diseases | |||||||||
| Polyclonal Tregs | Autologous | T1D | Isolation/enrichment and ex vivo expansion | I | 12 | No AEs | 8/12 clinical remission | ISRCTN06128462 | Marek-Trzonkowska et al., 2014 [39] |
| Polyclonal Tregs | Autologous | T1D | Isolation/enrichment and ex vivo expansion | I | 14 | Well-tolerated. No cell therapy-related high-grade AEs | Not powered to detect improvement in metabolic function | NCT01210664 | Bluestone et al., 2015 [24] |
| Polyclonal Tregs | Autologous | T1D | Ex vivo expansion | II, randomized placebo-controlled double blind | 110 | Well-tolerated | No improvement in the preservation of C-peptide levels vs. placebo | NCT02691247 | Caladrius Biosciences, 2019 [40] |
| Polyclonal Tregs | UCB | T1D | Isolation/enrichment and ex vivo expansion | I/II, randomized, parallel assignment, open label | Recruiting | NCT02932826 | |||
| Combinational: polyclonal Tregs + low-dose IL-2 | Autologous | T1D | Isolation/enrichment and ex vivo expansion | I | 7 | Off-target effect of low-dose IL-2 (dramatic reduction in C-peptide production and potential shift of the immune balance toward activation rather than tolerance)—terminated | No preservation or improvement of C-peptide production | NCT02772679 | Dong et al., 2021 [41] |
| Combinational: polyclonal Tregs + anti-CD20 | Autologous | T1D | Isolation/enrichment and ex vivo expansion | I/II, randomized, three-arm, open-label, single-blinded | 36 paediatric (Tregs only n = 13, Tregs + rituximab n = 12, control n = 11) | AEs in 80% of pts (combined group and Tregs only group). AEs, such as infections, needed special surveillance | Tregs+anti-CD20 were superior than Tregs in controlling recent-onset T1DM regarding C-peptide levels and remission | TregVAC2.0; EudraCT: 2014-004319-35 | Zieliński et al., 2022 [42] |
| Combinational: polyclonal Tregs + Liraglutide | UCB | T1D | Isolation/enrichment and ex vivo expansion | I/II, randomized, parallel assignment, open label | Recruiting | NCT03011021 | |||
| Polyclonal Tregs | Autologous | MS | Tregs for iv: isolation/enrichment and ex vivo expansion Tregs for IT: isolation/enrichment | 1b/2a (randomized to iv or IT Treg administration) | 14 (iv n = 11, IT n = 3) | No severe AEs | 5/11 relapses (iv-treated), 0/3 relapses (IT-treated). The statistical results may be underpowered due to the low number of patients | EudraCT 2014–004320-22 | Chwojnick et al., 2021 [43] |
| Polyclonal Tregs | Autologous | Autoimmune hepatitis | Isolation/enrichment and ex vivo expansion | I/II | Unknown status | NCT02704338 | |||
| Polyclonal Tregs | Autologous | Active cutaneous lupus | Isolation/enrichment and ex vivo expansion | I | 1 | Terminated due to participant recruitment feasibility | Stable clinical status | NCT02428309 | Dall’Era et al., 2019 [28] |
| Polyclonal Tregs | Autologous | Active Pemphigus | Isolation/enrichment and ex vivo expansion | I | 5 | Terminated due to recruitment issues and the impact of the coronavirus infectious disease 19 (COVID-19) pandemic | NCT03239470 | ||
| Ag-specific, ovalbumin-specific type 1 Tregs (ova-Tregs) | Autologous | Refractory Crohn’s disease | Isolation/enrichment and ex vivo expansion | I/IIa | 29 enrolled, 20 treated | Well-tolerated, good safety profile for this small patient cohort—significant AEs primarily related to the gastrointestinal system and the underlying CD | 8/20 (40%) total clinical improvement and 6/8 (75%) clinical response in the low-dose group (reducing dose-dependent efficacy) | NCT02327221 | Desreumaux et al., 2012 [44] |
| Polyclonal Tregs | Autologous | Crohn’s disease | Isolation/enrichment and ex vivo expansion | I | Recruiting | NCT03185000 | |||
| Tregs for solid organ transplantation | |||||||||
| Donor-alloantigen-specific Tregs | Autologous | Liver transplantation | Ex vivo expansion | I/IIa | 10 | Good safety profile | 10/10 normal graft function and histology. 7/10 successful cessation of immunosuppressive drugs. 3/10 required conventional low-dose immunotherapy | n/a | Todo et al., 2016 [45] |
| Donor-alloantigen-specific Tregs | Autologous | Liver transplantation | Ex vivo expansion | I | 15 | Terminated as it could not be completed within the grant timeline | NCT02188719 (darTregs) in Liver Transplantation (deLTa) | ||
| Donor-alloantigen-specific Tregs | Autologous | Liver transplantation | Isolation/enrichment and ex vivo expansion | I | Unknown status | NCT01624077 | |||
| Donor-alloantigen-specific Tregs | Autologous | Liver transplantation | Isolation/enrichment and ex vivo expansion | I/II | 15 | Not sufficiently powered to assess safety or efficacy (only n = 5 finally received Tregs) | NCT02474199 (ARTEMIS) | Tang Q et al., 2022 [46] | |
| Polyclonal Tregs | Autologous | Liver transplantation | Ex vivo expansion | I/II | 9 (3 received 106 Tregs/kg, 6 received 4.5 × 106 Tregs/kg) | Good safety profile | 6/6 of the high-dose-treated demonstrated reduced donor-specific T cell responses | NCT02166177 (ThRIL) | Sánchez-Fueyo et al., 2020 [47] |
| Donor-alloantigen-specific Tregs | Autologous | Liver transplantation | Isolation/enrichment and ex vivo expansion | I/II | Active, not recruiting | NCT03577431(ITN073ST) | |||
| HLA-A∗02-CAR Tregs | Autologous | Liver transplantation | Ex vivo expansion and genetic engineering | I/II | Recruiting | NCT05234190 (LIBERATE) | |||
| HLA-A∗02-CAR Tregs | Autologous | Kidney transplantation | Ex vivo expansion and genetic engineering | I/II | Recruiting | NCT04817774 (Steadfast) | Schreeb et al., 2022 [48] | ||
| Polyclonal Tregs | Autologous | Kidney transplantation | Isolation/enrichment and ex vivo expansion | I | 3 | Well-tolerated | 2/3 improvement in follow-up biopsies | NCT02088931 (TASKp pilot trial) | Chandran et al., 2017 [49] |
| Polyclonal Tregs | Autologous | Kidney transplantation | Ex vivo expansion | I | 9 | Good safety profile | All pts survived for at least 2 years | NCT02145325 (TRACT trial) | Mathew et al., 2018 [50] |
| Polyclonal Tregs | Autologous | Kidney transplantation | Isolation/enrichment and ex vivo expansion | n/a | Recruiting | NCT03284242 | |||
| Combinational: polyclonal Tregs+ donor bone marrow cells + Tocilizumab | Autologous | Kidney transplantation | Isolation/enrichment and ex vivo expansion | I/IIa | Active, not recruiting | NCT03867617 (Trex001) | Oberbauer et al., 2021 [51] | ||
| Polyclonal Tregs | Autologous | Kidney transplantation | Isolation/enrichment and ex vivo expansion | I/II | Unknown status | NCT01446484 (RSMU-001) | |||
| Polyclonal vs. donor-specific Tregs | Autologous | Kidney transplantation | Ex vivo expansion | I/II randomized open-label | n/a | Completed. No results posted yet | NCT02711826 (TASK, CTOT-21) | ||
| Polyclonal and donor-antigen reactive Tregs, tolerogenic dendritic cell and regulatory macrophage cells | Autologous | Kidney transplantation | Isolation/enrichment and/or ex vivo expansion | 7 phase I/II trials | 66 cell-treated group vs. 38 reference-group | Good safety profile | Lower infection rates; rates of biopsy-confirmed acute rejection (BCAR) comparable between the standard immunosuppressive group and the cell-based therapy group. Successfully weaned off immunosuppression within the first year post-transplantation to monotherapy in nearly all cell-treated patients | NCT02371434, NCT02129881 (polyclonal Treg), NCT02244801, NCT02091232 (donor-antigen reactive Treg), NCT02252055 (tolerogenic dendritic cell), NCT02085629 (regulatory mac rophage cell), NCT01656135 (reference group) (ONE study) | Sawitzki et al., 2020 [52] |
| Combinational: total lymphoid irradiation (TLI), total body irradiation (TBI), anti-thymocyte globulin (ATG), donor HSCs and polyclonal Tregs | Autologous | Kidney transplantation | Ex vivo expansion | I | Recruiting | NCT03943238 | |||
| Polyclonal Tregs | Autologous | Kidney transplantation | Ex vivo expansion | IIb, randomized | Recruiting | ISRCTN11038572 (Two study) | Brook et al., 2022 [53] | ||
| Polyclonal Tregs | Autologous | Heart transplantation | Isolation/enrichment and ex vivo expansion | I/II, randomized | Recruiting | NCT04924491 (THYTECH) | Bernaldo-de-Quirós et al., 2022 [54] | ||
| Polyclonal Tregs | Autologous | Islet transplantation | Isolation/enrichment and ex vivo expansion | I | Active, not recruiting | NCT03444064 | |||
| Tregs for COVID-19 | |||||||||
| Polyclonal Tregs | Allogeneic, UCB | COVID-19 | Isolation/enrichment and ex vivo expansion | I, randomized, double-blinded, placebo-controlled clinical trial | 45 (15 pts placebo, 15 pts 100 × 106 Tregs, 15 pts 300 × 106, 3 doses Tregs) | Good safety profile | No definitive conclusions with respect to efficacy due to to the low number of patients | NCT04468971 | Gladstone et al., 2023 [55] |
| Tregs for GvHD | |||||||||
| Polyclonal HLA-G + induced T-regulatory cells (iG-Tregs) | Allogeneic, HLA-identical sibling donor-derived | GvHD prophylaxis | Ex vivo expansion | I/II | Recruiting | EUDRACT-2021-006367-26 | Lysandrou et al., 2023 [56] | ||
| Polyclonal Tregs | Allogeneic, HLA-matched sibling donor-derived | GvHD treatment | Isolation/enrichment and ex vivo expansion | I | 2 | Temporal control of grade IV acute GvHD refractory to all other immunosuppressants used/significant alleviation of chronic GvHD accompanied by reduced pharmacologic immunosuppression | NKEBN/458-310/2008 | Trzonkowski et al., 2009 [57] | |
| Polyclonal Tregs | Allogeneic, partially HLA-matched third UCB | GvHD prophylaxis | Isolation/enrichment | I | 23 | No infusional toxicities | No adverse effect in terms of infection, relapse, or early mortality/decreased incidence of grade II–IV acute GVHD vs. identically treated historical controls | NCT00602693 | Brunstein et al., 2011 [58] |
| Polyclonal Tregs | Allogeneic, HSC donor-derived | Severe refractory GvHD treatment | Isolation/enrichment | I/II | Completed. No results posted yet | NCT02749084 | |||
| Polyclonal Tregs | Allogeneic, UCB donor-derived | GvHD prophylaxis | Not specified | II | 3 | 2/3 AEs/Treg-cell infusion toxicity | 2/3 grade II-IV acute GvHD; 1/3 bacterial infection; 2/3 viral infection. Terminated due to the consideration of new technology for the product | NCT02991898 | |
| Polyclonal Tregs | Allogeneic, HLA-matched sibling donor-derived | Steroid dependent/refractory chronic GvHD treatment | Not specified | I | Completed. No results posted yet | NCT01911039 | |||
| Combinational: polyclonal Tregs + low-dose IL-2 | Allogeneic, HSC donor-derived | Steroid refractory chronic GvHD treatment | Isolation/enrichment | I | 25 | Good safety profile | 5/25 (20%) PR; 10/25 (40%) stable disease | NCT01937468 | Whangbo et al., 2022 [59] |
| Polyclonal Tregs | Allogeneic, HSC donor-derived | Steroid refractory chronic GvHD treatment | Isolation/enrichment | I/II | Unknown status | NCT02385019 | |||
| Combinational: polyclonal Tregs + IL-2 + rapamycin | Allogeneic, HSC donor-derived | Chronic GvHD treatment | Isolation/enrichment | II | Teriminated due to slow recruitment | NCT01903473 | |||
| Combinational: polyclonal Tregs + Tcon | Allogeneic, HSC donor-derived | GvHD prophylaxis + GvL augmentation in pts with high-risk hematological malignancies undergoing allogeneic myeloablative (MA) HCT with a T cell-depleted graft | Isolation/enrichment | I/II | Interim results: 12 (initial group: 5 pts with frozen Tregs, modified groupI:7 pts with fresh Tregs and single-agent GVHD prophylaxis) | No infusion reaction | Initial group: 2/5 grade II GvHD; modified group: 0/7 GvHD | NCT01660607 | Meyer et al., 2019 [60] |
| Polyclonal, fucosylated Tregs | Allogeneic UCB-derived | GvHD prophylaxis | Isolation/enrichment and ex vivo expansion | I | 5 | No infusion reaction | 5/5 ≥grade II acute GVHD. No longterm complications for 4/5 alive pts | NCT02423915 | Kellner et al., 2018 [61] |
| Alloantigen-specific Tr1 cells | Allogeneic,HSC donor-derived | GvHD prophylaxis | Ex vivo expansion | I | 3 (preliminary results) | No AEs post infusion | 3/3 alive, disease-free and acute GvHD-free at 1 year post-HCT | NCT03198234 | Chen et al., 2021 [62] |
| Polyclonal Tregs | Allogeneic,HSC donor-derived | GvHD prophylaxis | Isolation/enrichment and ex vivo expansion | I | 14 | No severe infusional toxicities | Pts receiving sirolimus/MMF: 2/2 grade III acute GvHD pts receiving CSA/MMF: 5/12 acute GvHD grade II-III, 6/12 chronic GvHD | NCT01634217 | MacMillan et al., 2021 [63] |
| Polyclonal Tregs | Allogeneic, HSC donor-derived | Steroid refractory chronic GvHD treatment | Isolation/enrichment | II | Recruiting | NCT05095649 | |||
| CD6-CAR Tregs | Allogeneic,HSC donor-derived | Chronic GvHD treatment | Ex vivo expansion and genetic engineering | I | Not yet recruiting | NCT05993611 | |||
2. Specificity of Tolerance
Generating Antigen-Specific Tregs to Overcome Polyclonal Treg Limitations
3. Treg Functional Stability versus Plasticity
Stabilizing Treg Phenotype to Overcome Plasticity
4. Inhibitory Treg Signaling by the Tumor Microenvironment
Functional Treg Enhancement against Inflammatory Cytokine Signaling
5. Tissue Homeostatic Repair
Promoting Tissue Homeostatic Regeneration by Treg Cells
6. Site-Specific Treg Cell Migration
Enhancing Treg Cell Recruitment In Vivo
7. Survival and Persistence
Improving Treg Survival and Persistence
8. Treg Safety Considerations
Optimizing Treg Safety
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Edinger, M.; Hoffmann, P.; Ermann, J.; Drago, K.; Garrison Fathman, C.; Strober, S.; Negrin, R.S. CD4+CD25+ Regulatory T Cells Preserve Graft-versus-Tumor Activity While Inhibiting Graft-versus-Host Disease after Bone Marrow Transplantation. Nat. Med. 2003, 9, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.L.; Trenado, A.; Vasey, D.; Klatzmann, D.; Salomon, B.L. CD4+CD25+ Immunoregulatory T Cells: New Therapeutics for Graft-versus-Host Disease. J. Exp. Med. 2002, 196, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Benoist, C.; Bluestone, J.A.; Campbell, D.J.; Ghosh, S.; Hori, S.; Jiang, S.; Kuchroo, V.K.; Mathis, D.; Roncarolo, M.G.; et al. Regulatory T Cells: Recommendations to Simplify the Nomenclature. Nat. Immunol. 2013, 14, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Wyss, L.; Stadinski, B.D.; King, C.G.; Schallenberg, S.; Mccarthy, N.I.; Lee, J.Y.; Kretschmer, K.; Terracciano, L.M.; Anderson, G.; Surh, C.D.; et al. Affinity for Self Antigen Selects Treg Cells with Distinct Functional Properties. Nat. Immunol. 2016, 17, 1093–1101. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. J. Immunol. 2017, 198, 981–985. [Google Scholar] [CrossRef]
- Zeng, H.; Zhang, R.; Jin, B.; Chen, L. Type 1 Regulatory T Cells: A New Mechanism of Peripheral Immune Tolerance. Cell. Mol. Immunol. 2015, 12, 566–571. [Google Scholar] [CrossRef]
- Weiner, H.L.; da Cunha, A.P.; Quintana, F.; Wu, H. Oral Tolerance. Immunol. Rev. 2011, 241, 241–259. [Google Scholar] [CrossRef]
- Mishra, S. CD8+ Regulatory T Cell—A Mystery to Be Revealed. Front. Immunol. 2021, 12, 708874. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Baeyens, A.; You, S.; Elhage, R.; Fourcade, G.; Gregoire, S.; Cagnard, N.; Carpentier, W.; Tang, Q.; Bluestone, J.; et al. IL-2 Reverses Established Type 1 Diabetes in NOD Mice by a Local Effect on Pancreatic Regulatory T Cells. J. Exp. Med. 2010, 207, 1871–1878. [Google Scholar] [CrossRef]
- Kosmaczewska, A. Low-Dose Interleukin-2 Therapy: A Driver of an Imbalance between Immune Tolerance and Autoimmunity. Int. J. Mol. Sci. 2014, 15, 18574–18592. [Google Scholar] [CrossRef] [PubMed]
- Hartemann, A.; Bensimon, G.; Payan, C.A.; Jacqueminet, S.; Bourron, O.; Nicolas, N.; Fonfrede, M.; Rosenzwajg, M.; Bernard, C.; Klatzmann, D. Low-Dose Interleukin 2 in Patients with Type 1 Diabetes: A Phase 1/2 Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Diabetes Endocrinol. 2013, 1, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.E.; Walters, S.; Kohler, R.E.; Mrkvan, T.; Boyman, O.; Surh, C.D.; Grey, S.T.; Sprent, J. In Vivo Expansion of t Reg Cells with Il-2-Mab Complexes: Induction of Resistance to Eae and Long-Term Acceptance of Islet Allografts without Immunosuppression. J. Exp. Med. 2009, 206, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Baker, J.; Leveson-Gower, D.B.; Smith, A.T.; Sega, E.I.; Negrin, R.S. Rapamycin and IL-2 Reduce Lethal Acute Graft-versus-Host Disease Associated with Increased Expansion of Donor Type CD4+CD25 +Foxp3+ Regulatory T Cells. Blood 2011, 118, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.I.; Koreth, J.; Kim, H.T.; Bascug, G.; McDonough, S.; Kawano, Y.; Murase, K.; Cutler, C.; Ho, V.T.; Alyea, E.P.; et al. Low-Dose Interleukin-2 Therapy Restores Regulatory T Cell Homeostasis in Patients with Chronic Graft-versus-Host Disease. Sci. Transl. Med. 2013, 5, 179ra43. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Stabilini, A.; Migliavacca, B.; Horejs-Hoeck, J.; Kaupper, T.; Roncarolo, M.-G. Rapamycin Promotes Expansion of Functional CD4+CD25+FOXP3+ Regulatory T Cells of Both Healthy Subjects and Type 1 Diabetic Patients. J. Immunol. 2006, 177, 8338–8347. [Google Scholar] [CrossRef] [PubMed]
- Trinath, J.; Hegde, P.; Sharma, M.; Maddur, M.S.; Rabin, M.; Vallat, J.M.; Magy, L.; Balaji, K.N.; Kaveri, S.V.; Bayry, J. Intravenous Immunoglobulin Expands Regulatory T Cells via Induction of Cyclooxygenase-2-Dependent Prostaglandin E2 in Human Dendritic Cells. Blood 2013, 122, 1419–1427. [Google Scholar] [CrossRef]
- Bayry, J.; Sibéril, S.; Triebel, F.; Tough, D.F.; Kaveri, S.V. Rescuing CD4+CD25+ Regulatory T-Cell Functions in Rheumatoid Arthritis by Cytokine-Targeted Monoclonal Antibody Therapy. Drug Discov. Today 2007, 12, 548–552. [Google Scholar] [CrossRef]
- Nadkarni, S.; Mauri, C.; Ehrenstein, M.R. Anti-TNF-α Therapy Induces a Distinct Regulatory T Cell Population in Patients with Rheumatoid Arthritis via TGF-β. J. Exp. Med. 2007, 204, 33–39. [Google Scholar] [CrossRef]
- Marfil-Garza, B.A.; Pawlick, R.L.; Szeto, J.; Kroger, C.; Tahiliani, V.; Hefler, J.; Dadheech, N.; Seavey, M.M.; Wolf, J.; Jasuja, R.R.; et al. Tumor Necrosis Factor Receptor Superfamily Member 25 (TNFRSF25) Agonists in Islet Transplantation: Endogenous in Vivo Regulatory T Cell Expansion Promotes Prolonged Allograft Survival. Am. J. Transplant. 2022, 22, 1101–1114. [Google Scholar] [CrossRef]
- Wolf, D.; Schreiber, T.H.; Tryphonopoulos, P.; Li, S.; Tzakis, A.G.; Ruiz, P.; Podack, E.R. Tregs Expanded in Vivo by TNFRSF25 Agonists Promote Cardiac Allograft Survival. Transplantation 2012, 94, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, T.H.; Wolf, D.; Tsai, M.S.; Chirinos, J.; Deyev, V.V.; Gonzalez, L.; Malek, T.R.; Levy, R.B.; Podack, E.R. Therapeutic Treg Expansion in Mice by TNFRSF25 Prevents Allergic Lung Inflammation. J. Clin. Investig. 2010, 120, 3629–3640. [Google Scholar] [CrossRef] [PubMed]
- Orozco, G.; Gupta, M.; Gedaly, R.; Marti, F. Untangling the Knots of Regulatory T Cell Therapy in Solid Organ Transplantation. Front. Immunol. 2022, 13, 883855. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 Diabetes Immunotherapy Using Polyclonal Regulatory T Cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef] [PubMed]
- Rezaei Kahmini, F.; Shahgaldi, S.; Azimi, M.; Mansourabadi, A.H. Emerging Therapeutic Potential of Regulatory T (Treg) Cells for Rheumatoid Arthritis: New Insights and Challenges. Int. Immunopharmacol. 2022, 108, 108858. [Google Scholar] [CrossRef]
- Kohm, A.P.; Carpentier, P.A.; Anger, H.A.; Miller, S.D. Cutting Edge: CD4+CD25+ Regulatory T Cells Suppress Antigen-Specific Autoreactive Immune Responses and Central Nervous System Inflammation During Active Experimental Autoimmune Encephalomyelitis. J. Immunol. 2002, 169, 4712–4716. [Google Scholar] [CrossRef]
- Gladstone, D.E.; Kim, B.S.; Mooney, K.; Karaba, A.H.; D’Alessio, F.R. Regulatory T Cells for Treating Patients with COVID-19 and Acute Respiratory Distress Syndrome: Two Case Reports. Ann. Intern. Med. 2020, 173, 852–853. [Google Scholar] [CrossRef]
- Dall’Era, M.; Pauli, M.L.; Remedios, K.; Taravati, K.; Sandova, P.M.; Putnam, A.L.; Lares, A.; Haemel, A.; Tang, Q.; Hellerstein, M.; et al. Adoptive Treg Cell Therapy in a Patient With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 431–440. [Google Scholar] [CrossRef]
- Hippen, K.L.; Hefazi, M.; Larson, J.H.; Blazar, B.R. Emerging Translational Strategies and Challenges for Enhancing Regulatory T Cell Therapy for Graft-versus-Host Disease. Front. Immunol. 2022, 13, 926550. [Google Scholar] [CrossRef]
- Yang, L.; Wang, G.; Xia, H. Molecular Mechanism for Impaired Suppressive Function of Tregs in Autoimmune Diseases: A Summary of Cell-Intrinsic and Cell-Extrinsic Factors. J. Cell. Mol. Med. 2020, 24, 11056–11063. [Google Scholar] [CrossRef]
- Whitehouse, G.; Gray, E.; Mastoridis, S.; Merritt, E.; Kodela, E.; Yang, J.H.M.; Danger, R.; Mairal, M.; Christakoudi, S.; Lozano, J.J.; et al. IL-2 Therapy Restores Regulatory T-Cell Dysfunction Induced by Calcineurin Inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, 7083–7088. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Wang, Q.; Guo, W.; Pei, X.; Niu, Q.; Liu, M.; Liu, Y.; Chen, S.; Feng, S.; He, Y.; et al. Loss of Lkb1 Impairs Treg Function and Stability to Aggravate Graft-versus-Host Disease after Bone Marrow Transplantation. Cell. Mol. Immunol. 2020, 17, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Povoleri, G.A.M.; Scottá, C.; Nova-Lamperti, E.A.; John, S.; Lombardi, G.; Afzali, B. Thymic versus Induced Regulatory T Cells-Who Regulates the Regulators? Front. Immunol. 2013, 4, 169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lee, J.S.; Gartlan, K.H.; Schuster, I.S.; Comerford, I.; Varelias, A.; Ullah, M.A.; Vuckovic, S.; Koyama, M.; Kuns, R.D.; et al. Eomesodermin Promotes the Development of Type 1 Regulatory T (TR1) Cells. Sci. Immunol. 2017, 2, eaah7152. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, N.; Chen, L.; Fang, L. Tr1 Cells as a Key Regulator for Maintaining Immune Homeostasis in Transplantation. Front. Immunol. 2021, 12, 671579. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.H.; Chiang, B.L. Regulatory T Cells Induced by B Cells: A Novel Subpopulation of Regulatory T Cells. J Biomed Sci 2017, 24, 86. [Google Scholar] [CrossRef]
- Vieyra-Lobato, M.R.; Vela-Ojeda, J.; Montiel-Cervantes, L.; López-Santiago, R.; Moreno-Lafont, M.C. Description of CD8+ Regulatory T Lymphocytes and Their Specific Intervention in Graft-versus-Host and Infectious Diseases, Autoimmunity, and Cancer. J. Immunol. Res. 2018, 2018, 3758713. [Google Scholar] [CrossRef]
- Mishra, S.; Liao, W.; Liu, Y.; Yang, M.; Ma, C.; Wu, H.; Zhao, M.; Zhang, X.; Qiu, Y.; Lu, Q.; et al. TGF-β and Eomes Control the Homeostasis of CD8+regulatory T Cells. J. Exp. Med. 2021, 218, e20200030. [Google Scholar] [CrossRef]
- Marek-Trzonkowska, N.; Myśliwiec, M.; Dobyszuk, A.; Grabowska, M.; Derkowska, I.; Juścińska, J.; Owczuk, R.; Szadkowska, A.; Witkowski, P.; Młynarski, W.; et al. Therapy of Type 1 Diabetes with CD4+CD25highCD127-Regulatory T Cells Prolongs Survival of Pancreatic Islets—Results of One Year Follow-Up. Clin. Immunol. 2014, 153, 23–30. [Google Scholar] [CrossRef]
- Caladrius Biosciences, Inc. Caladrius Biosciences Reports Top-Line Data for the Phase 2a Sanford Project: T-Rex Trial of CLBS03 for Recent Onset Type 1 Diabetes; Caladrius Biosciences, Inc.: Basking Ridge, NJ, USA, 2019. [Google Scholar]
- Dong, S.; Hiam-Galvez, K.J.; Mowery, C.T.; Herold, K.C.; Gitelman, S.E.; Esensten, J.H.; Liu, W.; Lares, A.P.; Leinbach, A.S.; Lee, M.; et al. The Effect of Low-Dose IL-2 and Treg Adoptive Cell Therapy in Patients with Type 1 Diabetes. JCI Insight 2021, 6, e147474. [Google Scholar] [CrossRef]
- Zieliński, M.; Żalińska, M.; Iwaszkiewicz-Grześ, D.; Gliwiński, M.; Hennig, M.; Jaźwińska-Curyłło, A.; Kamińska, H.; Sakowska, J.; Wołoszyn-Durkiewicz, A.; Owczuk, R.; et al. Combined Therapy with CD4+CD25highCD127− T Regulatory Cells and Anti-CD20 Antibody in Recent-Onset Type 1 Diabetes Is Superior to Monotherapy: Randomized Phase I/II Trial. Diabetes Obes. Metab. 2022, 24, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Chwojnicki, K.; Iwaszkiewicz-Grześ, D.; Jankowska, A.; Zieliński, M.; Łowiec, P.; Gliwiński, M.; Grzywińska, M.; Kowalczyk, K.; Konarzewska, A.; Glasner, P.; et al. Administration of CD4+CD25highCD127−FoxP3+ Regulatory T Cells for Relapsing-Remitting Multiple Sclerosis: A Phase 1 Study. BioDrugs 2021, 35, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hébuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and Efficacy of Antigen-Specific Regulatory T-Cell Therapy for Patients with Refractory Crohn’s Disease. Gastroenterology 2012, 143, 1207–1217.e2. [Google Scholar] [CrossRef] [PubMed]
- Todo, S.; Yamashita, K.; Goto, R.; Zaitsu, M.; Nagatsu, A.; Oura, T.; Watanabe, M.; Aoyagi, T.; Suzuki, T.; Shimamura, T.; et al. A Pilot Study of Operational Tolerance with a Regulatory T-cell-based Cell Therapy in Living Donor Liver Transplantation. Hepatology 2016, 64, 632–643. [Google Scholar] [CrossRef]
- Tang, Q.; Leung, J.; Peng, Y.; Sanchez-Fueyo, A.; Lozano, J.-J.; Lam, A.; Lee, K.; Greenland, J.R.; Hellerstein, M.; Fitch, M.; et al. Selective Decrease of Donor-Reactive T Regs after Liver Transplantation Limits Treg Therapy for Promoting Allograft Tolerance in Humans. Sci. Transl. Med. 2022, 14, eabo2628. [Google Scholar] [CrossRef]
- Sánchez-Fueyo, A.; Whitehouse, G.; Grageda, N.; Cramp, M.E.; Lim, T.Y.; Romano, M.; Thirkell, S.; Lowe, K.; Fry, L.; Heward, J.; et al. Applicability, Safety, and Biological Activity of Regulatory T Cell Therapy in Liver Transplantation. Am. J. Transplant. 2020, 20, 1125–1136. [Google Scholar] [CrossRef]
- Schreeb, K.; Culme-Seymour, E.; Ridha, E.; Dumont, C.; Atkinson, G.; Hsu, B.; Reinke, P. Study Design: Human Leukocyte Antigen Class I Molecule A∗02-Chimeric Antigen Receptor Regulatory T Cells in Renal Transplantation. Kidney Int. Rep. 2022, 7, 1258–1267. [Google Scholar] [CrossRef]
- Chandran, S.; Tang, Q.; Sarwal, M.; Laszik, Z.G.; Putnam, A.L.; Lee, K.; Leung, J.; Nguyen, V.; Sigdel, T.; Tavares, E.C.; et al. Polyclonal Regulatory T Cell Therapy for Control of Inflammation in Kidney Transplants. Am. J. Transplant. 2017, 17, 2945–2954. [Google Scholar] [CrossRef]
- Mathew, J.M.; H-Voss, J.; LeFever, A.; Konieczna, I.; Stratton, C.; He, J.; Huang, X.; Gallon, L.; Skaro, A.; Ansari, M.J.; et al. A Phase i Clinical Trial with Ex Vivo Expanded Recipient Regulatory T Cells in Living Donor Kidney Transplants. Sci. Rep. 2018, 8, 7428. [Google Scholar] [CrossRef]
- Oberbauer, R.; Edinger, M.; Berlakovich, G.; Kalhs, P.; Worel, N.; Heinze, G.; Wolzt, M.; Lion, T.; Wekerle, T. A Prospective Controlled Trial to Evaluate Safety and Efficacy of in Vitro Expanded Recipient Regulatory T Cell Therapy and Tocilizumab Together With Donor Bone Marrow Infusion in HLA-Mismatched Living Donor Kidney Transplant Recipients (Trex001). Front. Med. 2021, 7, 634260. [Google Scholar] [CrossRef]
- Sawitzki, B.; Harden, P.N.; Reinke, P.; Moreau, A.; Hutchinson, J.A.; Game, D.S.; Tang, Q.; Guinan, E.C.; Battaglia, M.; Burlingham, W.J.; et al. Regulatory Cell Therapy in Kidney Transplantation (The ONE Study): A Harmonised Design and Analysis of Seven Non-Randomised, Single-Arm, Phase 1/2A Trials. Lancet 2020, 395, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Brook, M.O.; Hester, J.; Petchey, W.; Rombach, I.; Dutton, S.; Bottomley, M.J.; Black, J.; Abdul-Wahab, S.; Bushell, A.; Lombardi, G.; et al. Transplantation Without Overimmunosuppression (TWO) Study Protocol: A Phase 2b Randomised Controlled Single-Centre Trial of Regulatory T Cell Therapy to Facilitate Immunosuppression Reduction in Living Donor Kidney Transplant Recipients. BMJ Open 2022, 12, 893576. [Google Scholar] [CrossRef] [PubMed]
- Bernaldo-de-Quirós, E.; Cózar, B.; López-Esteban, R.; Clemente, M.; Gil-Jaurena, J.M.; Pardo, C.; Pita, A.; Pérez-Caballero, R.; Camino, M.; Gil, N.; et al. A Novel GMP Protocol to Produce High-Quality Treg Cells From the Pediatric Thymic Tissue to Be Employed as Cellular Therapy. Front. Immunol. 2022, 13, 893576. [Google Scholar] [CrossRef] [PubMed]
- Gladstone, D.E.; D’Alessio, F.; Howard, C.; Lyu, M.-A.; Mock, J.R.; Gibbs, K.W.; Abrams, D.; Huang, M.; Zeng, K.; Herlihy, J.P.; et al. Randomized, Double-Blinded, Placebo-Controlled Trial of Allogeneic Cord Blood T-Regulatory Cells for Treatment of COVID-19 ARDS. Blood Adv. 2023, 7, 3075–3079. [Google Scholar] [CrossRef] [PubMed]
- Lysandrou, M.; Stamou, P.; Kefala, D.; Pierides, C.; Kyriakou, M.; Savvopoulos, N.; Christofi, P.; Papadopoulou, A.; Yannaki, E.; Costeas, P.; et al. Hypomethylation-Induced Regulatory Programs in T Cells Unveiled by Transcriptomic Analyses. Front. Immunol. 2023, 14, 1235661. [Google Scholar] [CrossRef]
- Trzonkowski, P.; Bieniaszewska, M.; Juścińska, J.; Dobyszuk, A.; Krzystyniak, A.; Marek, N.; Myśliwska, J.; Hellmann, A. First-in-Man Clinical Results of the Treatment of Patients with Graft versus Host Disease with Human Ex Vivo Expanded CD4+CD25+CD127- T Regulatory Cells. Clin. Immunol. 2009, 133, 22–26. [Google Scholar] [CrossRef]
- Brunstein, C.G.; Miller, J.S.; Cao, Q.; McKenna, D.H.; Hippen, K.L.; Curtsinger, J.; DeFor, T.; Levine, B.L.; June, C.H.; Rubinstein, P.; et al. Infusion of Ex Vivo Expanded T Regulatory Cells in Adults Transplanted with Umbilical Cord Blood: Safety Profile and Detection Kinetics. Blood 2011, 117, 1061–1070. [Google Scholar] [CrossRef]
- Whangbo, J.S.; Nikiforow, S.; Kim, H.T.; Wahl, J.; Reynolds, C.G.; Rai, S.C.; Kim, S.; Burden, A.; Alho, A.C.; Lacerda, J.F.; et al. A Phase 1 Study of Donor Regulatory T-Cell Infusion plus Low-Dose Interleukin-2 for Steroid-Refractory Chronic Graft-vs-Host Disease. Blood Adv. 2022, 6, 5786–5796. [Google Scholar] [CrossRef]
- Meyer, E.H.; Laport, G.; Xie, B.J.; MacDonald, K.; Heydari, K.; Sahaf, B.; Tang, S.W.; Baker, J.; Armstrong, R.; Tate, K.; et al. Transplantation of Donor Grafts with Defined Ratio of Conventional and Regulatory T Cells in HLA-Matched Recipients. JCI Insight 2019, 4, e127244. [Google Scholar] [CrossRef]
- Kellner, J.N.; Delemarre, E.M.; Yvon, E.; Nierkens, S.; Boelens, J.J.; Mcniece, I.; Olson, A.; Nieto, Y.; Ciurea, S.; Popat, U.; et al. Third Party, Umbilical Cord Blood Derived Regulatory T-Cells for Prevention of Graft versus Host Disease in Allogeneic Hematopoietic Stem Cell Transplantation: Feasibility, Safety and Immune Reconstitution. Oncotarget 2018, 9, 35611–35622. [Google Scholar] [CrossRef]
- Chen, P.P.; Cepika, A.-M.; Agarwal-Hashmi, R.; Saini, G.; Uyeda, M.J.; Louis, D.M.; Cieniewicz, B.; Narula, M.; Amaya Hernandez, L.C.; Harre, N.; et al. Alloantigen-Specific Type 1 Regulatory T Cells Suppress through CTLA-4 and PD-1 Pathways and Persist Long-Term in Patients. Sci. Transl. Med. 2021, 13, eabf5264c. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, M.L.; Hippen, K.L.; McKenna, D.H.; Kadidlo, D.; Sumstad, D.; Defor, T.E.; Brunstein, C.G.; Holtan, S.G.; Miller, J.S.; Warlick, E.D.; et al. First-in-Human Phase 1 Trial of Induced Regulatory T Cells for Graft-versus-Host Disease Prophylaxis in HLA-Matched Siblings. Blood Adv. 2021, 5, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Brunstein, C.G.; Blazar, B.R.; Miller, J.S.; Cao, Q.; Hippen, K.L.; McKenna, D.H.; Curtsinger, J.; McGlave, P.B.; Wagner, J.E. Adoptive Transfer of Umbilical Cord Blood-Derived Regulatory T Cells and Early Viral Reactivation. Biol. Blood Marrow Transplant. 2013, 19, 1271–1273. [Google Scholar] [CrossRef] [PubMed]
- Noyes, D.; Bag, A.; Oseni, S.; Semidey-Hurtado, J.; Cen, L.; Sarnaik, A.A.; Sondak, V.K.; Adeegbe, D. Tumor-Associated Tregs Obstruct Antitumor Immunity by Promoting T Cell Dysfunction and Restricting Clonal Diversity in Tumor-Infiltrating CD8+ T Cells. J. Immunother. Cancer 2022, 10, e004605. [Google Scholar] [CrossRef] [PubMed]
- Thornton, A.M.; Shevach, E.M. Suppressor Effector Function of CD4+CD25+ Immunoregulatory T Cells Is Antigen Nonspecific. J. Immunol. 2000, 164, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.; Boeld, T.J.; Eder, R.; Huehn, J.; Floess, S.; Wieczorek, G.; Olek, S.; Dietmaier, W.; Andreesen, R.; Edinger, M. Loss of FOXP3 Expression in Natural Human CD4+ CD25+ Regulatory T Cells upon Repetitive in Vitro Stimulation. Eur. J. Immunol. 2009, 39, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Henriksen, K.J.; Bi, M.; Finger, E.B.; Szot, G.; Ye, J.; Masteller, E.L.; McDevitt, H.; Bonyhadi, M.; Bluestone, J.A. In Vitro-Expanded Antigen-Specific Regulatory T Cells Suppress Autoimmune Diabetes. J. Exp. Med. 2004, 199, 1455–1465. [Google Scholar] [CrossRef]
- Tarbell, K.V.; Yamazaki, S.; Olson, K.; Toy, P.; Steinman, R.M. CD25+ CD4+ T Cells, Expanded with Dendritic Cells Presenting a Single Autoantigenic Peptide, Suppress Autoimmune Diabetes. J. Exp. Med. 2004, 199, 1467–1477. [Google Scholar] [CrossRef]
- Sagoo, P.; Ali, N.; Garg, G.; Nestle, F.O.; Lechler, R.I.; Lombardi, G. Human Regulatory T Cells with Alloantigen Specificity Are More Potent Inhibitors of Alloimmune Skin Graft Damage than Polyclonal Regulatory T Cells. Sci. Transl. Med. 2011, 3, 83ra42. [Google Scholar] [CrossRef]
- Golshayan, D.; Jiang, S.; Tsang, J.; Garin, M.I.; Mottet, C.; Lechler, R.I. In Vitro-Expanded Donor Alloantigen-Specific CD4+CD25 + Regulatory T Cells Promote Experimental Transplantation Tolerance. Blood 2007, 109, 827–835. [Google Scholar] [CrossRef]
- Kim, Y.C.; Zhang, A.H.; Su, Y.; Rieder, S.A.; Rossi, R.J.; Ettinger, R.A.; Pratt, K.P.; Shevach, E.M.; Scott, D.W. Engineered Antigen-Specific Human Regulatory T Cells: Immunosuppression of FVIII-Specific T- and B-Cell Responses. Blood 2015, 125, 1107–1115. [Google Scholar] [CrossRef]
- Mahic, M.; Henjum, K.; Yaqub, S.; Bjørnbeth, B.A.; Torgersen, K.M.; Taskén, K.; Aandahl, E.M. Generation of Highly Suppressive Adaptive CD8+CD25+FOXP3+ Regulatory T Cells by Continuous Antigen Stimulation. Eur. J. Immunol. 2008, 38, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Safinia, N.; Scotta, C.; Vaikunthanathan, T.; Lechler, R.I.; Lombardi, G. Regulatory T Cells: Serious Contenders in the Promise for Immunological Tolerance in Transplantation. Front. Immunol. 2015, 6, 438. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, M.; Mikami, N.; Ohkura, N.; Kawakami, R.; Kitagawa, Y.; Sugimoto, A.; Hirota, K.; Nakamura, N.; Ujihara, S.; Kurosaki, T.; et al. Conversion of Antigen-Specific Effector/Memory T Cells into Foxp3-Expressing Tregcells by Inhibition of CDK8/19. Sci. Immunol. 2019, 4, eaaw2707. [Google Scholar] [CrossRef] [PubMed]
- DiPaolo, R.J.; Brinster, C.; Davidson, T.S.; Andersson, J.; Glass, D.; Shevach, E.M. Autoantigen-Specific TGFβ-Induced Foxp3+ Regulatory T Cells Prevent Autoimmunity by Inhibiting Dendritic Cells from Activating Autoreactive T Cells. J. Immunol. 2007, 179, 4685–4693. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Hehlgans, T.; Feuerer, M. Engineered Treg Cells as Putative Therapeutics against Inflammatory Diseases and Beyond. Trends Immunol. 2023, 44, 468–483. [Google Scholar] [CrossRef]
- Hull, C.M.; Nickolay, L.E.; Estorninho, M.; Richardson, M.W.; Riley, J.L.; Peakman, M.; Maher, J.; Tree, T.I.M. Generation of Human Islet-Specific Regulatory T Cells by TCR Gene Transfer. J. Autoimmun. 2017, 79, 63–73. [Google Scholar] [CrossRef]
- Kim, Y.C.; Zhang, A.H.; Yoon, J.; Culp, W.E.; Lees, J.R.; Wucherpfennig, K.W.; Scott, D.W. Engineered MBP-Specific Human Tregs Ameliorate MOG-Induced EAE through IL-2-Triggered Inhibition of Effector T Cells. J. Autoimmun. 2018, 92, 77–86. [Google Scholar] [CrossRef]
- Stephens, L.A.; Malpass, K.H.; Anderton, S.M. Curing CNS Autoimmune Disease with Myelin-Reactive Foxp3+ Treg. Eur. J. Immunol. 2009, 39, 1108–1117. [Google Scholar] [CrossRef]
- Karim, M.; Feng, G.; Wood, K.J.; Bushell, A.R. CD25+CD4+ Regulatory T Cells Generated by Exposure to a Model Protein Antigen Prevent Allograft Rejection: Antigen-Specific Reactivation in Vivo Is Critical for Bystander Regulation. Blood 2005, 105, 4871–4877. [Google Scholar] [CrossRef]
- Wright, G.P.; Notley, C.A.; Xue, S.A.; Bendle, G.M.; Holler, A.; Schumacher, T.N.; Ehrenstein, M.R.; Stauss, H.J. Adoptive Therapy with Redirected Primary Regulatory T Cells Results in Antigen-Specific Suppression of Arthritis. Proc. Natl. Acad. Sci. USA 2009, 106, 19078–19083. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Heinrichs, J.; Haarberg, K.; Semple, K.; Veerapathran, A.; Liu, C.; Anasetti, C.; Yu, X.-Z. HY-Specific Induced Regulatory T Cells Display High Specificity and Efficacy in the Prevention of Acute Graft-versus-Host Disease. J. Immunol. 2015, 195, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.S.; Tanriver, Y.; Jiang, S.; Xue, S.A.; Ratnasothy, K.; Chen, D.; Stauss, H.J.; Bucy, R.P.; Lombardi, G.; Lechler, R. Conferring Indirect Allospecificity on CD4+CD25+ Tregs by TCR Gene Transfer Favors Transplantation Tolerance in Mice. J. Clin. Investig. 2008, 118, 3619–3628. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Waks, T.; Eshhar, Z. Redirection of Regulatory T Cells With Predetermined Specificity for the Treatment of Experimental Colitis in Mice. Gastroenterology 2008, 134, 2014–2024. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Adam, N.; Waks, T.; Eshhar, Z. Amelioration of Colitis by Genetically Engineered Murine Regulatory T Cells Redirected by Antigen-Specific Chimeric Receptor. Gastroenterology 2009, 136, 1721–1731. [Google Scholar] [CrossRef]
- Blat, D.; Zigmond, E.; Alteber, Z.; Waks, T.; Eshhar, Z. Suppression of Murine Colitis and Its Associated Cancer by Carcinoembryonic Antigen-Specific Regulatory T Cells. Mol. Ther. 2014, 22, 1018–1028. [Google Scholar] [CrossRef]
- Wu, D.; Wong, M.Q.; Vent-Schmidt, J.; Boardman, D.A.; Steiner, T.S.; Levings, M.K. A Method for Expansion and Retroviral Transduction of Mouse Regulatory T Cells. J Immunol Methods 2021, 488, 112931. [Google Scholar] [CrossRef]
- Fransson, M.; Piras, E.; Burman, J.; Nilsson, B.; Essand, M.; Lu, B.F.; Harris, R.A.; Magnusson, P.U.; Brittebo, E.; Loskog, A.S.I. CAR/FoxP3-Engineered T Regulatory Cells Target the CNS and Suppress EAE upon Intranasal Delivery. J. Neuroinflamm. 2012, 9, 112. [Google Scholar] [CrossRef]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T Cells Engineered with a Novel Insulin-Specific Chimeric Antigen Receptor as a Candidate Immunotherapy for Type 1 Diabetes. J. Autoimmun. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Skuljec, J.; Chmielewski, M.; Happle, C.; Habener, A.; Busse, M.; Abken, H.; Hansen, G. Chimeric Antigen Receptor-Redirected Regulatory T Cells Suppress Experimental Allergic Airway Inflammation, a Model of Asthma. Front. Immunol. 2017, 8, 1125. [Google Scholar] [CrossRef]
- Yoon, J.; Schmidt, A.; Zhang, A.H.; Königs, C.; Kim, Y.C.; Scott, D.W. FVIII-Specific Human Chimeric Antigen Receptor T-Regulatory Cells Suppress T- and B-Cell Responses to FVIII. Blood 2017, 129, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.A.J.; Rosado-Sánchez, I.; Novakovsky, G.E.; Fung, V.C.W.; Huang, Q.; McIver, E.; Sun, G.; Gillies, J.; Speck, M.; Orban, P.C.; et al. Functional Effects of Chimeric Antigen Receptor Co-Receptor Signaling Domains in Human Regulatory T Cells. Sci. Transl. Med. 2020, 12, eaaz3866. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.G.; Hoeppli, R.E.; Huang, Q.; Gillies, J.; Luciani, D.S.; Orban, P.C.; Broady, R.; Levings, M.K. Alloantigen-Specific Regulatory T Cells Generated with a Chimeric Antigen Receptor. J. Clin. Investig. 2016, 126, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Pierini, A.; Iliopoulou, B.P.; Peiris, H.; Pérez-Cruz, M.; Baker, J.; Hsu, K.; Gu, X.; Zheng, P.P.; Erkers, T.; Tang, S.W.; et al. T Cells Expressing Chimeric Antigen Receptor Promote Immune Tolerance. JCI Insight 2017, 2, e92865. [Google Scholar] [CrossRef] [PubMed]
- Muller, Y.D.; Ferreira, L.M.R.; Ronin, E.; Ho, P.; Nguyen, V.; Faleo, G.; Zhou, Y.; Lee, K.; Leung, K.K.; Skartsis, N.; et al. Precision Engineering of an Anti-HLA-A2 Chimeric Antigen Receptor in Regulatory T Cells for Transplant Immune Tolerance. Front. Immunol. 2021, 12, 686439. [Google Scholar] [CrossRef] [PubMed]
- Boardman, D.A.; Philippeos, C.; Fruhwirth, G.O.; Ibrahim, M.A.A.; Hannen, R.F.; Cooper, D.; Marelli-Berg, F.M.; Watt, F.M.; Lechler, R.I.; Maher, J.; et al. Expression of a Chimeric Antigen Receptor Specific for Donor HLA Class I Enhances the Potency of Human Regulatory T Cells in Preventing Human Skin Transplant Rejection. Am. J. Transplant. 2017, 17, 931–943. [Google Scholar] [CrossRef]
- Noyan, F.; Zimmermann, K.; Hardtke-Wolenski, M.; Knoefel, A.; Schulde, E.; Geffers, R.; Hust, M.; Huehn, J.; Galla, M.; Morgan, M.; et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am. J. Transplant. 2017, 17, 917–930. [Google Scholar] [CrossRef]
- Wagner, J.C.; Ronin, E.; Ho, P.; Peng, Y.; Tang, Q. Anti-HLA-A2-CAR Tregs Prolong Vascularized Mouse Heterotopic Heart Allograft Survival. Am. J. Transplant. 2022, 22, 2237–2245. [Google Scholar] [CrossRef]
- Rawlings, D.J.; Metzler, G.; Wray-Dutra, M.; Jackson, S.W. Altered B Cell Signalling in Autoimmunity. Nat. Rev. Immunol. 2017, 17, 421–436. [Google Scholar] [CrossRef]
- Imura, Y.; Ando, M.; Kondo, T.; Ito, M.; Yoshimura, A. CD19-Targeted CAR Regulatory T Cells Suppress B Cell Pathology without GvHD. JCI Insight 2020, 5, e136185. [Google Scholar] [CrossRef]
- Mackensen, A.; Müller, F.; Mougiakakos, D.; Böltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Author Correction: Anti-CD19 CAR T Cell Therapy for Refractory Systemic Lupus Erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Rana, J.; Perry, D.J.; Kumar, S.R.P.; Muñoz-Melero, M.; Saboungi, R.; Brusko, T.M.; Biswas, M. CAR- and TRuC-Redirected Regulatory T Cells Differ in Capacity to Control Adaptive Immunity to FVIII. Mol. Ther. 2021, 29, 2660–2676. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhao, J.; Song, Y. Engineering Switchable and Programmable Universal CARs for CAR T Therapy. J. Hematol. Oncol. 2019, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- Koristka, S.; Kegler, A.; Bergmann, R.; Arndt, C.; Feldmann, A.; Albert, S.; Cartellieri, M.; Ehninger, A.; Ehninger, G.; Middeke, J.M.; et al. Engrafting Human Regulatory T Cells with a Flexible Modular Chimeric Antigen Receptor Technology. J Autoimmun 2018, 90, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Pohl, A.D.P.; Venkatesha, S.H.; Zhang, A.H.; Scott, D.W. Suppression of FVIII-Specific Memory B Cells by Chimeric BAR Receptor-Engineered Natural Regulatory T Cells. Front. Immunol. 2020, 11, 693. [Google Scholar] [CrossRef] [PubMed]
- Akatsuka, Y. TCR-Like CAR-T Cells Targeting MHC-Bound Minor Histocompatibility Antigens. Front. Immunol. 2020, 11, 257. [Google Scholar] [CrossRef] [PubMed]
- Abdeladhim, M.; Zhang, A.H.; Kropp, L.E.; Lindrose, A.R.; Venkatesha, S.H.; Mitre, E.; Scott, D.W. Engineered Ovalbumin-Expressing Regulatory T Cells Protect against Anaphylaxis in Ovalbumin-Sensitized Mice. Clin. Immunol. 2019, 207, 49–54. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Therapy of Autoimmune Disease. Science 2017, 353, 179–184. [Google Scholar] [CrossRef]
- Ramírez-Chacón, A.; Betriu-Méndez, S.; Bartoló-Ibars, A.; González, A.; Martí, M.; Juan, M. Ligand-Based CAR-T Cell: Different Strategies to Drive T Cells in Future New Treatments. Front. Immunol. 2022, 13, 932559. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. J. Immunol. 2017, 198, 986–992. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Vignali, D.A.A.; Rudensky, A.Y.; Niec, R.E.; Waldmann, H. The Plasticity and Stability of Regulatory T Cells. Nat. Rev. Immunol. 2013, 13, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Qiu, R.; Zhou, L.; Ma, Y.; Zhou, L.; Liang, T.; Shi, L.; Long, J.; Yuan, D. Regulatory T Cell Plasticity and Stability and Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2020, 58, 52–70. [Google Scholar] [CrossRef] [PubMed]
- Dupage, M.; Bluestone, J.A. Harnessing the Plasticity of CD4+ T Cells to Treat Immune-Mediated Disease. Nat. Rev. Immunol. 2016, 16, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Bailey-Bucktrout, S.L.; Jeker, L.T.; Penaranda, C.; Martínez-Llordella, M.; Ashby, M.; Nakayama, M.; Rosenthal, W.; Bluestone, J.A. Instability of the Transcription Factor Foxp3 Leads to the Generation of Pathogenic Memory T Cells in Vivo. Nat. Immunol. 2009, 10, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Bucktrout, S.L.; Martinez-Llordella, M.; Zhou, X.; Anthony, B.; Rosenthal, W.; Luche, H.; Fehling, H.J.; Bluestone, J.A. Self-Antigen-Driven Activation Induces Instability of Regulatory T Cells during an Inflammatory Autoimmune Response. Immunity 2013, 39, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-Hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic Conversion of Foxp3 + T Cells into TH17 Cells in Autoimmune Arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Hua, J.; Inomata, T.; Chen, Y.; Foulsham, W.; Stevenson, W.; Shiang, T.; Bluestone, J.A.; Dana, R. Pathological Conversion of Regulatory T Cells Is Associated with Loss of Allotolerance. Sci. Rep. 2018, 8, 7059. [Google Scholar] [CrossRef]
- Komatsu, N.; Mariotti-Ferrandiz, M.E.; Wang, Y.; Malissen, B.; Waldmann, H.; Hori, S. Heterogeneity of Natural Foxp3+ T Cells: A Committed Regulatory T-Cell Lineage and an Uncommitted Minor Population Retaining Plasticity. Proc. Natl. Acad. Sci. USA 2009, 106, 1903–1908. [Google Scholar] [CrossRef]
- Floess, S.; Freyer, J.; Siewert, C.; Baron, U.; Olek, S.; Polansky, J.; Schlawe, K.; Chang, H.D.; Bopp, T.; Schmitt, E.; et al. Epigenetic Control of the Foxp3 Locus in Regulatory T Cells. PLoS Biol 2007, 5, e38. [Google Scholar] [CrossRef]
- McGovern, J.; Holler, A.; Thomas, S.; Stauss, H.J. Forced Fox-P3 Expression Can Improve the Safety and Antigen-Specific Function of Engineered Regulatory T Cells. J. Autoimmun. 2022, 132, 102888. [Google Scholar] [CrossRef]
- Janson, P.C.J.; Winerdal, M.E.; Marits, P.; Thörn, M.; Ohlsson, R.; Winqvist, O. FOXP3 Promoter Demethylation Reveals the Committed Treg Population in Humans. PLoS ONE 2008, 3, e1612. [Google Scholar] [CrossRef] [PubMed]
- Baron, U.; Floess, S.; Wieczorek, G.; Baumann, K.; Grützkau, A.; Dong, J.; Thiel, A.; Boeld, T.J.; Hoffmann, P.; Edinger, M.; et al. DNA Demethylation in the Human FOXP3 Locus Discriminates Regulatory T Cells from Activated FOXP3+ Conventional T Cells. Eur. J. Immunol. 2007, 37, 2378–2389. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kanamori, M.; Someya, K.; Nakatsukasa, H.; Yoshimura, A. Stabilization of Foxp3 Expression by CRISPR-DCas9-Based Epigenome Editing in Mouse Primary T Cells. Epigenetics Chromatin 2017, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Forstnerič, V.; Oven, I.; Ogorevc, J.; Lainšček, D.; Praznik, A.; Lebar, T.; Jerala, R.; Horvat, S. CRISPRa-Mediated FOXP3 Gene Upregulation in Mammalian Cells. Cell Biosci. 2019, 9, 93. [Google Scholar] [CrossRef]
- Polansky, J.K.; Floess, S.; Freyer, J.; Hamann, A.; Huehn, J. Epigenetic regulation of Foxp3 expression in regulatory T cells. Epigenetics Autoimmune Dis. 2009, 182, 21–38. [Google Scholar] [CrossRef]
- Polansky, J.K.; Kretschmer, K.; Freyer, J.; Floess, S.; Garbe, A.; Baron, U.; Olek, S.; Hamann, A.; von Boehmer, H.; Huehn, J. DNA Methylation Controls Foxp3 Gene Expression. Eur. J. Immunol. 2008, 38, 1654–1663. [Google Scholar] [CrossRef]
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Wang, L.; Li, B.; Greene, M.I.; Wells, A.D.; Hancock, W.W. Histone Deacetylase Inhibitors and Transplantation. Curr. Opin. Immunol. 2007, 19, 589–595. [Google Scholar] [CrossRef][Green Version]
- Tao, R.; De Zoeten, E.F.; Özkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase Inhibition Promotes the Generation and Function of Regulatory T Cells. Nat. Med. 2007, 13, 1299–1307. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Song, M.H.; Oh, K.I. Vitamin C Facilitates Demethylation of the Foxp3 Enhancer in a Tet-Dependent Manner. J. Immunol. 2016, 196, 2119–2131. [Google Scholar] [CrossRef]
- Yue, X.; Trifari, S.; Äijö, T.; Tsagaratou, A.; Pastor, W.A.; Zepeda-Martínez, J.A.; Lio, C.W.J.; Li, X.; Huang, Y.; Vijayanand, P.; et al. Control of Foxp3 Stability through Modulation of TET Activity. J. Exp. Med. 2016, 213, 377–397. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Chi, H. Metabolic Control of Treg Cell Stability, Plasticity, and Tissue-Specific Heterogeneity. Front. Immunol. 2019, 10, 2716. [Google Scholar] [CrossRef] [PubMed]
- Colamatteo, A.; Carbone, F.; Bruzzaniti, S.; Galgani, M.; Fusco, C.; Maniscalco, G.T.; Di Rella, F.; de Candia, P.; De Rosa, V. Molecular Mechanisms Controlling Foxp3 Expression in Health and Autoimmunity: From Epigenetic to Post-Translational Regulation. Front. Immunol. 2020, 10, 3136. [Google Scholar] [CrossRef] [PubMed]
- Joudi, A.M.; Reyes Flores, C.P.; Singer, B.D. Epigenetic Control of Regulatory T Cell Stability and Function: Implications for Translation. Front. Immunol. 2022, 13, 861607. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, Y.; Ohkura, N.; Kidani, Y.; Vandenbon, A.; Hirota, K.; Kawakami, R.; Yasuda, K.; Motooka, D.; Nakamura, S.; Kondo, M.; et al. Guidance of Regulatory T Cell Development by Satb1-Dependent Super-Enhancer Establishment. Nat. Immunol. 2017, 18, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Chen, Y.; Dai, R.; Bian, S.; Xue, W.; Zhu, Y.; Li, Z.; Yang, Y.; Zhang, Y.; Zhang, J.; et al. Chromatin Organizer SATB1 Controls the Cell Identity of CD4+ CD8+ Double-Positive Thymocytes by Regulating the Activity of Super-Enhancers. Nat. Commun. 2022, 13, 5554. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Sakaguchi, S. Transcriptional and Epigenetic Basis of Treg Cell Development and Function: Its Genetic Anomalies or Variations in Autoimmune Diseases. Cell Res. 2020, 30, 465–474. [Google Scholar] [CrossRef]
- Gao, Y.; Tang, J.; Chen, W.; Li, Q.; Nie, J.; Lin, F.; Wu, Q.; Chen, Z.; Gao, Z.; Fan, H.; et al. Inflammation Negatively Regulates FOXP3 and Regulatory T-Cell Function via DBC1. Proc. Natl. Acad. Sci. USA 2015, 112, E3246–E3254. [Google Scholar] [CrossRef]
- Iamsawat, S.; Daenthanasanmak, A.; Voss, J.H.; Nguyen, H.; Bastian, D.; Liu, C.; Yu, X.-Z. Stabilization of Foxp3 by Targeting JAK2 Enhances Efficacy of CD8 Induced Regulatory T Cells in the Prevention of Graft-versus-Host Disease. J. Immunol. 2018, 201, 2812–2823. [Google Scholar] [CrossRef]
- vanLoosdregt, J.; Fleskens, V.; Fu, J.; Brenkman, A.B.; Bekker, C.P.J.; Pals, C.E.G.M.; Meerding, J.; Berkers, C.R.; Barbi, J.; Gröne, A.; et al. Stabilization of the Transcription Factor Foxp3 by the Deubiquitinase USP7 Increases Treg-Cell-Suppressive Capacity. Immunity 2013, 39, 259–271. [Google Scholar] [CrossRef]
- Chen, Z.; Barbi, J.; Bu, S.; Yang, H.Y.; Li, Z.; Gao, Y.; Jinasena, D.; Fu, J.; Lin, F.; Chen, C.; et al. The Ubiquitin Ligase Stub1 Negatively Modulates Regulatory T Cell Suppressive Activity by Promoting Degradation of the Transcription Factor Foxp3. Immunity 2013, 39, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Josefowicz, S.; Chaudhry, A.; Peng, X.P.; Forbush, K.; Rudensky, A.Y. Role of Conserved Non-Coding DNA Elements in the Foxp3 Gene in Regulatory T-Cell Fate. Nature 2010, 463, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Seng, A.; Krausz, K.L.; Pei, D.; Koestler, D.C.; Fischer, R.T.; Yankee, T.M.; Markiewicz, M.A. Coexpression of FOXP3 and a Helios Isoform Enhances the Effectiveness of Human Engineered Regulatory T Cells. Blood Adv. 2020, 4, 1325–1339. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Xia, C.; Morel, L. IL-6 Produced by Dendritic Cells from Lupus-Prone Mice Inhibits CD4+CD25+ T Cell Regulatory Functions. J. Immunol. 2007, 178, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Valencia, X.; Stephens, G.; Goldbach-Mansky, R.; Wilson, M.; Shevach, E.M.; Lipsky, P.E. TNF Downmodulates the Function of Human CD4+CD25hi T-Regulatory Cells. Blood 2006, 108, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Bergmann, C.; Szczepanski, M.; Gooding, W.; Johnson, J.T.; Whiteside, T.L. A Unique Subset of CD4+CD25highFoxp3+ T Cells Secreting Interleukin-10 and Transforming Growth Factor-Β1 Mediates Suppression in the Tumor Microenvironment. Clin. Cancer Res. 2007, 13, 4345–4354. [Google Scholar] [CrossRef] [PubMed]
- Pasare, C.; Medzhitov, R. Toll Pathway-Dependent Blockade of CD4+CD25+ T Cell-Mediated Suppression by Dendritic Cells. Science 2003, 299, 1033–1036. [Google Scholar] [CrossRef]
- Crellin, N.K.; Garcia, R.V.; Levings, M.K. Altered Activation of AKT Is Required for the Suppressive Function of Human CD4 CD25 T Regulatory Cells. Blood 2007, 109, 2014–2022. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Larkin, J.; Minor, D.; Angelo, S.D.; Neyns, B.; Smylie, M.; Miller, W.H.; Gutzmer, R.; Linette, G.; Chmielowski, B.; Lao, C.D.; et al. Overall Survival in Patients With Advanced Melanoma Who Received Nivolumab Versus Investigator’s Choice Chemotherapy in CheckMate 037: A Randomized, Controlled, Open-Label Phase III Trial. J. Clin. Oncol. 2017, 36, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chikuma, S.; Hori, S.; Fagarasan, S.; Honjo, T. Nonoverlapping Roles of PD-1 and FoxP3 in Maintaining Immune Tolerance in a Novel Autoimmune Pancreatitis Mouse Model. Proc. Natl. Acad. Sci. USA 2016, 113, 8490–8495. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in Regulatory T Cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic Self-Tolerance Maintained by Cd25+Cd4+Regulatory T Cells Constitutively Expressing Cytotoxic T Lymphocyte–Associated Antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ Regulatory T Cells Amplified by PD-1 Blockade Promote Hyperprogression of Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008. [Google Scholar] [CrossRef]
- Kim, C.G.; Kim, K.H.; Pyo, K.H.; Xin, C.F.; Hong, M.H.; Ahn, B.C.; Kim, Y.; Choi, S.J.; Yoon, H.I.; Lee, J.G.; et al. Hyperprogressive Disease during PD-1/PD-L1 Blockade in Patients with Non-Small-Cell Lung Cancer. Ann. Oncol. 2019, 30, 1104–1113. [Google Scholar] [CrossRef]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 Combination Blockade Expands Infiltrating T Cells and Reduces Regulatory T and Myeloid Cells within B16 Melanoma Tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef]
- Zanin-Zhorov, A.; Ding, Y.; Kumari, S.; Attur, M.; Hippen, K.L.; Brown, M.; Blazar, B.R.; Abramson, S.B.; Lafaille, J.J.; Dustin, M.L. Protein Kinase C-θ Mediates Negative Feedback on Regulatory T Cell Function. Science 2010, 328, 372–376. [Google Scholar] [CrossRef]
- Altman, A.; Villalba, M. Protein Kinase C-θ (PKCθ): It’s All about Location, Location, Location. Immunol. Rev. 2003, 192, 53–63. [Google Scholar] [CrossRef]
- Ozay, E.I.; Shanthalingam, S.; Sherman, H.L.; Torres, J.A.; Osborne, B.A.; Tew, G.N.; Minter, L.M. Cell-Penetrating Anti-Protein Kinase C Theta Antibodies Act Intracellularly to Generate Stable, Highly Suppressive Regulatory T Cells. Mol. Ther. 2020, 28, 1987–2006. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Leung, J.; Hu, C.; Laszik, Z.G.; Tang, Q.; Vincenti, F.G. Interleukin-6 Blockade with Tocilizumab Increases Tregs and Reduces T Effector Cytokines in Renal Graft Inflammation: A Randomized Controlled Trial. Am. J. Transplant. 2021, 21, 2543–2554. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, M.R.; Evans, J.G.; Singh, A.; Moore, S.; Warnes, G.; Isenberg, D.A.; Mauri, C. Compromised Function of Regulatory T Cells in Rheumatoid Arthritis and Reversal by Anti-TNFα Therapy. J. Exp. Med. 2004, 200, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Burga, R.A.; Yvon, E.; Chorvinsky, E.; Fernandes, R.; Cruz, C.R.; Bollard, C.M. Engineering the TGFb Receptor to Enhance the Therapeutic Potential of Natural Killer Cells as an Immunotherapy for Neuroblastoma. Clin. Cancer Res. 2019, 25, 4400–4412. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guo, L.; Song, Y.; Zhang, Y.; Lin, D.; Hu, B.; Mei, Y.; Sandikin, D.; Liu, H. Augmented Anti-Tumor Activity of NK-92 Cells Expressing Chimeric Receptors of TGF-ΒR II and NKG2D. Cancer Immunol. Immunother. 2017, 66, 537–548. [Google Scholar] [CrossRef]
- Foster, A.E.; Dotti, G.; Lu, A.; Khalil, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M.; Bollard, C.M. Antitumor Activity of EBV-Specific T Lymphocytes Transduced with a Dominant Negative TGF-β Receptor. J. Immunother. 2008, 31, 500–505. [Google Scholar] [CrossRef]
- Bollard, C.M.; Tripic, T.; Cruz, C.R.; Dotti, G.; Gottschalk, S.; Torrano, V.; Dakhova, O.; Carrum, G.; Ramos, C.A.; Liu, H.; et al. Tumor-Specific t-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients with Relapsed Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 1128–1139. [Google Scholar] [CrossRef]
- Mohseni, Y.R.; Saleem, A.; Tung, S.L.; Dudreuilh, C.; Lang, C.; Peng, Q.; Volpe, A.; Adigbli, G.; Cross, A.; Hester, J.; et al. Chimeric Antigen Receptor-Modified Human Regulatory T Cells That Constitutively Express IL-10 Maintain Their Phenotype and Are Potently Suppressive. Eur. J. Immunol. 2021, 51, 2522–2530. [Google Scholar] [CrossRef]
- Bézie, S.; Picarda, E.; Ossart, J.; Tesson, L.; Usal, C.; Renaudin, K.; Anegon, I.; Guillonneau, C. IL-34 Is a Treg-Specific Cytokine and Mediates Transplant Tolerance. J. Clin. Investig. 2015, 125, 3952–3964. [Google Scholar] [CrossRef]
- Zaiss, D.M.; Minutti, C.M.; Knipper, J.A. Immune- and Non-Immune-Mediated Roles of Regulatory T-Cells during Wound Healing. Immunology 2019, 157, 190–197. [Google Scholar] [CrossRef]
- Dial, C.F.; Tune, M.K.; Doerschuk, C.M.; Mock, J.R. Foxp31 Regulatory t Cell Expression of Keratinocyte Growth Factor Enhances Lung Epithelial Proliferation. Am. J. Respir. Cell Mol. Biol. 2017, 57, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A Special Population of Regulatory T Cells Potentiates Muscle Repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Mock, J.R.; Garibaldi, B.T.; Aggarwal, N.R.; Jenkins, J.; Limjunyawong, N.; Singer, B.D.; Chau, E.; Rabold, R.; Files, D.C.; Sidhaye, V.; et al. Foxp3 + Regulatory T Cells Promote Lung Epithelial Proliferation. Mucosal. Immunol. 2014, 7, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.; Hofmann, U.D.W.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T Cells Improve Healing after Myocardial Infarction by Modulating Monocyte/Macrophage Differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, K.Y.; Tam, R.C.Y.; Chan, V.W.; Lan, H.Y.; Hori, S.; Zhou, B.; Lui, K.O. Regulatory T-Cells Regulate Neonatal Heart Regeneration by Potentiating Cardiomyocyte Proliferation in a Paracrine Manner. Theranostics 2019, 9, 4324–4341. [Google Scholar] [CrossRef] [PubMed]
- Povoleri, G.A.M.; Nova-Lamperti, E.; Scottà, C.; Fanelli, G.; Chen, Y.C.; Becker, P.D.; Boardman, D.; Costantini, B.; Romano, M.; Pavlidis, P.; et al. Human Retinoic Acid–Regulated CD161 + Regulatory T Cells Support Wound Repair in Intestinal Mucosa. Nat. Immunol. 2018, 19, 1403–1414. [Google Scholar] [CrossRef]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef]
- Muñoz-Rojas, A.R.; Mathis, D. Tissue Regulatory T Cells: Regulatory Chameleons. Nat. Rev. Immunol. 2021, 21, 597–611. [Google Scholar] [CrossRef]
- Dombrowski, Y.; O’Hagan, T.; DIttmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T Cells Promote Myelin Regeneration in the Central Nervous System. Nat. Neurosci. 2017, 20, 674–680. [Google Scholar] [CrossRef]
- Shime, H.; Odanaka, M.; Tsuiji, M.; Matoba, T.; Imai, M.; Yasumizu, Y.; Uraki, R.; Minohara, K.; Watanabe, M.; Bonito, A.J.; et al. Proenkephalin+ Regulatory T Cells Expanded by Ultraviolet B Exposure Maintain Skin Homeostasis with a Healing Function. Proc. Natl. Acad. Sci. USA 2020, 117, 20696–20705. [Google Scholar] [CrossRef]
- Delacher, M.; Simon, M.; Sanderink, L.; Hotz-Wagenblatt, A.; Wuttke, M.; Schambeck, K.; Schmidleithner, L.; Bittner, S.; Pant, A.; Ritter, U.; et al. Single-Cell Chromatin Accessibility Landscape Identifies Tissue Repair Program in Human Regulatory T Cells. Immunity 2021, 54, 702–720.e17. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.J.; MacDonald, K.N.; Pesenacker, A.M.; Juvet, S.C.; Morishita, K.A.; Bressler, B.; Pan, J.G.; Sidhu, S.S.; Rioux, J.D.; Levings, M.K. Innate Control of Tissue-Reparative Human Regulatory T Cells. J. Immunol. 2019, 202, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Tarbell, K.V. Arming Treg Cells at the Inflammatory Site. Immunity 2009, 30, 322–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Schröppel, B.; Lal, G.; Jakubzick, C.; Mao, X.; Chen, D.; Yin, N.; Jessberger, R.; Ochando, J.C.; Ding, Y.; et al. Regulatory T Cells Sequentially Migrate from Inflamed Tissues to Draining Lymph Nodes to Suppress the Alloimmune Response. Immunity 2009, 30, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; Broxmeyer, H.E.; Kim, C.H. Regulation of Trafficking Receptor Expression in Human Forkhead Box P3+ Regulatory T Cells1. J. Immunol. 2006, 177, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Hoeppli, R.E.; MacDonald, K.N.; Leclair, P.; Fung, V.C.W.; Mojibian, M.; Gillies, J.; Rahavi, S.M.R.; Campbell, A.I.M.; Gandhi, S.K.; Pesenacker, A.M.; et al. Tailoring the Homing Capacity of Human Tregs for Directed Migration to Sites of Th1-Inflammation or Intestinal Regions. Am. J. Transplant. 2019, 19, 62–76. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Raimondi, G.; Glowacki, A.J.; Hall, S.J.; Maskarinec, D.; Thorne, S.H.; Thomson, A.W.; Little, S.R. Bioinspired Controlled Release of CCL22 Recruits Regulatory T Cells in Vivo. Adv. Mater. 2012, 24, 4735–4738. [Google Scholar] [CrossRef]
- Fisher, J.D.; Zhang, W.; Balmert, S.C.; Aral, A.M.; Acharya, A.P.; Kulahci, Y.; Li, J.; Turnquist, H.R.; Thomson, A.W.; Solari, M.G.; et al. In Situ Recruitment of Regulatory T Cells Promotes Donor-Specific Tolerance in Vascularized Composite Allotransplantation. Sci. Adv. 2020, 6, eaax8429. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Feng, B.S.; Zhang, H.; Yang, G.; Jin, Q.R.; Luo, X.Q.; Ma, N.; Huang, Q.M.; Yang, L.T.; Zhang, G.H.; et al. Modulating Oxidative Stress Counteracts Specific Antigen-Induced Regulatory T-Cell Apoptosis in Mice. Eur. J. Immunol. 2021, 51, 1748–1761. [Google Scholar] [CrossRef]
- Taams, L.S.; Smith, J.; Rustin, M.H.; Salmon, M.; Poulter, L.W.; Akbar, A.N. Human Anergic/Suppressive CD4+CD25+ T Cells: A Highly Differentiated and Apoptosis-Prone Population. Eur. J. Immunol. 2001, 31, 1122–1131. [Google Scholar] [CrossRef]
- MacDonald, K.N.; Piret, J.M.; Levings, M.K. Methods to Manufacture Regulatory T Cells for Cell Therapy. Clin. Exp. Immunol. 2019, 197, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Ou, K.; Hamo, D.; Schulze, A.; Roemhild, A.; Kaiser, D.; Gasparoni, G.; Salhab, A.; Zarrinrad, G.; Amini, L.; Schlickeiser, S.; et al. Strong Expansion of Human Regulatory T Cells for Adoptive Cell Therapy Results in Epigenetic Changes Which May Impact Their Survival and Function. Front. Cell Dev. Biol. 2021, 9, 751590. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Gautron, A.-S.; Dominguez-Villar, M.; de Marcken, M.; Hafler, D.A. Enhanced Suppressor Function of TIM-3+FoxP3+ Regulatory T Cells. Eur. J. Immunol. 2014, 44, 2703–2711. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Fierabracci, A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front. Immunol. 2018, 9, 2374. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, C.; Ward-Hartstonge, K.; Mi, T.; Lin, D.T.S.; Huang, Q.; Brown, A.; Edwards, K.; Novakovsky, G.E.; Qi, C.N.; Kobor, M.S.; et al. Tonic-Signaling Chimeric Antigen Receptors Drive Human Regulatory T Cell Exhaustion. Proc. Natl. Acad. Sci. USA 2023, 120, e2219086120. [Google Scholar] [CrossRef]
- Thornton, A.M.; Donovan, E.E.; Piccirillo, C.A.; Shevach, E.M. Cutting Edge: IL-2 Is Critically Required for the In Vitro Activation of CD4+CD25+ T Cell Suppressor Function. J. Immunol. 2004, 172, 6519–6523. [Google Scholar] [CrossRef]
- Bayer, A.L.; Yu, A.; Adeegbe, D.; Malek, T.R. Essential Role for Interleukin-2 for CD4+CD25+ T Regulatory Cell Development during the Neonatal Period. J. Exp. Med. 2005, 201, 769–777. [Google Scholar] [CrossRef]
- Koreth, J.; Matsuoka, K.; Kim, H.T.; McDonough, S.M.; Bindra, B.; Alyea, E.P.; Armand, P.; Cutler, C.; Ho, V.T.; Treister, N.S.; et al. Interleukin-2 and Regulatory T Cells in Graft-versus-Host Disease. N. Engl. J. Med. 2011, 365, 2055–2066. [Google Scholar] [CrossRef]
- He, J.; Zhang, X.; Wei, Y.; Sun, X.; Chen, Y.; Deng, J.; Jin, Y.; Gan, Y.; Hu, X.; Jia, R.; et al. Low-Dose Interleukin-2 Treatment Selectively Modulates CD4+ T Cell Subsets in Patients with Systemic Lupus Erythematosus. Nat. Med. 2016, 22, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, D.; Rosenzwajg, M.; Joly, F.; Six, A.; Carrat, F.; Thibault, V.; Sene, D.; Klatzmann, P.C.D. Regulatory T-Cell Responses to Low-Dose Interleukin-2 in HCV-Induced Vasculitis. N. Engl. J. Med. 2011, 365, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Bollard, C.M.; Carlsten, M.; Melenhorst, J.J.; Biancotto, A.; Wang, E.; Chen, J.; Kotliarov, Y.; Cheung, F.; Xie, Z.; et al. Ultra-Low Dose Interleukin-2 Promotes Immune-Modulating Function of Regulatory t Cells and Natural Killer Cells in Healthy Volunteers. Mol. Ther. 2014, 22, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 Consecutive Patients With Metastatic Melanoma or Renal Cell Cancer Using High-Dose Bolus Interleukin 2. JAMA 1994, 271, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Lotze, M.T.; Yang, J.C.; Aebersold, P.M.; Linehan, W.M.; Seipp, C.A.; White, D.E. Experience with the Use of High-Dose Interleukin-2 in the Treatment of 652 Cancer Patients. Ann. Surg. 1989, 210, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.Y.; Martinez-Llordella, M.; Kodela, E.; Gray, E.; Heneghan, M.A.; Sanchez-Fueyo, A. Low-Dose Interleukin-2 for Refractory Autoimmune Hepatitis. Hepatology 2018, 68, 1649–1652. [Google Scholar] [CrossRef]
- Ward, N.C.; Yu, A.; Moro, A.; Ban, Y.; Chen, X.; Hsiung, S.; Keegan, J.; Arbanas, J.M.; Loubeau, M.; Thankappan, A.; et al. IL-2/CD25: A Long-Acting Fusion Protein That Promotes Immune Tolerance by Selectively Targeting the IL-2 Receptor on Regulatory T Cells. J. Immunol. 2018, 201, 2579–2592. [Google Scholar] [CrossRef]
- Kennedy-Nasser, A.A.; Ku, S.; Castillo-Caro, P.; Hazrat, Y.; Wu, M.F.; Liu, H.; Melenhorst, J.; Barrett, A.J.; Ito, S.; Foster, A.; et al. Ultra Low-Dose IL-2 for GVHD Prophylaxis after Allogeneic Hematopoietic Stem Cell Transplantation Mediates Expansion of Regulatory t Cells without Diminishing Antiviral and Antileukemic Activity. Clin. Cancer Res. 2014, 20, 2215–2225. [Google Scholar] [CrossRef]
- Von Spee-Mayer, C.; Siegert, E.; Abdirama, D.; Rose, A.; Klaus, A.; Alexander, T.; Enghard, P.; Sawitzki, B.; Hiepe, F.; Radbruch, A.; et al. Low-Dose Interleukin-2 Selectively Corrects Regulatory T Cell Defects in Patients with Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2016, 75, 1407–1415. [Google Scholar] [CrossRef]
- Humrich, J.Y.; Von Spee-Mayer, C.; Siegert, E.; Alexander, T.; Hiepe, F.; Radbruch, A.; Burmester, G.R.; Riemekasten, G. Rapid Induction of Clinical Remission by Low-Dose Interleukin-2 in a Patient with Refractory SLE. Ann. Rheum. Dis. 2015, 74, 791–792. [Google Scholar] [CrossRef]
- Speeckaert, R.; Lambert, J.; Van Geel, N. Clinical Significance of Serum Soluble CD Molecules to Assess Disease Activity in Vitiligo. JAMA Dermatol. 2016, 152, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Rosenzwajg, M.; Salet, R.; Lorenzon, R.; Tchitchek, N.; Roux, A.; Bernard, C.; Carel, J.C.; Storey, C.; Polak, M.; Beltrand, J.; et al. Low-Dose IL-2 in Children with Recently Diagnosed Type 1 Diabetes: A Phase I/II Randomised, Double-Blind, Placebo-Controlled, Dose-Finding Study. Diabetologia 2020, 63, 1808–1821. [Google Scholar] [CrossRef] [PubMed]
- Le Duff, F.; Bouaziz, J.D.; Fontas, E.; Ticchioni, M.; Viguier, M.; Dereure, O.; Reygagne, P.; Montaudié, H.; Lacour, J.P.; Monestier, S.; et al. Low-Dose IL-2 for Treating Moderate to Severe Alopecia Areata: A 52-Week Multicenter Prospective Placebo-Controlled Study Assessing Its Impact on T Regulatory Cell and NK Cell Populations. J. Investig. Dermatol. 2021, 141, 933–936.e6. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.Y.; Perpiñán, E.; Londoño, M.C.; Miquel, R.; Ruiz, P.; Kurt, A.S.; Kodela, E.; Cross, A.R.; Berlin, C.; Hester, J.; et al. Low Dose Interleukin-2 Selectively Expands Circulating Regulatory T Cells but Fails to Promote Liver Allograft Tolerance in Humans. J. Hepatol. 2023, 78, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Furlan, S.N.; Singh, K.; Lopez, C.; Tkachev, V.; Hunt, D.J.; Hibbard, J.; Betz, K.M.; Blazar, B.R.; Trapnell, C.; Kean, L.S. IL-2 Enhances Ex Vivo-Expanded Regulatory T-Cell Persistence after Adoptive Transfer. Blood Adv. 2020, 4, 1594–1605. [Google Scholar] [CrossRef] [PubMed]
- Ratnasothy, K.; Jacob, J.; Tung, S.; Boardman, D.; Lechler, R.I.; Sanchez-Fueyo, A.; Martinez-Llordella, M.; Lombardi, G. IL-2 Therapy Preferentially Expands Adoptively Transferred Donor-Specific Tregs Improving Skin Allograft Survival. Am. J. Transplant. 2019, 19, 2092–2100. [Google Scholar] [CrossRef]
- Meguri, Y.; Asano, T.; Yoshioka, T.; Iwamoto, M.; Ikegawa, S.; Sugiura, H.; Kishi, Y.; Nakamura, M.; Sando, Y.; Kondo, T.; et al. Responses of Regulatory and Effector T-Cells to Low-Dose Interleukin-2 Differ Depending on the Immune Environment after Allogeneic Stem Cell Transplantation. Front. Immunol. 2022, 13, 891925. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective Targeting of Engineered T Cells Using Orthogonal IL-2 Cytokine-Receptor Complexes. Science 2018, 359, 1037–1042. [Google Scholar] [CrossRef]
- Hirai, T.; Ramos, T.L.; Lin, P.Y.; Simonetta, F.; Su, L.L.; Picton, L.K.; Baker, J.; Lin, J.X.; Li, P.; Seo, K.; et al. Selective Expansion of Regulatory T Cells Using an Orthogonal IL-2/IL-2 Receptor System Facilitates Transplantation Tolerance. J. Clin. Investig. 2021, 131, e139991. [Google Scholar] [CrossRef]
- Zhang, Q.; Hresko, M.E.; Picton, L.K.; Su, L.; Hollander, M.J.; Nunez-Cruz, S.; Zhang, Z.; Assenmacher, C.A.; Sockolosky, J.T.; Garcia, K.C.; et al. A Human Orthogonal IL-2 and IL-2Rβ System Enhances CAR T Cell Expansion and Antitumor Activity in a Murine Model of Leukemia. Sci. Transl. Med. 2021, 13, abg6986. [Google Scholar] [CrossRef]
- Ramos, T.L.; Bolivar-Wagers, S.; Jin, S.; Thangavelu, G.; Simonetta, F.; Lin, P.-Y.; Hirai, T.; Saha, A.; Koehn, B.; Su, L.L.; et al. Prevention of Acute GVHD Using an Orthogonal IL-2/IL-2Rβ System to Selectively Expand Regulatory T Cells in Vivo. Blood 2023, 141, 1337–1352. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.; Berdugo, Y.A.; Tree, T. IL-2-Based Approaches to Treg Enhancement. Clin. Exp. Immunol. 2023, 211, 149–163. [Google Scholar] [CrossRef] [PubMed]
- VanDyke, D.; Iglesias, M.; Tomala, J.; Young, A.; Smith, J.; Perry, J.A.; Gebara, E.; Cross, A.R.; Cheung, L.S.; Dykema, A.G.; et al. Engineered Human Cytokine/Antibody Fusion Proteins Expand Regulatory T Cells and Confer Autoimmune Disease Protection. Cell Rep. 2022, 41, 111478. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.; Henschel, P.; Simon, D.; Riet, T.; Falk, C.; Hardtke-Wolenski, M.; Wedemeyer, H.; Noyan, F.; Jaeckel, E. Membrane-Bound IL-2 Improves the Expansion, Survival, and Phenotype of CAR Tregs and Confers Resistance to Calcineurin Inhibitors. Front. Immunol. 2022, 13, 1005582. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Barreras, H.; Bader, C.S.; Copsel, S.; Lightbourn, C.O.; Pfeiffer, B.J.; Altman, N.H.; Podack, E.R.; Komanduri, K.V.; Levy, R.B. Marked in Vivo Donor Regulatory T Cell Expansion via Interleukin-2 and TL1A-Ig Stimulation Ameliorates Graft-versus-Host Disease but Preserves Graft-versus-Leukemia in Recipients after Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2017, 23, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Mavers, M.; Simonetta, F.; Nishikii, H.; Ribado, J.V.; Maas-Bauer, K.; Alvarez, M.; Hirai, T.; Turkoz, M.; Baker, J.; Negrin, R.S. Activation of the DR3-TL1A Axis in Donor Mice Leads to Regulatory T Cell Expansion and Activation with Reduction in Graft-versus-Host Disease. Front. Immunol. 2019, 10, 1624. [Google Scholar] [CrossRef] [PubMed]
- Copsel, S.; Wolf, D.; Kale, B.; Barreras, H.; Lightbourn, C.O.; Bader, C.S.; Alperstein, W.; Altman, N.H.; Komanduri, K.V.; Levy, R.B. Very Low Numbers of CD4+ FoxP3+ Tregs Expanded in Donors via TL1A-Ig and Low-Dose IL-2 Exhibit a Distinct Activation/Functional Profile and Suppress GVHD in a Preclinical Model. Biol. Blood Marrow Transplant. 2018, 24, 1788–1794. [Google Scholar] [CrossRef]
- Kim, B.S.; Nishikii, H.; Baker, J. Treatment with Agonistic DR3 Antibody Results in Expansion of Donor Tregs and Reduced Graft-versus-Host Disease. Transplantation 2015, 99, 2005–2006. [Google Scholar] [CrossRef]
- Passerini, L.; Allan, S.E.; Battaglia, M.; Di Nunzio, S.; Alstad, A.N.; Levings, M.K.; Roncarolo, M.G.; Bacchetta, R. STAT5-Signaling Cytokines Regulate the Expression of FOXP3 in CD4+CD25+ Regulatory T Cells and CD4+ CD25- Effector T Cells. Int. Immunol. 2008, 20, 421–431. [Google Scholar] [CrossRef]
- Vogtenhuber, C.; Bucher, C.; Highfill, S.L.; Koch, L.K.; Goren, E.; Panoskaltsis-Mortari, A.; Taylor, P.A.; Farrar, M.A.; Blazar, B.R. Constitutively Active Stat5b in CD4+ T Cells Inhibits Graft-versus-Host Disease Lethality Associated with Increased Regulatory T-Cell Potency and Decreased T Effector Cell Responses. Blood 2010, 116, 466–474. [Google Scholar] [CrossRef]
- Koukoulias, K.; Papayanni, P.G.; Georgakopoulou, A.; Alvanou, M.; Laidou, S.; Kouimtzidis, A.; Pantazi, C.; Gkoliou, G.; Vyzantiadis, T.A.; Spyridonidis, A.; et al. “Cerberus” T Cells: A Glucocorticoid-Resistant, Multi-Pathogen Specific T Cell Product to Fight Infections in Severely Immunocompromised Patients. Front. Immunol. 2021, 11, 608701. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, A.; Alvanou, M.; Karavalakis, G.; Tzannou, I.; Yannaki, E. Pathogen-Specific T Cells: Targeting Old Enemies and New Invaders in Transplantation and Beyond. Hemasphere 2023, 7, E809. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, K.T.; Jadlowsky, J.K.; Hwang, W.T.; Suhoski-Davis, M.; Gonzalez, V.E.; Kulikovskaya, I.; Gupta, M.; Lacey, S.F.; Plesa, G.; Chew, A.; et al. Retroviral and Lentiviral Safety Analysis of Gene-Modified T Cell Products and Infused HIV and Oncology Patients. Mol. Ther. 2018, 26, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-Long Safety and Function of Retroviral-Modified Chimeric Antigen Receptor T Cells. Sci. Transl. Med. 2012, 4, 132ra53. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.-L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A Serious Adverse Event after Successful Gene Therapy for X-Linked Severe Combined Immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.P.; Wilson, J.M.; Batshaw, M.L. Fatal Systemic Inflammatory Response Syndrome in a Ornithine Transcarbamylase Deficient Patient Following Adenoviral Gene Transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef]
- Chandler, R.J.; Sands, M.S.; Venditti, C.P. Recombinant Adeno-Associated Viral Integration and Genotoxicity: Insights from Animal Models. Hum. Gene Ther. 2017, 28, 314–322. [Google Scholar] [CrossRef]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High Levels of AAV Vector Integration into CRISPR-Induced DNA Breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.A.; Merricks, E.P.; Long, C.T.; et al. A Long-Term Study of AAV Gene Therapy in Dogs with Hemophilia A Identifies Clonal Expansions of Transduced Liver Cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A Highly Compact Epitope-Based Marker/Suicide Gene for Easier and Safer T-Cell Therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chang, W.-C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A Transgene-Encoded Cell Surface Polypeptide for Selection, in Vivo Tracking, and Ablation of Engineered Cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef]
- Papayanni, P.-G.; Psatha, N.; Christofi, P.; Li, X.-G.; Melo, P.; Volpin, M.; Montini, E.; Liu, M.; Kaltsounis, G.; Yiangou, M.; et al. Investigating the Barrier Activity of Novel, Human Enhancer-Blocking Chromatin Insulators for Hematopoietic Stem Cell Gene Therapy. Hum. Gene Ther. 2021, 32, 1186–1199. [Google Scholar] [CrossRef]
- Yee, J.K. Off-Target Effects of Engineered Nucleases. FEBS J. 2016, 283, 3239–3248. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Majeed, S.; Hoque, M.Z.; Ahmad, I. Latest Developed Strategies to Minimize the Off-Target Effects in CRISPR-Cas-Mediated Genome Editing. Cells 2020, 9, 1608. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of Off-Target Effects of CRISPR/Cas-Derived RNA-Guided Endonucleases and Nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery Strategies of the CRISPR-Cas9 Gene-Editing System for Therapeutic Applications. J. Control. Release 2017, 266, 17–26. [Google Scholar] [CrossRef]
- Gogishvili, T.; Langenhorst, D.; Lühder, F.; Elias, F.; Elflein, K.; Dennehy, K.M.; Gold, R.; Hünig, T. Rapid Regulatory T-Cell Response Prevents Cytokine Storm in CD28 Superagonist Treated Mice. PLoS ONE 2009, 4, e4643. [Google Scholar] [CrossRef]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Bin Emran, T.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T Cells (Tregs) and Their Therapeutic Potential against Autoimmune Disorders–Advances and Challenges. Hum. Vaccin Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef]
- Galani, I.E.; Wendel, M.; Stojanovic, A.; Jesiak, M.; Müller, M.M.; Schellack, C.; Suri-Payer, E.; Cerwenka, A. Regulatory T Cells Control Macrophage Accumulation and Activation in Lymphoma. Int. J. Cancer 2010, 127, 1131–1140. [Google Scholar] [CrossRef]
- Serafini, M.; Manganini, M.; Borleri, G.; Bonamino, M.; Imberti, L.; Biondi, A.; Golay, J.; Rambaldi, A.; Introna, M. Characterization of CD20-Transduced T Lymphocytes as an Alternative Suicide Gene Therapy Approach for the Treatment of Graft-Versus-Host Disease. Hum. Gene Ther. 2004, 15, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, X.; Zhang, F.; Zhang, X.; Tang, F.; Han, Z.; Li, Y. TCR-T Immunotherapy: The Challenges and Solutions. Front. Oncol. 2022, 11, 794183. [Google Scholar] [CrossRef] [PubMed]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T Cell Specificity towards Leukemia by Zinc Finger Nucleases and Lentiviral Gene Transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-Mediated TCR Replacement Generates Superior Anticancer Transgenic t Cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Mastaglio, S.; Genovese, P.; Magnani, Z.; Ruggiero, E.; Landoni, E.; Camisa, B.; Schiroli, G.; Provasi, E.; Lombardo, A.; Reik, A.; et al. NY-ESO-1 TCR Single Edited Stem and Central Memory T Cells to Treat Multiple Myeloma without Graft-versus-Host Disease. Blood 2017, 130, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Fujiwara, H.; Okamoto, S.; An, J.; Nagai, K.; Shirakata, T.; Mineno, J.; Kuzushima, K.; Shiku, H.; Yasukawa, M. Novel Adoptive T-Cell Immunotherapy Using a WT1-Specific TCR Vector Encoding Silencers for Endogenous TCRs Shows Marked Antileukemia Reactivity and Safety. Blood 2011, 118, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Li, Y.F.; El-Gamil, M.; Robbins, P.F.; Rosenberg, S.A.; Morgan, R.A. Enhanced Antitumor Activity of T Cells Engineered to Express T-Cell Receptors with a Second Disulfide Bond. Cancer Res. 2007, 67, 3898–3903. [Google Scholar] [CrossRef]
- Gorovits, B.; Koren, E. Immunogenicity of Chimeric Antigen Receptor T-Cell Therapeutics. BioDrugs 2019, 33, 275–284. [Google Scholar] [CrossRef]
- Dawson, N.A.J.; Lamarche, C.; Hoeppli, R.E.; Bergqvist, P.; Fung, V.C.W.; McIver, E.; Huang, Q.; Gillies, J.; Speck, M.; Orban, P.C.; et al. Systematic Testing and Specificity Mapping of Alloantigen-Specific Chimeric Antigen Receptors in Regulatory T Cells. JCI Insight 2019, 4, e123672. [Google Scholar] [CrossRef]
- Tuomela, K.; Salim, K.; Levings, M.K. Eras of Designer Tregs: Harnessing Synthetic Biology for Immune Suppression. Immunol. Rev. 2023, 320, 250–267. [Google Scholar] [CrossRef]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.A.; Clegg, D.O.; et al. HLA-E-Expressing Pluripotent Stem Cells Escape Allogeneic Responses and Lysis by NK Cells. Nat. Biotechnol. 2017, 35, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Kudriaeva, A.; Miftakhova, R.; Rybalov, A.; Bulatov, E. Recent Advances in the Development of Bioreactors for Manufacturing of Adoptive Cell Immunotherapies. Bioengineering 2022, 9, 808. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christofi, P.; Pantazi, C.; Psatha, N.; Sakellari, I.; Yannaki, E.; Papadopoulou, A. Promises and Pitfalls of Next-Generation Treg Adoptive Immunotherapy. Cancers 2023, 15, 5877. https://doi.org/10.3390/cancers15245877
Christofi P, Pantazi C, Psatha N, Sakellari I, Yannaki E, Papadopoulou A. Promises and Pitfalls of Next-Generation Treg Adoptive Immunotherapy. Cancers. 2023; 15(24):5877. https://doi.org/10.3390/cancers15245877
Chicago/Turabian StyleChristofi, Panayiota, Chrysoula Pantazi, Nikoleta Psatha, Ioanna Sakellari, Evangelia Yannaki, and Anastasia Papadopoulou. 2023. "Promises and Pitfalls of Next-Generation Treg Adoptive Immunotherapy" Cancers 15, no. 24: 5877. https://doi.org/10.3390/cancers15245877
APA StyleChristofi, P., Pantazi, C., Psatha, N., Sakellari, I., Yannaki, E., & Papadopoulou, A. (2023). Promises and Pitfalls of Next-Generation Treg Adoptive Immunotherapy. Cancers, 15(24), 5877. https://doi.org/10.3390/cancers15245877