
Mobilization of Circulating Tumor Cells after Short- and Long-Term FOLFIRINOX and GEM/nab-PTX Chemotherapy in Xenograft Mouse Models of Human Pancreatic Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines
2.3. Animals
2.4. Animal Experiment Using Pancreatic Cancer Xenograft Mouse CTC Models
2.5. Blood Sampling
2.6. CTC Isolation from Mouse Blood by Filtration-Based Microfluidic Platform
2.7. Immunocytological and Cytological Staining of CTCs
2.8. Histological and Immunohistochemical Staining of Primary Tumor Tissues
2.9. Statistical Analyses
3. Results
3.1. Sequential Changes in CTCs after Single-Dose FFX and GnP Chemotherapy in the SUIT-2 Mouse Tumor Models
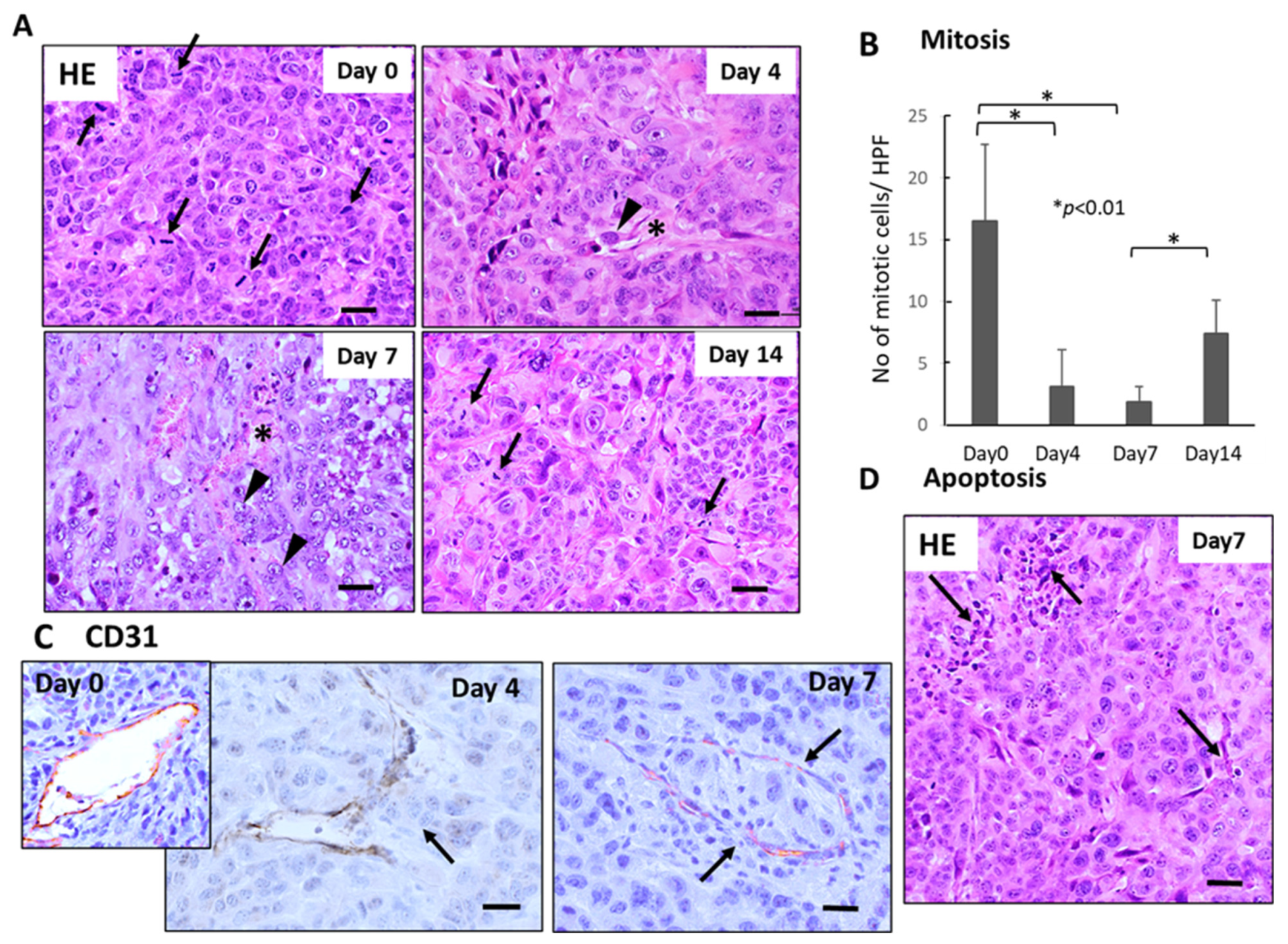
3.2. Histological Changes in the Primary Tumor Tissues(SUIT-2) after Single-Dose FFX and GnP Chemotherapy
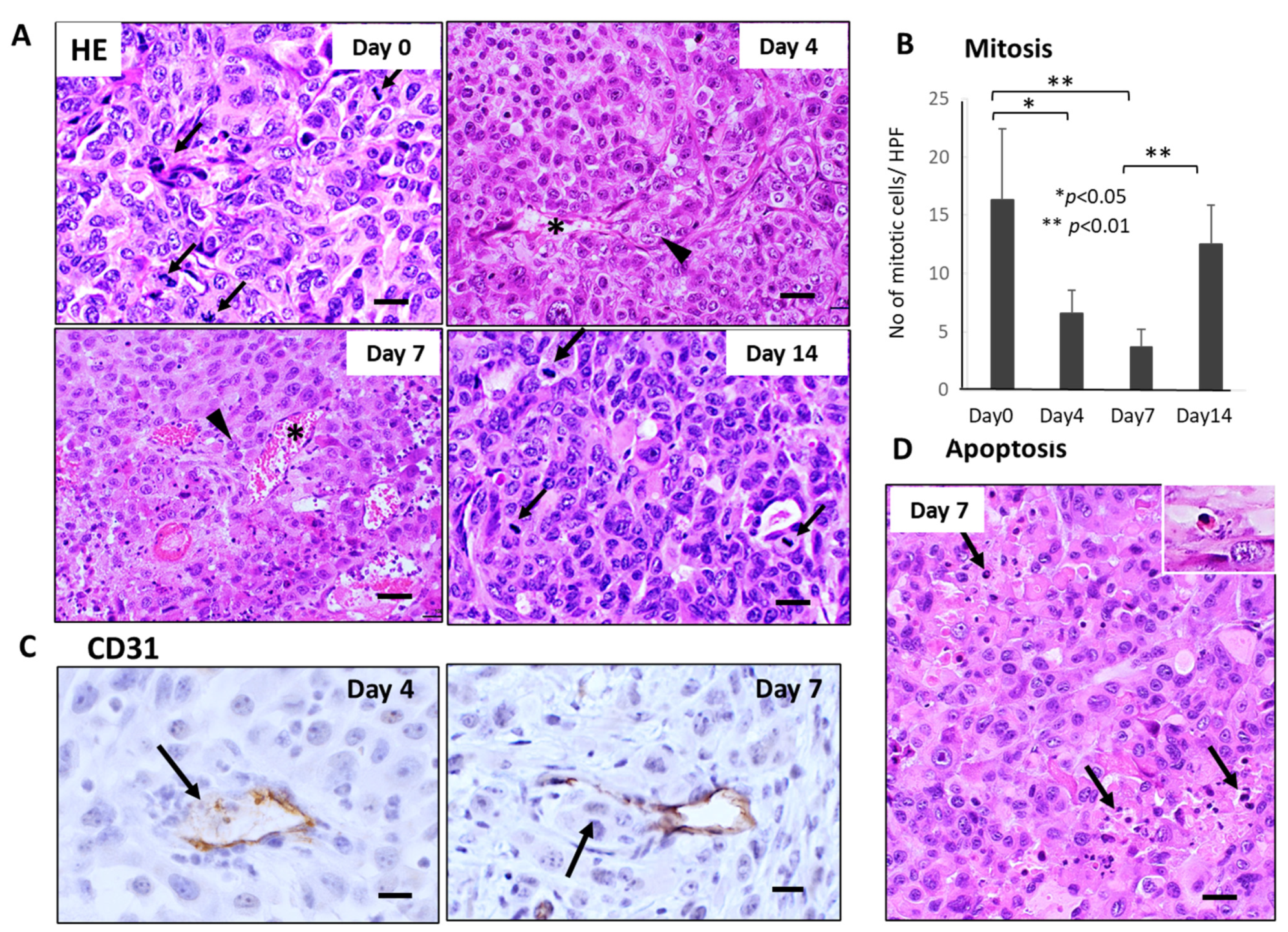
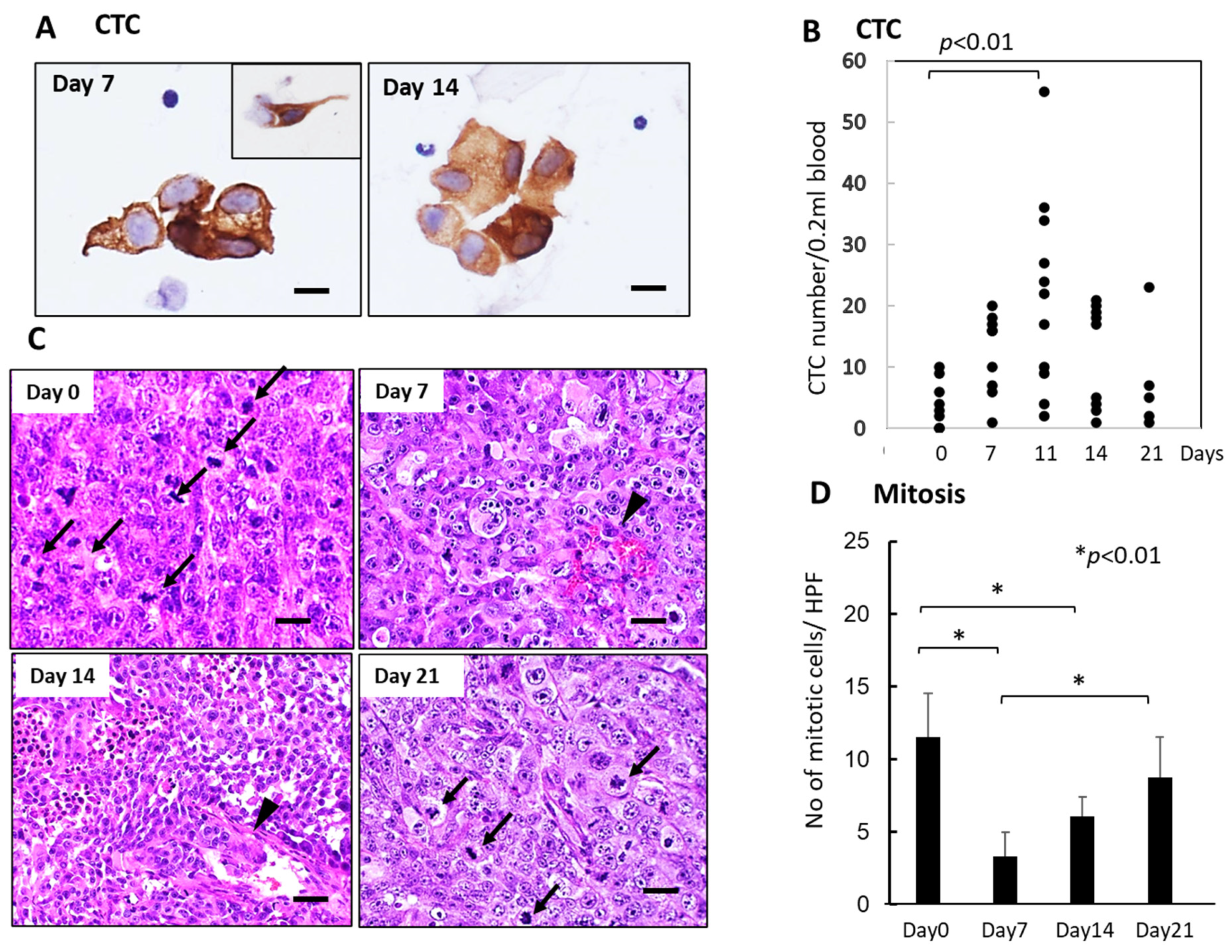
3.3. Sequential Changes in CTCs and Histology of the Primary Tumor Tissues after Single-Dose FFX Chemotherapy in the KP-4 Mouse Tumor Model
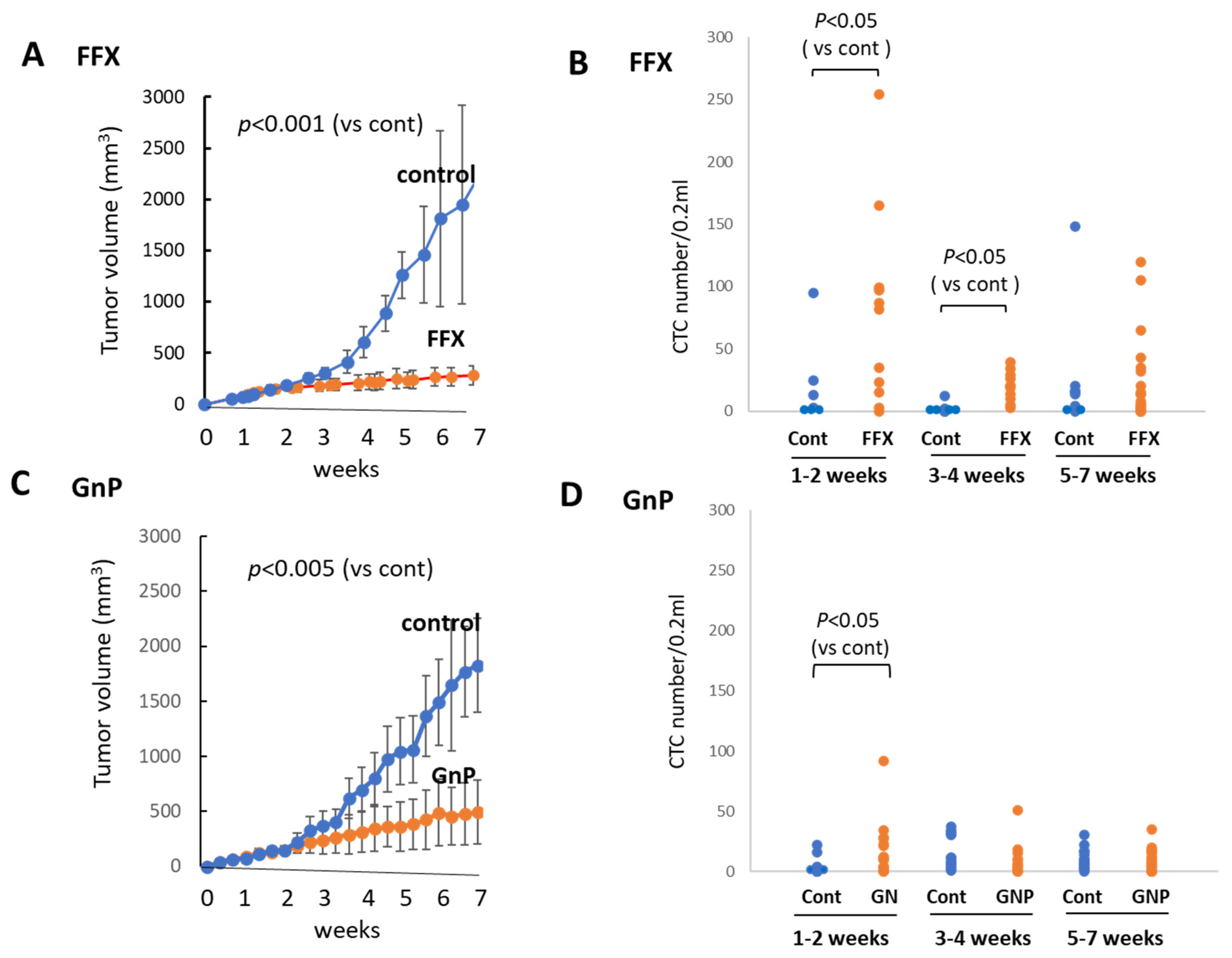
3.4. Sequential Changes in the CTC Numbers after Long-Term, Repetitive FFX and GnP Chemotherapy
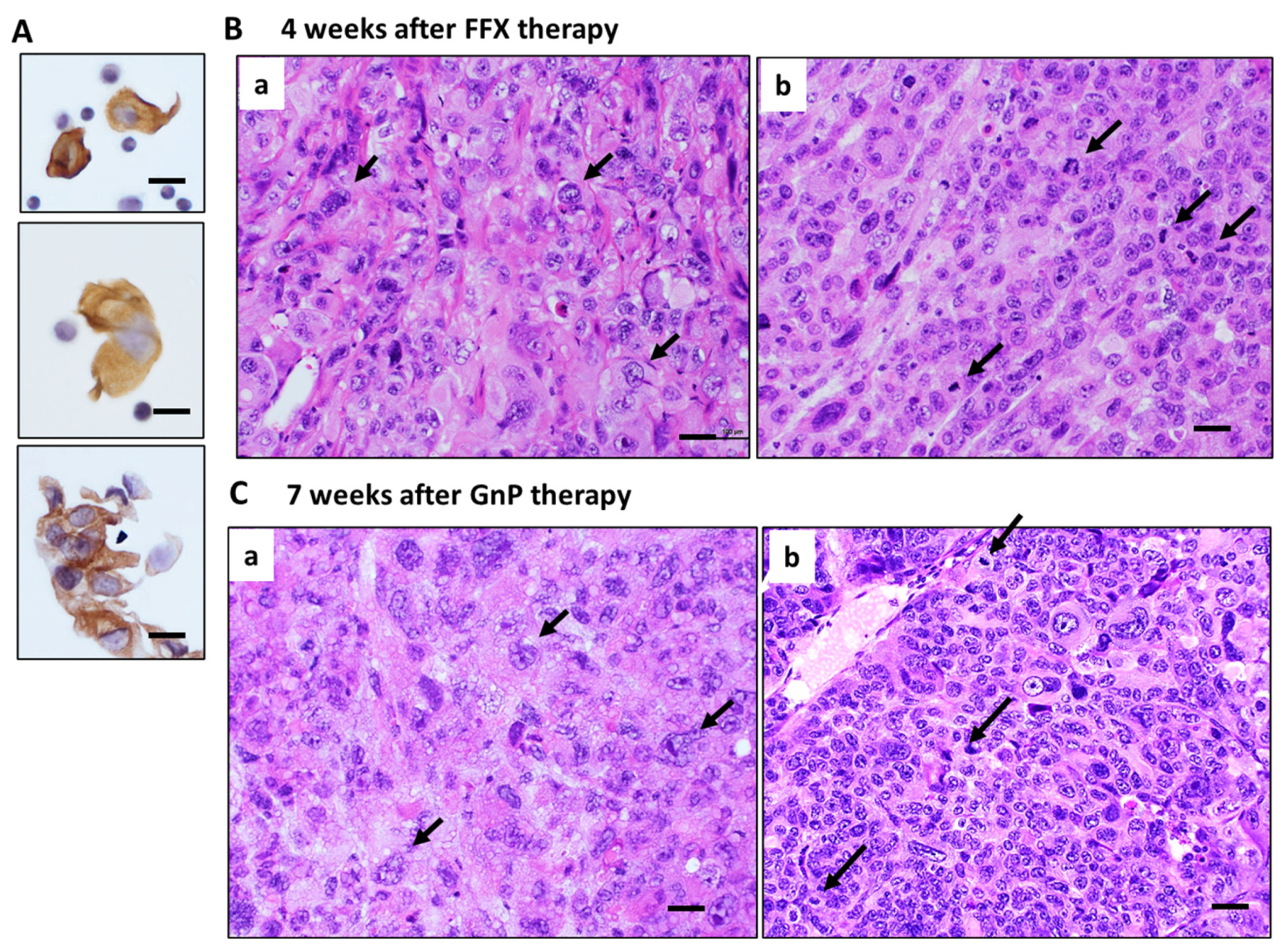
3.5. Histological Changes in Primary Tumor Tissues after Long-Term FFX and GnP Chemotherapy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vasseur, A.; Kiavue, N.; Bidard, F.-C.; Pierga, J.-Y.; Cabel, L. Clinical utility of circulating tumor cells. Mol. Oncol. 2021, 15, 1647–1666. [Google Scholar] [CrossRef]
- Lawrence, R.; Watters, M.; Davies, C.R.; Pantel, K.; Lu, Y.J. Circulating tumor cells for early detection of clinical relevant cancer. Nat. Rev. Clin. Oncol. 2023, 20, 487–500. [Google Scholar] [CrossRef]
- Nikanjam, M.; Kato, S.; Kurzrock, R. Liquid biopsy: Current technology and clinical applications. J. Hematol. Oncol. 2022, 15, 131. [Google Scholar] [CrossRef]
- Freed, I.M.; Kasi, A.; Fateru, O.; Hu, M.; Gonzalez, P.; Weatherington, N.; Pathak, H.; Hyter, S.; Sun, W.; Al-Rajabi, R.; et al. Circulating tumor cell subpopulations predict treatment outcome in Pancreatic ductal adenocarcinoma (PDAC) patients. Cells 2023, 12, 2266. [Google Scholar] [CrossRef] [PubMed]
- Grigoryeva, E.S.; Tashireva, L.A.; Alifanov, V.V.; Savelieva, O.E.; Vtorushin, S.V.; Zavyalova, M.V.; Cherdyntseva, N.V.; Perelmuter, V.M. The novel association of early apoptotic circulating tumor cells with treatment outcomes in breast cancer patients. Int. J. Mol. Sci. 2022, 23, 9475. [Google Scholar] [CrossRef]
- Adachi, Y.; Yoshimura, M.; Nishida, K.; Usuki, H.; Shibata, K.; Hattori, M.; Kondo, N.; Yatabe, Y.; Iwata, H.; Kikumori, T.; et al. Acute phase dynamics of circulating tumor cells after paclitaxel and doxorubicin chemotherapy in breast cancer mouse models. Breast Cancer Res. Treat. 2018, 167, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Nakanishi, H.; Yoshimura, M.; Ito, S.; Sakao, Y.; Kodera, Y.; Yatabe, Y.; Kaneda, N. Dynamics of circulating tumor cells early after targeting therapy to human EGFR-mutated lung cancers and HER2 gene-amplified gastric cancers in mice. Anticancer. Res. 2019, 39, 4711–4720. [Google Scholar] [CrossRef]
- Henley, S.J.; Ward, E.M.; Scott, S.; Ma, J.; Anderson, R.N.; Firth, A.U.; Thomas, C.C.; Islami, F.; Weir, H.K.; Lewis, D.R.; et al. Annual Report to the Nation on the Status of Cancer, part I: National Cancer Statistics. Cancer 2020, 126, 2225–2249. [Google Scholar] [CrossRef] [PubMed]
- Loveday, B.; Lipton, L.; Thomson, B. Pancreatic cancer: An update on diagnosis and management. Aust. J. Gen. Pract. 2019, 48, 826–831. [Google Scholar] [CrossRef]
- Ohgi, K.; Sugiura, T.; Okamura, Y.; Ashida, R.; Yamada, M.; Otsuka, S.; Todaka, A.; Uesaka, K. Long-term adjuvant chemotherapy after resection for pancreatic cancer patients with positive peritoneal lavage cytology. Langenbecks Arch. Surg. 2023, 408, 165. [Google Scholar] [CrossRef]
- Eshmuminov, D.; Aminjonov, B.; Palm, R.F.; Malleo, G.; Schmocker, R.K.; Abdallah, R.; Yoo, C.; Shaib, W.L.; Schneider, M.A.; Rangelova, E.; et al. FOLFIRINOX or gemcitabine-based chemotherapy for borderline resectable and locally advanced pancreatic cancer: A multi-institutional, patient-level, meta-analysis and systematic review. Ann. Surg. Oncol. 2023, 30, 4417–4428. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Yokoyama, Y.; Fujii, T.; Yamada, S.; Takami, H.; Kawashima, H.; Ohno, E.; Ishikawa, T.; Maeda, O.; Hiroshi Ogawa, H.; et al. Results of a phase II study on the use of neoadjuvant chemotherapy (FOLFIRINOX or GEM/nab-PTX) for borderline-resectable pancreatic cancer (NUPAT-01). Ann. Sur. 2022, 275, 1043–1049. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Yeo, D.; Bastian, A.; Strauss, H.; Saxena, P.; Grimison, P.; Rasko, J.E.J. Exploring the clinical utility of pancreatic cancer circulating tumor cells. Int. J. Mol. Sci. 2022, 23, 1671. [Google Scholar] [CrossRef] [PubMed]
- Okubo, K.; Uenosono, Y.; Arigami, T.; Mataki, Y.; Matsushita, D.; Yanagita, S.; Kurahara, H.; Sakoda, M.; Kijima, Y.; Maemura, K.; et al. Clinical impact of circulating tumor cells and therapy response in pancreatic cancer. Eur. J. Surg. Oncol. 2017, 43, 1050–1055. [Google Scholar] [CrossRef]
- Ito, Y.; Inoue, E.; Matsui, Y.; Kobuchi, S.; Moyama, C.; Amagase, K.; Yoshimura, M.; Ikehara, Y.; Nakata, S.; Nakanishi, H. Cytology-based Detection of circulating tumour cells in human pancreatic cancer xenograft models with KRAS mutation. Anticancer Res. 2020, 40, 6781–6789. [Google Scholar] [CrossRef]
- Iwamura, T.; Katsuki, T.; Ide, K. Establishment and characterization of a human pancreatic cancer cell line (SUIT-2) producing carcinoembryonic antigen and carbohydrate antigen 19-9. Jpn. J. Cancer Res. 1987, 78, 54–62. [Google Scholar] [PubMed]
- Nishi, Y.; Haji, M.; Takayanagi, R.; Iguchi, H.; Shimazoe, J.; Hirata, T.; Nawata, H. Establishment and characterization of pthrp-producing human pancreatic-cancer cell-line. Int. J. Oncol. 1994, 5, 33–39. [Google Scholar] [CrossRef]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.H.; Glennie, M.J.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef]
- Eliane, J.P.; Repollet, M.; Luker, K.E.; Brown, M.; Rae, J.M.; Dontu, G.; Schott, A.F.; Wicha, M.; Doyle, G.V.; Hayes, D.F.; et al. Monitoring serial changes in circulating human breast cancer cells in murine xenograft models. Cancer Res. 2008, 68, 5529–5532. [Google Scholar] [CrossRef]
- Tsutsuyama, M.; Nakanishi, H.; Yoshimura, M.; Oshiro, T.; Kinoshita, T.; Komori, K.; Shimizu, Y.; Ichinosawa, Y.; Kinuta, S.; Wajima, K.; et al. Detection of circulating tumor cells in drainage venous blood from colorectal cancer patients using a new filtration and cytology-based automated platform. PLoS ONE 2019, 14, e0212221. [Google Scholar] [CrossRef] [PubMed]
- Gall, T.M.H.; Jacob, J.; Frampton, A.E.; Krell, J.; Kyriakides, C.; Castellano, L.; Stebbing, J.; Jiao, L.R. Reduced dissemination of circulating tumor cells with no-touch isolation surgical technique in patients with pancreatic cancer. JAMA Surg. 2014, 149, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.A.; Anderson, R.L.; Russell, P.A.; Cox, R.A.; Ivashkevich, A.; Swierczak, A.; Doherty, J.P.; Jacobs, D.H.; Smith, J.; Siva, S.; et al. Mobilization of viable tumor cells into the circulation during radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.A.; Anderson, R.L.; Narayan, K.; MacManus, M.P. Does the mobilization of circulating tumour cells during cancer therapy cause metastasis? Nat. Rev. Clin. Oncol. 2017, 14, 32–44. [Google Scholar] [CrossRef]
- Inhestern, J.; Oertel, K.; Stemmann, V.; Schmalenberg, H.; Dietz, A.; Rotter, N.; Veit, J.; Gorner, M.; Sudhoff, H.; Junghanss, C.; et al. Prognostic Role of Circulating Tumor Cells during Induction Chemotherapy Followed by Curative Surgery Combined with Postoperative Radiotherapy in Patients with Locally Advanced Oral and Oropharyngeal Squamous Cell Cancer. PLoS ONE 2015, 10, e0132901. [Google Scholar] [CrossRef]
- Pore, A.A.; Dhanasekara, C.S.; Navaid, H.B.; Vanapalli, S.A.; Rahman, R.L. Comprehensive profiling of cancer-associated cells in the blood of breast cancer patients undergoing neoadjuvant chemotherapy to predict pathological complete response. Bioengineering 2023, 10, 485. [Google Scholar] [CrossRef]
- Ortiz-Otero, N.; Marshall, J.R.; Lash, B.; King, M.R. Chemotherapy-induced release of circulating tumor cells into the blood stream in collective migration with cancer associated fibroblast in metastatic cancer patients. BMC Cancer 2020, 20, 873. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, Y.; Kobuchi, S.; Kawakita, A.; Tosaka, K.; Matsunaga, Y.; Yoshioka, S.; Jonan, S.; Amagase, K.; Hashimoto, K.; Kanda, M.; et al. Mobilization of Circulating Tumor Cells after Short- and Long-Term FOLFIRINOX and GEM/nab-PTX Chemotherapy in Xenograft Mouse Models of Human Pancreatic Cancer. Cancers 2023, 15, 5482. https://doi.org/10.3390/cancers15225482
Ito Y, Kobuchi S, Kawakita A, Tosaka K, Matsunaga Y, Yoshioka S, Jonan S, Amagase K, Hashimoto K, Kanda M, et al. Mobilization of Circulating Tumor Cells after Short- and Long-Term FOLFIRINOX and GEM/nab-PTX Chemotherapy in Xenograft Mouse Models of Human Pancreatic Cancer. Cancers. 2023; 15(22):5482. https://doi.org/10.3390/cancers15225482
Chicago/Turabian StyleIto, Yukako, Shinji Kobuchi, Amiri Kawakita, Kazuki Tosaka, Yume Matsunaga, Shoma Yoshioka, Shizuka Jonan, Kikuko Amagase, Katsunori Hashimoto, Mitsuro Kanda, and et al. 2023. "Mobilization of Circulating Tumor Cells after Short- and Long-Term FOLFIRINOX and GEM/nab-PTX Chemotherapy in Xenograft Mouse Models of Human Pancreatic Cancer" Cancers 15, no. 22: 5482. https://doi.org/10.3390/cancers15225482
APA StyleIto, Y., Kobuchi, S., Kawakita, A., Tosaka, K., Matsunaga, Y., Yoshioka, S., Jonan, S., Amagase, K., Hashimoto, K., Kanda, M., Saito, T., & Nakanishi, H. (2023). Mobilization of Circulating Tumor Cells after Short- and Long-Term FOLFIRINOX and GEM/nab-PTX Chemotherapy in Xenograft Mouse Models of Human Pancreatic Cancer. Cancers, 15(22), 5482. https://doi.org/10.3390/cancers15225482