
The Multifaceted Roles of Lamins in Lung Cancer and DNA Damage Response




Abstract
:Simple Summary
Abstract
1. Introduction
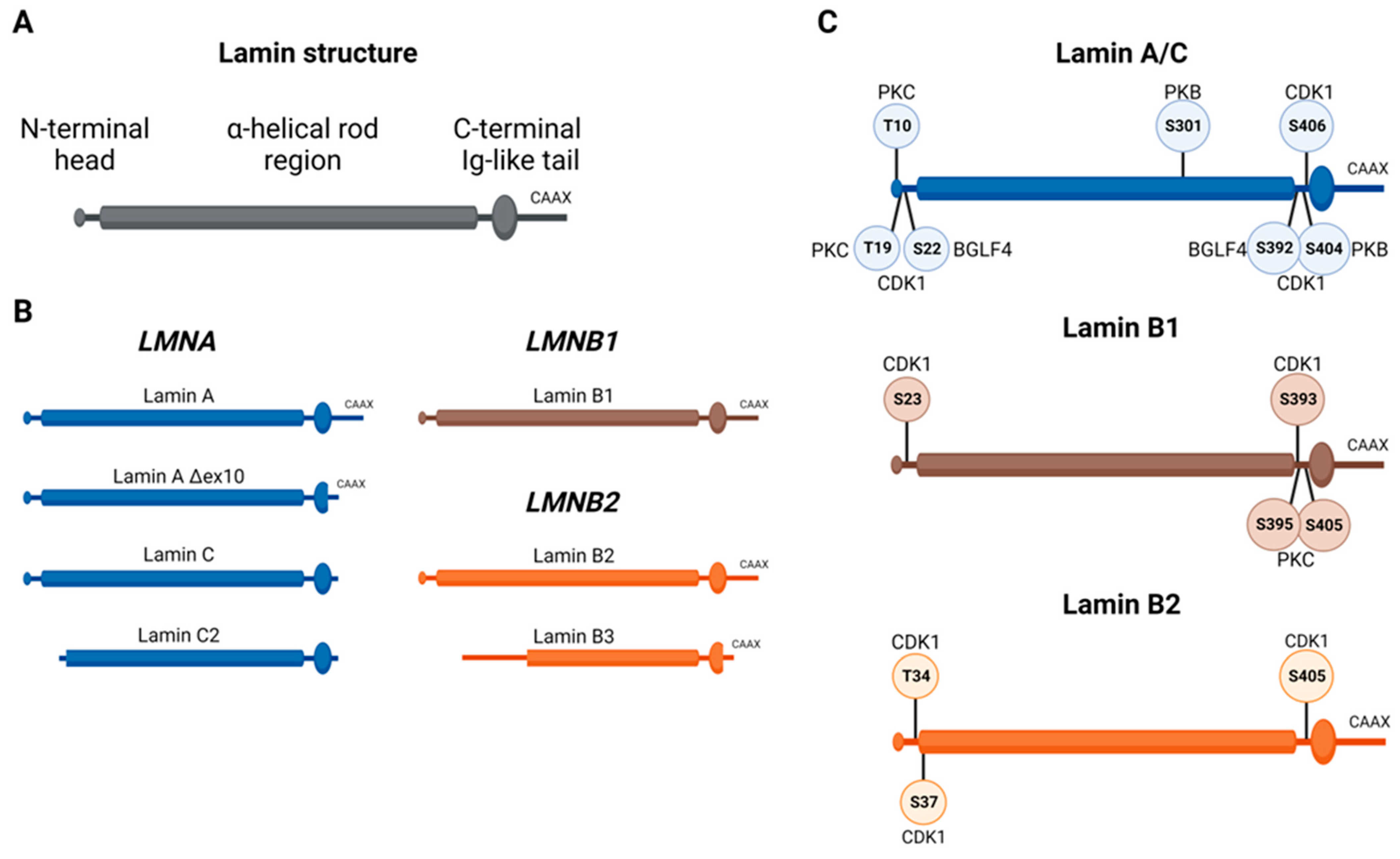
2. Physiological Role of Lamins and Their Regulation during Cell Cycle and Cell Division
3. Contribution of Lamins to the Maintenance of Genomic Stability and Their Role in the DNA Damage Response
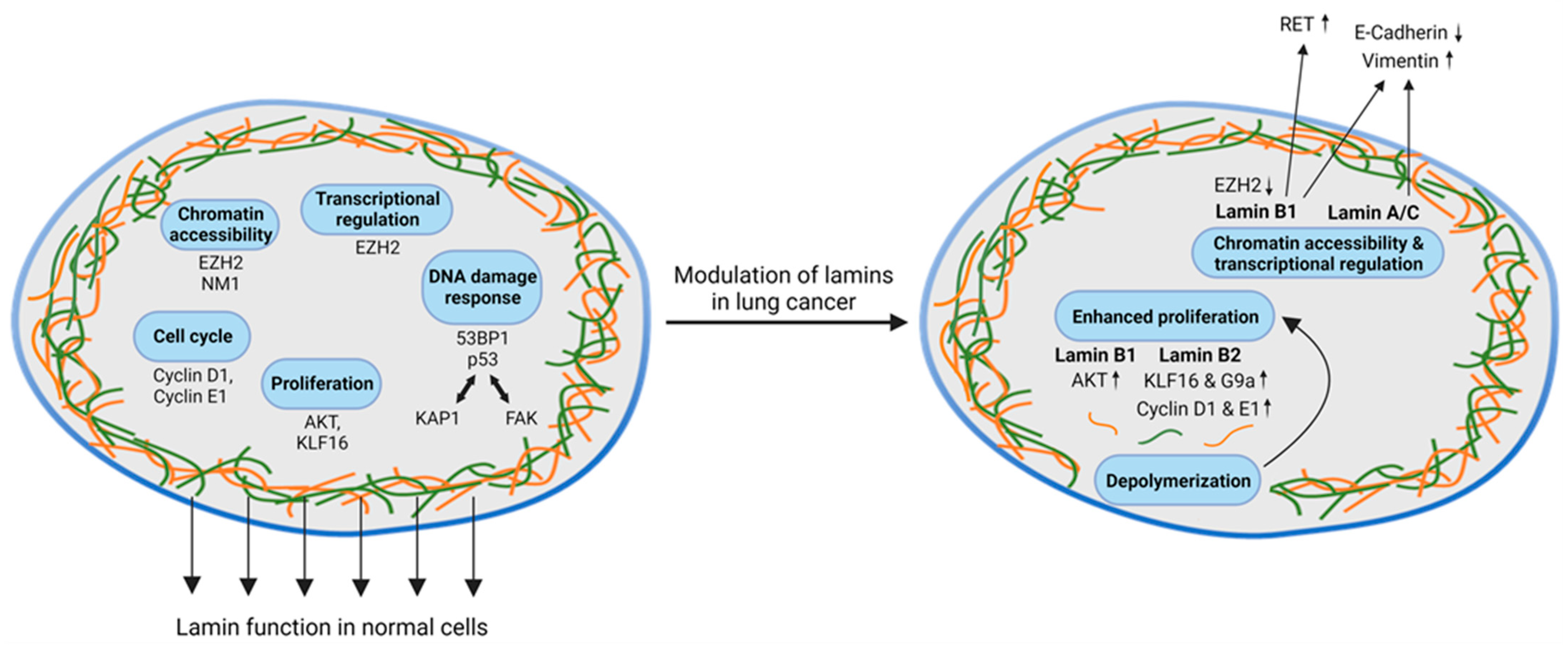
4. Role of Lamins in Lung Cancer
4.1. Role of Lamin Dysregulation in DNA Damage Response, Cell Cycle Transition, and DNA Repair
4.2. Lamin Dysregulation and Chromatin Organization in Lung Cancer
4.3. Impact of Lamin Dysregulation on Cellular Mobility, Plasticity, and EMT in Lung Cancer Cells and Lung Metastases
5. Discussion

{kind=link}
{kind=link}
| Cancer | Cell Line | Lamin Expression | Reference | ||
|---|---|---|---|---|---|
| Regulation | Reference Parameter | Impact | |||
| Ovarian Cancer | n = 1 | ↑* | Ovarian epithelial cells | / | [101] |
| Non-small cell lung cancer | n = 1 n = 1 | ↑ ↓ | Non-cancer cells Normal lung fibroblast cells | / / | [68] [69] |
| Small cell lung cancer | n = 3 | ↓ | NSCLC cell lines | / | [66] |
| Osteosarcoma | n = 2 | ↓ | Low aggressive cell line | Migration | [102] |
| Cancer | Tumor Tissue | Lamin Expression | Reference | ||
| Regulation | Reference Parameter | Impact | |||
| Prostate cancer | n = 46 n = 94 n = 94 | ↑* ↑* ↑ | Benign sample High- vs. low-grade tissue / | / Advanced stage Invasion | [103] [103] [104] |
| Glioblastoma multiforme | n = 152 | ↑ | / | Poor prognosis | [105] |
| Colorectal cancer | n = 656 | ↑ | / | Poor prognosis | [97] |
| Small cell lung cancer | n = 20 n = 33 | ↓ ↓ | NSCLC NSCLC | / / | [67] [66] |
| Breast cancer | n = 115 | ↓ | Non-cancerous tissue | Poor prognosis | [9] |
| Ovarian cancer | n = 108 | ↓ | Ovarian surface epithelial cells | / | [10] |
| Ewing Sarcoma | n = 64 | ↓ | Metastatic vs. primary tumor | Poor prognosis | [106] |
| Gastric cancer | n = 52 | ↓ | Normal tissue | Poor prognosis | [98] |
| Colon cancer | n = 35 n = 370 | ↓ ↓ | Colonic mucosa No recurrence | / Recurrence | [96] [95] |
| Cancer | Cell Line | Lamin Expression | Reference | ||
|---|---|---|---|---|---|
| Regulation | Reference Parameter | Impact | |||
| Ovarian cancer | n = 11 | ↑ | Ovarian surface epithelial cells | / | [10] |
| Hepatocellular carcinoma | n = 5 | ↑ | MIHA and HepG2 | / | [107] |
| Melanoma | n = 6 n = 1 n = 2 | ↑ ↑* ↓* | Normal epidermal melanocytes / / | / Migration Senescence | [55] [93] [55] |
| Prostate cancer | n = 1 | ↑* | / | Proliferation & invasion | [108] |
| Pancreatic cancer | n = 2 | ↓* | / | Migration & Invasion | [109] |
| Cancer | Tumor Tissue | Lamin Expression | Reference | ||
| Regulation | Reference Parameter | Impact | |||
| Ovarian cancer | n = 27 | ↑ | Benign tumors | / | [110] |
| Pancreatic cancer | n = 5 | ↑ | Normal pancreatic tissue | Poor prognosis | [109] |
| Hepatocellular carcinoma | n = 39 n = 364 + 229 | ↑ ↑ | Cirrhosis and non-diseased tissue Low metastasis score group | / Poor prognosis | [107] [111] |
| Clear cell renal cell carcinoma | n = 622 | ↑ | Normal renal tissue | Poor prognosis | [112] |
| Non-small cell lung cancer | n = 483 n = 139 | ↑ ↓ | Normal lung tissue Normal lung tissue | Poor prognosis Advanced stage | [71,72] [67] |
| Small cell lung cancer | n = 22 | ↓ | Normal lung tissue | / | [67] |
| Breast cancer | n = 115 | ↓ | Non-cancerous tissue | Poor prognosis | [9] |
| Colon cancer | n = 35 | ↓ | Normal colonic mucosa | / | [96] |
| Cancer | Cell Line | Lamin Expression | Reference | ||
|---|---|---|---|---|---|
| Regulation | Reference Parameter | Impact | |||
| Colorectal cancer | n = 5 | ↑ | Normal colon epithelial cell line | Proliferation | [94] |
| Cancer | Tumor Tissue | Lamin Expression | Reference | ||
| Regulation | Reference Parameter | Impact | |||
| Non-small cell lung cancer | n = 526 n = 20 n = 150 n = 135 | ↑ ↑ ↑ ↑ | Normal lung sample Adjacent normal lung tissue Low-grade tumor Normal lung tissue | Poor prognosis / Poor prognosis Poor prognosis | [73] [74] [74] [75] |
| Breast cancer | n = 82 n = 1085 | ↑ ↑ | Normal tissue Corresponding normal tissue | Poor prognosis / | [92] [92] |
| Ovarian cancer | n = 27 | ↑ | Benign tumor | / | [110] |
| Colorectal cancer | n = 226 | ↑ | Adjacent tissue | Poor prognosis | [94] |
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef]
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 6450–6454. [Google Scholar] [CrossRef]
- Machiels, B.M.; Zorenc, A.H.; Endert, J.M.; Kuijpers, H.J.; van Eys, G.J.; Ramaekers, F.C.; Broers, J.L. An alternative splicing product of the lamin A/C gene lacks exon 10. J. Biol. Chem. 1996, 271, 9249–9253. [Google Scholar] [CrossRef]
- Bouvier, D.; Hubert, J.; Seve, A.P.; Bouteille, M. Characterization of lamina-bound chromatin in the nuclear shell isolated from HeLa cells. Exp. Cell Res. 1985, 156, 500–512. [Google Scholar] [CrossRef]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of a-Type Lamin Expression Compromises Nuclear Envelope Integrity Leading to Muscular Dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef]
- Wong, X.; Stewart, C.L. The Laminopathies and the Insights They Provide into the Structural and Functional Organization of the Nucleus. Annu. Rev. Genomics Hum. Genet. 2020, 21, 263–288. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Wazir, U.; Ahmed, M.H.; Bridger, J.M.; Harvey, A.; Jiang, W.G.; Sharma, A.K.; Mokbel, K. The clinicopathological significance of lamin A/C, lamin B1 and lamin B receptor mRNA expression in human breast cancer. Cell. Mol. Biol. Lett. 2013, 18, 595–611. [Google Scholar] [CrossRef]
- Capo-chichi, C.D.; Cai, K.Q.; Simpkins, F.; Ganjei-Azar, P.; Godwin, A.K.; Xu, X.-X. Nuclear envelope structural defects cause chromosomal numerical instability and aneuploidy in ovarian cancer. BMC Med. 2011, 9, 28. [Google Scholar] [CrossRef]
- Evangelisti, C.; Rusciano, I.; Mongiorgi, S.; Ramazzotti, G.; Lattanzi, G.; Manzoli, L.; Cocco, L.; Ratti, S. The wide and growing range of lamin B-related diseases: From laminopathies to cancer. Cell. Mol. Life Sci. 2022, 79, 126. [Google Scholar] [CrossRef]
- Furukawa, K.; Inagaki, H.; Hotta, Y. Identification and cloning of an mRNA coding for a germ cell-specific A-type lamin in mice. Exp. Cell Res. 1994, 212, 426–430. [Google Scholar] [CrossRef]
- Furukawa, K.; Hotta, Y. cDNA cloning of a germ cell specific lamin B3 from mouse spermatocytes and analysis of its function by ectopic expression in somatic cells. EMBO J. 1993, 12, 97–106. [Google Scholar] [CrossRef]
- Shelton, K.R.; Egle, P.M.; Cochran, D.L. Nuclear envelope proteins: Identification of lamin B subtypes. Biochem. Biophys. Res. Commun. 1981, 103, 975–981. [Google Scholar] [CrossRef]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef]
- Heitlinger, E.; Peter, M.; Häner, M.; Lustig, A.; Aebi, U.; Nigg, E.A. Expression of chicken lamin B2 in Escherichia coli: Characterization of its structure, assembly, and molecular interactions. J. Cell Biol. 1991, 113, 485–495. [Google Scholar] [CrossRef]
- Shimi, T.; Pfleghaar, K.; Kojima, S.-i.; Pack, C.-G.; Solovei, I.; Goldman, A.E.; Adam, S.A.; Shumaker, D.K.; Kinjo, M.; Cremer, T.; et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4086. [Google Scholar] [CrossRef]
- Machowska, M.; Piekarowicz, K.; Rzepecki, R. Regulation of lamin properties and functions: Does phosphorylation do it all? Open Biol. 2015, 5, 150094. [Google Scholar] [CrossRef]
- Peter, M.; Nakagawa, J.; Dorée, M.; Labbé, J.C.; Nigg, E.A. In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 1990, 61, 591–602. [Google Scholar] [CrossRef]
- Hornbeck, P.; Huang, K.P.; Paul, W.E. Lamin B is rapidly phosphorylated in lymphocytes after activation of protein kinase C. Proc. Natl. Acad. Sci. USA 1988, 85, 2279–2283. [Google Scholar] [CrossRef]
- Obata, T.; Yaffe, M.B.; Leparc, G.G.; Piro, E.T.; Maegawa, H.; Kashiwagi, A.; Kikkawa, R.; Cantley, L.C. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J. Biol. Chem. 2000, 275, 36108–36115. [Google Scholar] [CrossRef]
- Peter, M.; Sanghera, J.S.; Pelech, S.L.; Nigg, E.A. Mitogen-activated protein kinases phosphorylate nuclear lamins and display sequence specificity overlapping that of mitotic protein kinase p34cdc2. Eur. J. Biochem. 1992, 205, 287–294. [Google Scholar] [CrossRef]
- Li, H.; Roux, S.J. Casein kinase II protein kinase is bound to lamina-matrix and phosphorylates lamin-like protein in isolated pea nuclei. Proc. Natl. Acad. Sci. USA 1992, 89, 8434–8438. [Google Scholar] [CrossRef]
- Clements, L.; Manilal, S.; Love, D.R.; Morris, G.E. Direct interaction between emerin and lamin A. Biochem. Biophys. Res. Commun. 2000, 267, 709–714. [Google Scholar] [CrossRef]
- Ranade, D.; Pradhan, R.; Jayakrishnan, M.; Hegde, S.; Sengupta, K. Lamin A/C and Emerin depletion impacts chromatin organization and dynamics in the interphase nucleus. BMC Mol. Cell Biol. 2019, 20, 11. [Google Scholar] [CrossRef]
- Heald, R.; McKeon, F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 1990, 61, 579–589. [Google Scholar] [CrossRef]
- Shtivelman, E.; Sussman, J.; Stokoe, D. A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr. Biol. 2002, 12, 919–924. [Google Scholar] [CrossRef]
- Cenni, V.; Bertacchini, J.; Beretti, F.; Lattanzi, G.; Bavelloni, A.; Riccio, M.; Ruzzene, M.; Marin, O.; Arrigoni, G.; Parnaik, V.; et al. Lamin A Ser404 is a nuclear target of Akt phosphorylation in C2C12 cells. J. Proteome Res. 2008, 7, 4727–4735. [Google Scholar] [CrossRef]
- Bertacchini, J.; Beretti, F.; Cenni, V.; Guida, M.; Gibellini, F.; Mediani, L.; Marin, O.; Maraldi, N.M.; de Pol, A.; Lattanzi, G.; et al. The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J. 2013, 27, 2145–2155. [Google Scholar] [CrossRef]
- Naeem, A.S.; Zhu, Y.; Di, W.L.; Marmiroli, S.; O’Shaughnessy, R.F.L. AKT1-mediated Lamin A/C degradation is required for nuclear degradation and normal epidermal terminal differentiation. Cell Death Differ. 2015, 22, 2123–2132. [Google Scholar] [CrossRef]
- Jeong, S.; Ahn, J.; Jo, I.; Kang, S.-M.; Park, B.-J.; Cho, H.-S.; Kim, Y.-H.; Ha, N.-C. Cyclin-dependent kinase 1 depolymerizes nuclear lamin filaments by disrupting the head-to-tail interaction of the lamin central rod domain. J. Biol. Chem. 2022, 298, 102256. [Google Scholar] [CrossRef]
- Neumann-Staubitz, P.; Kitsberg, D.; Buxboim, A.; Neumann, H. A method to map the interaction network of the nuclear lamina with genetically encoded photo-crosslinkers in vivo. Front. Chem. 2022, 10, 905794. [Google Scholar] [CrossRef]
- Kochin, V.; Shimi, T.; Torvaldson, E.; Adam, S.A.; Goldman, A.; Pack, C.-G.; Melo-Cardenas, J.; Imanishi, S.Y.; Goldman, R.D.; Eriksson, J.E. Interphase phosphorylation of lamin A. J. Cell Sci. 2014, 127, 2683–2696. [Google Scholar] [CrossRef]
- Dephoure, N.; Zhou, C.; Villén, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef]
- Kutay, U.; Jühlen, R.; Antonin, W. Mitotic disassembly and reassembly of nuclear pore complexes. Trends Cell Biol. 2021, 31, 1019–1033. [Google Scholar] [CrossRef]
- Dubińska-Magiera, M.; Kozioł, K.; Machowska, M.; Piekarowicz, K.; Filipczak, D.; Rzepecki, R. Emerin Is Required for Proper Nucleus Reassembly after Mitosis: Implications for New Pathogenetic Mechanisms for Laminopathies Detected in EDMD1 Patients. Cells 2019, 8, 240. [Google Scholar] [CrossRef]
- Tsai, M.-Y.; Wang, S.; Heidinger, J.M.; Shumaker, D.K.; Adam, S.A.; Goldman, R.D.; Zheng, Y. A mitotic lamin B matrix induced by RanGTP required for spindle assembly. Science 2006, 311, 1887–1893. [Google Scholar] [CrossRef]
- Ungricht, R.; Kutay, U. Mechanisms and functions of nuclear envelope remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 229–245. [Google Scholar] [CrossRef]
- McGowan, C.H.; Russell, P. The DNA damage response: Sensing and signaling. Curr. Opin. Cell Biol. 2004, 16, 629–633. [Google Scholar] [CrossRef]
- Roake, C.M.; Artandi, S.E. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Crabbe, L.; Cesare, A.J.; Kasuboski, J.M.; Fitzpatrick, J.A.J.; Karlseder, J. Human telomeres are tethered to the nuclear envelope during post-mitotic nuclear assembly. Cell Rep. 2012, 2, 1521–1529. [Google Scholar] [CrossRef]
- Pennarun, G.; Picotto, J.; Etourneaud, L.; Redavid, A.-R.; Certain, A.; Gauthier, L.R.; Fontanilla-Ramirez, P.; Busso, D.; Chabance-Okumura, C.; Thézé, B.; et al. Increase in lamin B1 promotes telomere instability by disrupting the shelterin complex in human cells. Nucleic Acids Res. 2021, 49, 9886–9905. [Google Scholar] [CrossRef]
- Okamoto, K.; Bartocci, C.; Ouzounov, I.; Diedrich, J.K.; Yates, J.R.; Denchi, E.L. A two-step mechanism for TRF2-mediated chromosome end protection. Nature 2013, 494, 502–505. [Google Scholar] [CrossRef]
- Sarthy, J.; Bae, N.S.; Scrafford, J.; Baumann, P. Human RAP1 inhibits non-homologous end joining at telomeres. EMBO J. 2009, 28, 3390–3399. [Google Scholar] [CrossRef]
- van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef]
- Qian, Y.; Chen, X. Senescence Regulation by the p53 Protein Family. Methods Mol. Biol. 2013, 965, 37–61. [Google Scholar] [CrossRef]
- Yoon, M.-H.; Kang, S.-M.; Lee, S.-J.; Woo, T.-G.; Oh, A.-Y.; Park, S.; Ha, N.-C.; Park, B.-J. p53 induces senescence through Lamin A/C stabilization-mediated nuclear deformation. Cell Death Dis. 2019, 10, 107. [Google Scholar] [CrossRef]
- Chuang, H.-H.; Wang, P.-H.; Niu, S.-W.; Zhen, Y.-Y.; Huang, M.-S.; Hsiao, M.; Yang, C.-J. Inhibition of FAK Signaling Elicits Lamin A/C-Associated Nuclear Deformity and Cellular Senescence. Front. Oncol. 2019, 9, 22. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Finch, R.; Kweh, F.; Massoll, N.A.; Campbell-Thompson, M.; Wallace, M.R.; Cance, W.G. p53 regulates FAK expression in human tumor cells. Mol. Carcinog. 2008, 47, 373–382. [Google Scholar] [CrossRef]
- Goelzer, M.; Dudakovic, A.; Olcum, M.; Sen, B.; Ozcivici, E.; Rubin, J.; van Wijnen, A.J.; Uzer, G. Lamin A/C Is Dispensable to Mechanical Repression of Adipogenesis. Int. J. Mol. Sci. 2021, 22, 6580. [Google Scholar] [CrossRef]
- Panatta, E.; Butera, A.; Celardo, I.; Leist, M.; Melino, G.; Amelio, I. p53 regulates expression of nuclear envelope components in cancer cells. Biol. Direct 2022, 17, 38. [Google Scholar] [CrossRef]
- Lämmerhirt, L.; Kappelmann-Fenzl, M.; Fischer, S.; Pommer, M.; Zimmermann, T.; Kluge, V.; Matthies, A.; Kuphal, S.; Bosserhoff, A.K. Knockdown of Lamin B1 and the Corresponding Lamin B Receptor Leads to Changes in Heterochromatin State and Senescence Induction in Malignant Melanoma. Cells 2022, 11, 2154. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Onorati, A.; Havas, A.P.; Lin, B.; Rajagopal, J.; Sen, P.; Adams, P.D.; Dou, Z. Upregulation of PD-L1 in Senescence and Aging. Mol. Cell. Biol. 2022, 42, e0017122. [Google Scholar] [CrossRef]
- Iwabuchi, K.; Bartel, P.L.; Li, B.; Marraccino, R.; Fields, S. Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl. Acad. Sci. USA 1994, 91, 6098–6102. [Google Scholar] [CrossRef]
- Bothmer, A.; Robbiani, D.F.; Feldhahn, N.; Gazumyan, A.; Nussenzweig, A.; Nussenzweig, M.C. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 2010, 207, 855–865. [Google Scholar] [CrossRef]
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Kim, J.-E.; Thompson, J.R.; Chen, J.; Mer, G. Structural Basis for the Methylation State-Specific Recognition of Histone H4-K20 by 53BP1 and Crb2 in DNA Repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Díaz, C.; Orthwein, A.; Leung, C.C.Y.; Huang, H.; Landry, M.-C.; Kitevski-LeBlanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef]
- Gibbs-Seymour, I.; Markiewicz, E.; Bekker-Jensen, S.; Mailand, N.; Hutchison, C.J. Lamin A/C-dependent interaction with 53BP1 promotes cellular responses to DNA damage. Aging Cell 2015, 14, 162–169. [Google Scholar] [CrossRef]
- Gonzalez-Suarez, I.; Redwood, A.B.; Perkins, S.M.; Vermolen, B.; Lichtensztejin, D.; Grotsky, D.A.; Morgado-Palacin, L.; Gapud, E.J.; Sleckman, B.P.; Sullivan, T.; et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009, 28, 2414–2427. [Google Scholar] [CrossRef]
- Etourneaud, L.; Moussa, A.; Rass, E.; Genet, D.; Willaume, S.; Chabance-Okumura, C.; Wanschoor, P.; Picotto, J.; Thézé, B.; Dépagne, J.; et al. Lamin B1 sequesters 53BP1 to control its recruitment to DNA damage. Sci. Adv. 2021, 7, eabb3799. [Google Scholar] [CrossRef]
- Chen, S.N.; Lombardi, R.; Karmouch, J.; Tsai, J.-Y.; Czernuszewicz, G.; Taylor, M.R.G.; Mestroni, L.; Coarfa, C.; Gurha, P.; Marian, A.J. DNA Damage Response/TP53 Pathway Is Activated and Contributes to the Pathogenesis of Dilated Cardiomyopathy Associated With LMNA (Lamin A/C) Mutations. Circ. Res. 2019, 124, 856–873. [Google Scholar] [CrossRef]
- Wang, J.; Kondo, T.; Nakazawa, T.; Oishi, N.; Mochizuki, K.; Katoh, R. Constitutional abnormality of nuclear membrane proteins in small cell lung carcinoma. Virchows Arch. 2019, 475, 407–414. [Google Scholar] [CrossRef]
- Jia, Y.; Vong, J.S.-L.; Asafova, A.; Garvalov, B.K.; Caputo, L.; Cordero, J.; Singh, A.; Boettger, T.; Günther, S.; Fink, L.; et al. Lamin B1 loss promotes lung cancer development and metastasis by epigenetic derepression of RET. J. Exp. Med. 2019, 216, 1377–1395. [Google Scholar] [CrossRef]
- Stefanello, S.T.; Luchtefeld, I.; Liashkovich, I.; Pethö, Z.; Azzam, I.; Bulk, E.; Rosso, G.; Döhlinger, L.; Hesse, B.; Oeckinghaus, A.; et al. Impact of the Nuclear Envelope on Malignant Transformation, Motility, and Survival of Lung Cancer Cells. Adv. Sci. 2021, 8, e2102757. [Google Scholar] [CrossRef]
- Rubporn, A.; Srisomsap, C.; Subhasitanont, P.; Chokchaichamnankit, D.; Chiablaem, K.; Svasti, J.; Sangvanich, P. Comparative proteomic analysis of lung cancer cell line and lung fibroblast cell line. Cancer Genom. Proteom. 2009, 6, 229–237. [Google Scholar]
- Hu, C.; Zhou, A.; Hu, X.; Xiang, Y.; Huang, M.; Huang, J.; Yang, D.; Tang, Y. LMNA Reduced Acquired Resistance to Erlotinib in NSCLC by Reversing the Epithelial-Mesenchymal Transition via the FGFR/MAPK/c-fos Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 13237. [Google Scholar] [CrossRef]
- Li, W.; Li, X.; Li, X.; Li, M.; Yang, P.; Wang, X.; Li, L.; Yang, B. Lamin B1 Overexpresses in Lung Adenocarcinoma and Promotes Proliferation in Lung Cancer Cells via AKT Pathway. Onco. Targets. Ther. 2020, 13, 3129–3139. [Google Scholar] [CrossRef]
- Li, J.; Sun, Z.; Cui, Y.; Qin, L.; Wu, F.; Li, Y.; Du, N.; Li, X. Knockdown of LMNB1 Inhibits the Proliferation of Lung Adenocarcinoma Cells by Inducing DNA Damage and Cell Senescence. Front. Oncol. 2022, 12, 913740. [Google Scholar] [CrossRef]
- Jiao, X.; Gao, W.; Ren, H.; Wu, Y.; Li, T.; Li, S.; Yan, H. Kruppel like factor 16 promotes lung adenocarcinoma progression by upregulating lamin B2. Bioengineered 2022, 13, 9482–9494. [Google Scholar] [CrossRef]
- Ma, Y.; Fei, L.; Zhang, M.; Zhang, W.; Liu, X.; Wang, C.; Luo, Y.; Zhang, H.; Han, Y. Lamin B2 binding to minichromosome maintenance complex component 7 promotes non-small cell lung carcinogenesis. Oncotarget 2017, 8, 104813–104830. [Google Scholar] [CrossRef]
- Zhang, M.-Y.; Han, Y.-C.; Han, Q.; Liang, Y.; Luo, Y.; Wei, L.; Yan, T.; Yang, Y.; Liu, S.-L.; Wang, E.-H. Lamin B2 promotes the malignant phenotype of non-small cell lung cancer cells by upregulating dimethylation of histone 3 lysine 9. Exp. Cell Res. 2020, 393, 112090. [Google Scholar] [CrossRef]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef]
- de Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef]
- White, D.E.; Negorev, D.; Peng, H.; Ivanov, A.V.; Maul, G.G.; Rauscher, F.J. KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res. 2006, 66, 11594–11599. [Google Scholar] [CrossRef]
- Sanchez, R.; Zhou, M.-M. The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 2011, 36, 364–372. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. The bromodomain interaction module. FEBS Lett. 2012, 586, 2692–2704. [Google Scholar] [CrossRef]
- Wang, C.; Ivanov, A.; Chen, L.; Fredericks, W.J.; Seto, E.; Rauscher, F.J.; Chen, J. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005, 24, 3279–3290. [Google Scholar] [CrossRef]
- Wang, C.; Rauscher, F.J.; Cress, W.D.; Chen, J. Regulation of E2F1 function by the nuclear corepressor KAP1. J. Biol. Chem. 2007, 282, 29902–29909. [Google Scholar] [CrossRef]
- Gesson, K.; Rescheneder, P.; Skoruppa, M.P.; von Haeseler, A.; Dechat, T.; Foisner, R. A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 2016, 26, 462–473. [Google Scholar] [CrossRef]
- Chuang, H.-H.; Huang, M.-S.; Zhen, Y.-Y.; Chuang, C.-H.; Lee, Y.-R.; Hsiao, M.; Yang, C.-J. FAK Executes Anti-Senescence via Regulating EZH2 Signaling in Non-Small Cell Lung Cancer Cells. Biomedicines 2022, 10, 1937. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Yu, Y.; Qi, J.; Xiong, J.; Jiang, L.; Cui, D.; He, J.; Chen, P.; Li, L.; Wu, C.; Ma, T.; et al. Epigenetic Co-Deregulation of EZH2/TET1 is a Senescence-Countering, Actionable Vulnerability in Triple-Negative Breast Cancer. Theranostics 2019, 9, 761–777. [Google Scholar] [CrossRef]
- Chibaya, L.; Murphy, K.C.; DeMarco, K.D.; Gopalan, S.; Liu, H.; Parikh, C.N.; Lopez-Diaz, Y.; Faulkner, M.; Li, J.; Morris, J.P.; et al. EZH2 inhibition remodels the inflammatory senescence-associated secretory phenotype to potentiate pancreatic cancer immune surveillance. Nat. Cancer 2023, 4, 872–892. [Google Scholar] [CrossRef]
- Mulligan, L.M. RET revisited: Expanding the oncogenic portfolio. Nat. Rev. Cancer 2014, 14, 173–186. [Google Scholar] [CrossRef]
- Otsuki, Y.; Saya, H.; Arima, Y. Prospects for new lung cancer treatments that target EMT signaling. Dev. Dyn. 2018, 247, 462–472. [Google Scholar] [CrossRef]
- Pascual-Reguant, L.; Blanco, E.; Galan, S.; Le Dily, F.; Cuartero, Y.; Serra-Bardenys, G.; Di Carlo, V.; Iturbide, A.; Cebrià-Costa, J.P.; Nonell, L.; et al. Lamin B1 mapping reveals the existence of dynamic and functional euchromatin lamin B1 domains. Nat. Commun. 2018, 9, 3420. [Google Scholar] [CrossRef]
- Zhao, C.-C.; Chen, J.; Zhang, L.-Y.; Liu, H.; Zhang, C.-G.; Liu, Y. Lamin B2 promotes the progression of triple negative breast cancer via mediating cell proliferation and apoptosis. Biosci. Rep. 2021, 41, BSR20203874. [Google Scholar] [CrossRef]
- Fracchia, A.; Asraf, T.; Salmon-Divon, M.; Gerlitz, G. Increased Lamin B1 Levels Promote Cell Migration by Altering Perinuclear Actin Organization. Cells 2020, 9, 2161. [Google Scholar] [CrossRef]
- Dong, C.-H.; Jiang, T.; Yin, H.; Song, H.; Zhang, Y.; Geng, H.; Shi, P.-C.; Xu, Y.-X.; Gao, H.; Liu, L.-Y.; et al. LMNB2 promotes the progression of colorectal cancer by silencing p21 expression. Cell Death Dis. 2021, 12, 331. [Google Scholar] [CrossRef]
- Belt, E.J.T.; Fijneman, R.J.A.; van den Berg, E.G.; Bril, H.; Delis-van Diemen, P.M.; Tijssen, M.; van Essen, H.F.; de Lange-de Klerk, E.S.M.; Beliën, J.A.M.; Stockmann, H.B.A.C.; et al. Loss of lamin A/C expression in stage II and III colon cancer is associated with disease recurrence. Eur. J. Cancer 2011, 47, 1837–1845. [Google Scholar] [CrossRef]
- Moss, S.; Krivosheyev, V.; de Souza, A.; Chin, K.; Gaetz, H.; Chaudhary, N.; Worman, H.; Holt, P. Decreased and aberrant nuclear lamin expression in gastrointestinal tract neoplasms. Gut 1999, 45, 723–729. [Google Scholar] [CrossRef]
- Willis, N.D.; Cox, T.R.; Rahman-Casañs, S.F.; Smits, K.; Przyborski, S.A.; van den Brandt, P.; van Engeland, M.; Weijenberg, M.; Wilson, R.G.; de Bruïne, A.; et al. Lamin A/C Is a Risk Biomarker in Colorectal Cancer. PLoS ONE 2008, 3, e2988. [Google Scholar] [CrossRef]
- Wu, Z.; Wu, L.; Weng, D.; Xu, D.; Geng, J.; Zhao, F. Reduced expression of lamin A/C correlates with poor histological differentiation and prognosis in primary gastric carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 8. [Google Scholar] [CrossRef]
- Roncato, F.; Regev, O.; Feigelson, S.W.; Yadav, S.K.; Kaczmarczyk, L.; Levi, N.; Drago-Garcia, D.; Ovadia, S.; Kizner, M.; Addadi, Y.; et al. Reduced Lamin A/C Does Not Facilitate Cancer Cell Transendothelial Migration but Compromises Lung Metastasis. Cancers 2021, 13, 2383. [Google Scholar] [CrossRef]
- Nishikawa, T.; Kuwano, Y.; Nakata, M.; Rokutan, K.; Nishida, K. Multiple G-quadruplexes in the LMNA promoter regulate LMNA variant 6 transcription and promote colon cancer cell growth. Biochim. Biophys. Acta Gene Regul. Mech. 2021, 1864, 194746. [Google Scholar] [CrossRef]
- Sengupta, D.; Sengupta, K. Elevated levels of lamin A promote HR and NHEJ-mediated repair mechanisms in etoposide-treated ovarian cancer cells. bioRxiv 2022. [Google Scholar] [CrossRef]
- Urciuoli, E.; D’Oria, V.; Petrini, S.; Peruzzi, B. Lamin A/C Mechanosensor Drives Tumor Cell Aggressiveness and Adhesion on Substrates With Tissue-Specific Elasticity. Front. Cell Dev. Biol. 2021, 9, 712377. [Google Scholar] [CrossRef]
- Skvortsov, S.; Schäfer, G.; Stasyk, T.; Fuchsberger, C.; Bonn, G.K.; Bartsch, G.; Klocker, H.; Huber, L.A. Proteomics profiling of microdissected low- and high-grade prostate tumors identifies Lamin A as a discriminatory biomarker. J. Proteome Res. 2011, 10, 259–268. [Google Scholar] [CrossRef]
- Kong, L.; Schäfer, G.; Bu, H.; Zhang, Y.; Zhang, Y.; Klocker, H. Lamin A/C protein is overexpressed in tissue-invading prostate cancer and promotes prostate cancer cell growth, migration and invasion through the PI3K/AKT/PTEN pathway. Carcinogenesis 2012, 33, 751–759. [Google Scholar] [CrossRef]
- Gatti, G.; Vilardo, L.; Musa, C.; Di Pietro, C.; Bonaventura, F.; Scavizzi, F.; Torcinaro, A.; Bucci, B.; Saporito, R.; Arisi, I.; et al. Role of Lamin A/C as Candidate Biomarker of Aggressiveness and Tumorigenicity in Glioblastoma Multiforme. Biomedicines 2021, 9, 1343. [Google Scholar] [CrossRef]
- Chiarini, F.; Paganelli, F.; Balestra, T.; Capanni, C.; Fazio, A.; Manara, M.C.; Landuzzi, L.; Petrini, S.; Evangelisti, C.; Lollini, P.-L.; et al. Lamin A and the LINC complex act as potential tumor suppressors in Ewing Sarcoma. Cell Death Dis. 2022, 13, 346. [Google Scholar] [CrossRef]
- Sun, S.; Xu, M.Z.; Poon, R.T.; Day, P.J.; Luk, J.M. Circulating Lamin B1 (LMNB1) biomarker detects early stages of liver cancer in patients. J. Proteome Res. 2010, 9, 70–78. [Google Scholar] [CrossRef]
- Hong, J.-H.; Liang, S.-T.; Wang, A.S.-S.; Yeh, C.-M.; Huang, H.-P.; Sun, C.-D.; Zhang, Z.-H.; Lu, S.-Y.; Chao, Y.-H.; Chen, C.-H.; et al. LMNB1, a potential marker for early prostate cancer progression. Am. J. Cancer Res. 2022, 12, 3390–3404. [Google Scholar]
- Li, L.; Du, Y.; Kong, X.; Li, Z.; Jia, Z.; Cui, J.; Gao, J.; Wang, G.; Xie, K. Lamin B1 Is a Novel Therapeutic Target of Betulinic Acid in Pancreatic Cancer. Clin. Cancer Res. 2013, 19, 4651–4661. [Google Scholar] [CrossRef]
- Bengtsson, S.; Krogh, M.; Szigyarto, C.A.-K.; Uhlen, M.; Schedvins, K.; Silfverswärd, C.; Linder, S.; Auer, G.; Alaiya, A.; James, P. Large-scale proteomics analysis of human ovarian cancer for biomarkers. J. Proteome Res. 2007, 6, 1440–1450. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, L.; Chen, J.; Xiao, W.; Liu, R.; Kan, H. Lamin B1 is a potential therapeutic target and prognostic biomarker for hepatocellular carcinoma. Bioengineered 2022, 13, 9211–9231. [Google Scholar] [CrossRef]
- Radspieler, M.M.; Schindeldecker, M.; Stenzel, P.; Försch, S.; Tagscherer, K.E.; Herpel, E.; Hohenfellner, M.; Hatiboglu, G.; Roth, W.; Macher-Goeppinger, S. Lamin-B1 is a senescence-associated biomarker in clear-cell renal cell carcinoma. Oncol. Lett. 2019, 18, 2654–2660. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janetzko, J.; Oeck, S.; Schramm, A. The Multifaceted Roles of Lamins in Lung Cancer and DNA Damage Response. Cancers 2023, 15, 5501. https://doi.org/10.3390/cancers15235501
Janetzko J, Oeck S, Schramm A. The Multifaceted Roles of Lamins in Lung Cancer and DNA Damage Response. Cancers. 2023; 15(23):5501. https://doi.org/10.3390/cancers15235501
Chicago/Turabian StyleJanetzko, Janina, Sebastian Oeck, and Alexander Schramm. 2023. "The Multifaceted Roles of Lamins in Lung Cancer and DNA Damage Response" Cancers 15, no. 23: 5501. https://doi.org/10.3390/cancers15235501
APA StyleJanetzko, J., Oeck, S., & Schramm, A. (2023). The Multifaceted Roles of Lamins in Lung Cancer and DNA Damage Response. Cancers, 15(23), 5501. https://doi.org/10.3390/cancers15235501