

Overcoming Immune Checkpoint Therapy Resistance with SHP2 Inhibition in Cancer and Immune Cells: A Review of the Literature and Novel Combinatorial Approaches

 ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
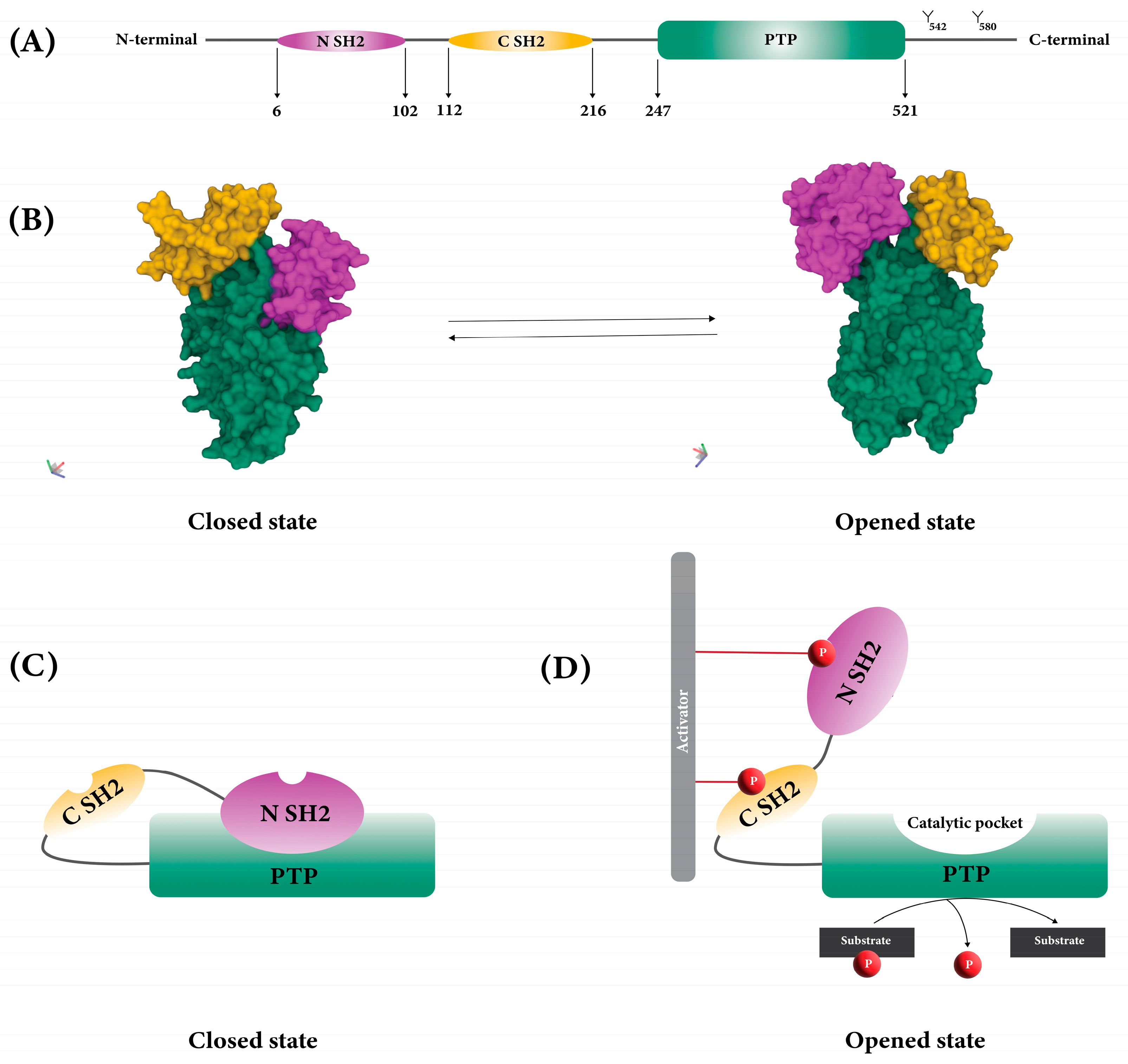
2. The Structure of SHP2
3. Immune Checkpoints
4. Immune Checkpoint Therapy
5. Immune Checkpoint Therapy Resistance Mechanisms
5.1. TME Changes
5.2. Loss of Antigen Expression
5.3. New Genetic Mutations
5.4. Upregulation of Inhibitory Molecules
5.5. Upregulation of Cancer Promoting and Immunosuppressive Signaling Pathways in Cancer and Immune Cells Which Involve in ICI Resistance
6. Role of SHP2 in Checkpoint Therapy Resistance in Immune and Cancer Cells
6.1. The Immune Suppressive Effects of SHP2 in the TME
6.2. The Role of SHP2 in Tumor Antigen Presentation in Cancer Cells
6.3. Role of SHP2 in Tumor-Promoting and Immunosuppressive Signaling Pathways in Cancer and Immune Cells
6.3.1. Immune Roles of SHP2 in Immunosuppressive Signaling Pathways
SHP2 in PD-1 Signaling, RAS/ERK and PI3K/AKT Signaling
SHP2 in CTLA-4 Signaling
SHP2 in BTLA Signaling
SHP2 in TCR Signaling and RAS/MAPK Signaling Pathways
SHP2 in BCR Signaling
SHP2 in NK Cell Signaling
TLR Signaling
SHP2 in JAK/STAT Pathway in Cytokine Receptor Signaling
6.3.2. Non-Immune Roles of SHP2 in Oncogenic Pathways (Tumor-Promoting) in Cancer Cells
RAS/ERK/SOS1
PI3K/AKT
JAK/STAT
7. SHP2 Inhibitors and Their Clinical Development in Oncology
7.1. SHP2 Inhibitors
7.2. SHP2 Inhibitors in the Clinic
7.2.1. TNO155
7.2.2. RMC-4630
7.2.3. Sodium Stibogluconate
7.2.4. JAB-3312 and JAB-3068
7.2.5. ERAS-601
7.2.6. BBP-398
7.2.7. RLY-1971
7.2.8. HBI-2376
7.2.9. BPI-442096
7.2.10. SH3809
8. Overcoming Drug Resistance in Solid Tumors: SHP2 Inhibitors in Combination with Immune Checkpoint Inhibitors
8.1. PD-1/PD-L1
8.2. RTK Inhibitors
8.3. MEK Inhibitors
8.4. ERK Inhibitors
8.5. ALK Inhibitors
8.6. CDK4/6 Inhibitors
8.7. BRAF Inhibitors
9. Future Perspectives
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schaffhausen, B. SH2 domain structure and function. Biochim. Biophys. Acta BBA Rev. Cancer 1995, 1242, 61–75. [Google Scholar] [CrossRef]
- Harris, S.J.; Parry, R.V.; Westwick, J.; Ward, S.G. Phosphoinositide lipid phosphatases: Natural regulators of phosphoinositide 3-kinase signaling in T lymphocytes. J. Biol. Chem. 2008, 283, 2465–2469. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, U. SHP-1 and SHP-2 in T cells: Two phosphatases functioning at many levels. Immunol. Rev. 2009, 228, 342–359. [Google Scholar] [CrossRef]
- Lu, W.; Gong, D.; Bar-Sagi, D.; Cole, P.A. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol. Cell 2001, 8, 759–769. [Google Scholar] [CrossRef]
- Song, Y.; Zhao, M.; Zhang, H.; Yu, B. Double-edged roles of protein tyrosine phosphatase SHP2 in cancer and its inhibitors in clinical trials. Pharmacol. Ther. 2022, 230, 107966. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Lim, S.O.; Yamaguchi, H. Oncogenic signaling pathways associated with immune evasion and resistance to immune checkpoint inhibitors in cancer. Semin. Cancer Biol. 2020, 65, 51–64. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Mechanisms of TGFβ-Induced Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 63. [Google Scholar] [CrossRef]
- Christofides, A.; Katopodi, X.-L.; Cao, C.; Karagkouni, D.; Aliazis, K.; Yenyuwadee, S.; Aksoylar, H.-I.; Pal, R.; Mahmoud, M.A.A.; Strauss, L.; et al. SHP-2 and PD-1-SHP-2 signaling regulate myeloid cell differentiation and antitumor responses. Nat. Immunol. 2023, 24, 55–68. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Dong, L.; Han, D.; Meng, X.; Xu, M.; Zheng, C.; Xia, Q. Activating Mutation of SHP2 Establishes a Tumorigenic Phonotype Through Cell-Autonomous and Non-Cell-Autonomous Mechanisms. Front. Cell Dev. Biol. 2021, 9, 630712. [Google Scholar] [CrossRef]
- Wang, Y.; Mohseni, M.; Grauel, A.; Diez, J.E.; Guan, W.; Liang, S.; Choi, J.E.; Pu, M.; Chen, D.; Laszewski, T.; et al. SHP2 blockade enhances anti-tumor immunity via tumor cell intrinsic and extrinsic mechanisms. Sci. Rep. 2021, 11, 1399. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wu, Z.; Zhao, M.; Zhang, R.; Li, M.; Sun, D.; Cheng, H.; Qi, X.; Shen, Y.; Xu, Q.; et al. Allosteric inhibition reveals SHP2-mediated tumor immunosuppression in colon cancer by single-cell transcriptomics. Acta Pharm. Sin. B 2022, 12, 149–166. [Google Scholar] [CrossRef]
- Liu, M.; Gao, S.; Elhassan, R.M.; Hou, X.B.; Fang, H. Strategies to overcome drug resistance using SHP2 inhibitors. Acta Pharm. Sin. B 2021, 11, 3908–3924. [Google Scholar] [CrossRef] [PubMed]
- Sarver, P.; Acker, M.; Bagdanoff, J.T.; Chen, Z.; Chen, Y.N.; Chan, H.; Firestone, B.; Fodor, M.; Fortanet, J.; Hao, H.; et al. 6-Amino-3-methylpyrimidinones as Potent, Selective, and Orally Efficacious SHP2 Inhibitors. J. Med. Chem. 2019, 62, 1793–1802. [Google Scholar] [CrossRef]
- Tao, Y.; Xie, J.; Zhong, Q.; Wang, Y.; Zhang, S.; Luo, F.; Wen, F.; Xie, J.; Zhao, J.; Sun, X.; et al. A novel partially open state of SHP2 points to a “multiple gear” regulation mechanism. J. Biol. Chem. 2021, 296, 100538. [Google Scholar] [CrossRef] [PubMed]
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal structure of the tyrosine phosphatase SHP-2. Cell 1998, 92, 441–450. [Google Scholar] [CrossRef]
- Darian, E.; Guvench, O.; Yu, B.; Qu, C.K.; MacKerell, A.D., Jr. Structural mechanism associated with domain opening in gain-of-function mutations in SHP2 phosphatase. Proteins 2011, 79, 1573–1588. [Google Scholar] [CrossRef]
- Zhao, R.; Fu, X.; Teng, L.; Li, Q.; Zhao, Z.J. Blocking the function of tyrosine phosphatase SHP-2 by targeting its Src homology 2 domains. J. Biol. Chem. 2003, 278, 42893–42898. [Google Scholar] [CrossRef]
- Cunnick, J.M.; Mei, L.; Doupnik, C.A.; Wu, J. Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J. Biol. Chem. 2001, 276, 24380–24387. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhang, R.; Yang, A.G.; Zheng, G. Diversity of immune checkpoints in cancer immunotherapy. Front. Immunol. 2023, 14, 1121285. [Google Scholar] [CrossRef] [PubMed]
- Park, R.; Winnicki, M.; Liu, E.; Chu, W.M. Immune checkpoints and cancer in the immunogenomics era. Brief. Funct. Genom. 2019, 18, 133–139. [Google Scholar] [CrossRef]
- Zhao, M.; Guo, W.; Wu, Y.; Yang, C.; Zhong, L.; Deng, G.; Zhu, Y.; Liu, W.; Gu, Y.; Lu, Y. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm. Sin. B 2019, 9, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Baldanzi, G. Immune Checkpoint Receptors Signaling in T Cells. Int. J. Mol. Sci. 2022, 23, 3529. [Google Scholar] [CrossRef] [PubMed]
- Kamran, K.; Feng, F. Immune checkpoint therapy modeling of PD-1/PD-L1 blockades reveals subtle difference in their response dynamics and potential synergy in combination. ImmunoInformatics 2021, 1–2, 100004. [Google Scholar] [CrossRef]
- Wang, M.; Yu, L.; Wei, X.; Wei, Y. Role of tumor gene mutations in treatment response to immune checkpoint blockades. Precis. Clin. Med. 2019, 2, 100–109. [Google Scholar] [CrossRef]
- Pulluri, B.; Kumar, A.; Shaheen, M.; Jeter, J.; Sundararajan, S. Tumor microenvironment changes leading to resistance of immune checkpoint inhibitors in metastatic melanoma and strategies to overcome resistance. Pharmacol. Res. 2017, 123, 95–102. [Google Scholar] [CrossRef]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef]
- Haibe, Y.; El Husseini, Z.; El Sayed, R.; Shamseddine, A. Resisting Resistance to Immune Checkpoint Therapy: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 6176. [Google Scholar] [CrossRef]
- Li, J.; Jie, H.B.; Lei, Y.; Gildener-Leapman, N.; Trivedi, S.; Green, T.; Kane, L.P.; Ferris, R.L. PD-1/SHP-2 Inhibits Tc1/Th1 Phenotypic Responses and the Activation of T Cells in the Tumor Microenvironment. Cancer Res. 2015, 75, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.S.; Srivastava, R.M.; Andrade Filho, P.A.; Egloff, A.M.; Wang, L.; Seethala, R.R.; Ferrone, S.; Ferris, R.L. SHP2 Is Overexpressed and Inhibits pSTAT1-Mediated APM Component Expression, T-cell Attracting Chemokine Secretion, and CTL Recognition in Head and Neck Cancer CellsSHP2 Causes pSTAT1-Dependent Immune Escape. Clin. Cancer Res. 2013, 19, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Nunès, J.A.; Vély, F. Natural Killer Cell Signaling Pathways. Science 2004, 306, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- Yusa, S.; Campbell, K.S. Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) can play a direct role in the inhibitory function of killer cell Ig-like receptors in human NK cells. J. Immunol. 2003, 170, 4539–4547. [Google Scholar] [CrossRef]
- Achkova, D.; Maher, J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem. Soc. Trans. 2016, 44, 333–341. [Google Scholar] [CrossRef]
- Veillette, A.; Chen, J. SIRPα–CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef]
- Cammann, C.; Israel, N.; Frentzel, S.; Jeron, A.; Topfstedt, E.; Schüler, T.; Simeoni, L.; Zenker, M.; Fehling, H.J.; Schraven, B.; et al. T cell-specific constitutive active SHP2 enhances T cell memory formation and reduces T cell activation. Front. Immunol. 2022, 13, 958616. [Google Scholar] [CrossRef]
- Schneider, H.; Rudd, C.E. Tyrosine phosphatase SHP-2 binding to CTLA-4: Absence of direct YVKM/YFIP motif recognition. Biochem. Biophys. Res. Commun. 2000, 269, 279–283. [Google Scholar] [CrossRef]
- Niogret, C.; Birchmeier, W.; Guarda, G. SHP-2 in Lymphocytes’ Cytokine and Inhibitory Receptor Signaling. Front. Immunol. 2019, 10, 2468. [Google Scholar] [CrossRef] [PubMed]
- Taku, O.; Akito, M.; Hiroyuki, N.; Tomohiro, K.; Tasuku, H. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef]
- Purdy, A.K.; Campbell, K.S. SHP-2 expression negatively regulates NK cell function. J. Immunol. 2009, 183, 7234–7243. [Google Scholar] [CrossRef] [PubMed]
- Lannoy, V.; Côté-Biron, A.; Asselin, C.; Rivard, N. Phosphatases in toll-like receptors signaling: The unfairly-forgotten. Cell Commun. Signal 2021, 19, 10. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal 2017, 15, 23. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Dance, M.; Montagner, A.; Salles, J.P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Tajan, M.; de Rocca Serra, A.; Valet, P.; Edouard, T.; Yart, A. SHP2 sails from physiology to pathology. Eur. J. Med. Genet. 2015, 58, 509–525. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Yang, W.; Kontaridis, M.I.; Bivona, T.G.; Wen, G.; Araki, T.; Luo, J.; Thompson, J.A.; Schraven, B.L.; Philips, M.R.; et al. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol. Cell 2004, 13, 341–355. [Google Scholar] [CrossRef]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Yart, A.; Dance, M.; Perret, B.; Salles, J.P.; Raynal, P. A novel role for Gab1 and SHP2 in epidermal growth factor-induced Ras activation. J. Biol. Chem. 2005, 280, 5350–5360. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Cheon, H.; Wang, Y. Responses to Cytokines and Interferons that Depend upon JAKs and STATs. Cold Spring Harb. Perspect. Biol. 2018, 10, a028555. [Google Scholar] [CrossRef]
- Shen, D.; Chen, W.; Zhu, J.; Wu, G.; Shen, R.; Xi, M.; Sun, H. Therapeutic potential of targeting SHP2 in human developmental disorders and cancers. Eur. J. Med. Chem. 2020, 190, 112117. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef]
- Chen, J.; Yu, W.M.; Bunting, K.D.; Qu, C.K. A negative role of SHP-2 tyrosine phosphatase in growth factor-dependent hematopoietic cell survival. Oncogene 2004, 23, 3659–3669. [Google Scholar] [CrossRef][Green Version]
- Ohtani, T.; Ishihara, K.; Atsumi, T.; Nishida, K.; Kaneko, Y.; Miyata, T.; Itoh, S.; Narimatsu, M.; Maeda, H.; Fukada, T.; et al. Dissection of signaling cascades through gp130 in vivo: Reciprocal roles for STAT3- and SHP2-mediated signals in immune responses. Immunity 2000, 12, 95–105. [Google Scholar] [CrossRef]
- Liu, Q.Q.; Qu, J.; Zhao, M.X.; Xu, Q.; Sun, Y. Targeting SHP2 as a promising strategy for cancer immunotherapy. Pharmacol. Res. 2020, 152, 104595. [Google Scholar] [CrossRef]
- Song, Y.; Yang, X.; Wang, S.; Zhao, M.; Yu, B. Crystallographic landscape of SHP2 provides molecular insights for SHP2 targeted drug discovery. Med. Res. Rev. 2022, 42, 1781–1821. [Google Scholar] [CrossRef]
- Hellmuth, K.; Grosskopf, S.; Lum, C.T.; Würtele, M.; Röder, N.; von Kries, J.P.; Rosario, M.; Rademann, J.; Birchmeier, W. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc. Natl. Acad. Sci. USA 2008, 105, 7275–7280. [Google Scholar] [CrossRef]
- Tang, K.; Jia, Y.N.; Yu, B.; Liu, H.M. Medicinal chemistry strategies for the development of protein tyrosine phosphatase SHP2 inhibitors and PROTAC degraders. Eur. J. Med. Chem. 2020, 204, 112657. [Google Scholar] [CrossRef]
- Sarmiento, M.; Wu, L.; Keng, Y.-F.; Song, L.; Luo, Z.; Huang, Z.; Wu, G.-Z.; Yuan, A.K.; Zhang, Z.-Y. Structure-based discovery of small molecule inhibitors targeted to protein tyrosine phosphatase 1B. J. Med. Chem. 2000, 43, 146–155. [Google Scholar] [CrossRef]
- Zhang, X.; He, Y.T.; Liu, S.J.; Yu, Z.H.; Jiang, Z.X.; Yang, Z.Y.; Dong, Y.S.; Nabinger, S.C.; Wu, L.; Gunawan, A.M.; et al. Salicylic Acid Based Small Molecule Inhibitor for the Oncogenic Src Homology-2 Domain Containing Protein Tyrosine Phosphatase-2 (SHP2). J. Med. Chem. 2010, 53, 2482–2493. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yu, B.; Xu, G.; Xu, W.-R.; Loh, M.L.; Tang, L.-D.; Qu, C.-K. Identification of Cryptotanshinone as an Inhibitor of Oncogenic Protein Tyrosine Phosphatase SHP2 (PTPN11). J. Med. Chem. 2013, 56, 7212–7221. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, H.R.; Pireddu, R.; Chen, L.W.; Luo, Y.T.; Sung, S.S.; Szymanski, A.M.; Yip, M.L.R.; Guida, W.C.; Sebti, S.M.; Wu, J.; et al. Inhibitors of Src homology-2 domain containing protein tyrosine phosphatase-2 (Shp2) based on oxindole scaffolds. J. Med. Chem. 2008, 51, 4948–4956. [Google Scholar] [CrossRef] [PubMed]
- Fodor, M.; Price, E.; Wang, P.; Lu, H.; Argintaru, A.; Chen, Z.; Glick, M.; Hao, H.X.; Kato, M.; Koenig, R.; et al. Dual Allosteric Inhibition of SHP2 Phosphatase. ACS Chem. Biol. 2018, 13, 647. [Google Scholar] [CrossRef]
- Lu, H.; Liu, C.; Hung Huynh, T.B.U.L.; LaMarche, M.J.; Mohseni, M.; Engelman, J.A.; Hammerman, P.S.; Caponigro, G.; Hao, H.-X. Resistance to allosteric SHP2 inhibition in FGFR-driven cancers through rapid feedback activation of FGFR. Oncotarget 2020, 11, 265. [Google Scholar] [CrossRef]
- Kerr, D.L.; Haderk, F.; Bivona, T.G. Allosteric SHP2 inhibitors in cancer: Targeting the intersection of RAS, resistance, and the immune microenvironment. Curr. Opin. Chem. Biol. 2021, 62, 1–12. [Google Scholar] [CrossRef]
- Garcia Fortanet, J.; Chen, C.H.; Chen, Y.N.; Chen, Z.; Deng, Z.; Firestone, B.; Fekkes, P.; Fodor, M.; Fortin, P.D.; Fridrich, C.; et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J. Med. Chem. 2016, 59, 7773. [Google Scholar] [CrossRef]
- Chen, Y.-N.P.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Wong, G.S.; Zhou, J.; Liu, J.B.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018, 24, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sama, I.; Ladumor, Y.; Kee, L.; Adderley, T.; Christopher, G.; Robinson, C.M.; Kano, Y.; Ohh, M.; Irwin, M.S. NRAS Status Determines Sensitivity to SHP2 Inhibitor Combination Therapies Targeting the RAS-MAPK Pathway in Neuroblastoma. Cancer Res. 2020, 80, 3413–3423. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pollard, K.; Allen, A.N.; Tomar, T.; Pijnenburg, D.; Yao, Z.; Rodriguez, F.J.; Pratilas, C.A. Combined Inhibition of SHP2 and MEK Is Effective in Models of NF1-Deficient Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2020, 80, 5367–5379. [Google Scholar] [CrossRef] [PubMed]
- Bagdanoff, J.T.; Chen, Z.; Acker, M.; Chen, Y.N.; Chan, H.; Dore, M.; Firestone, B.; Fodor, M.; Fortanet, J.; Hentemann, M.; et al. Optimization of Fused Bicyclic Allosteric SHP2 Inhibitors. J. Med. Chem. 2019, 62, 1781. [Google Scholar] [CrossRef]
- Raveendra-Panickar, D.; Finlay, D.; Layng, F.I.; Lambert, L.J.; Celeridad, M.; Zhao, M.; Barbosa, K.; De Backer, L.J.S.; Kwong, E.; Gosalia, P.; et al. Discovery of novel furanylbenzamide inhibitors that target oncogenic tyrosine phosphatase SHP2 in leukemia cells. J. Biol. Chem. 2022, 298, 101477. [Google Scholar] [CrossRef]
- LaMarche, M.J.; Acker, M.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C.H.-T.; Chen, Y.-N.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 13578–13594. [Google Scholar] [CrossRef]
- Liu, C.; Lu, H.; Wang, H.; Loo, A.; Zhang, X.; Yang, G.; Kowal, C.; Delach, S.; Wang, Y.; Goldoni, S.; et al. Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling. Clin. Cancer Res. 2021, 27, 342–354. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals. Dose Finding Study of TNO155 in Adult Patients with Advanced Solid Tumors; Novartis: Basel, Switzerland, 2023. [Google Scholar]
- Novartis Pharmaceuticals. Phase Ib Study of TNO155 in Combination with Spartalizumab or Ribociclib in Selected Malignancies; National Library of Medicine: Bethesda, MD, USA, 2019. [Google Scholar]
- Novartis Pharmaceuticals. A Study of Select Drug Combinations in Adult Patients with Advanced/Metastatic BRAF V600 Colorectal Cancer; National Library of Medicine: Bethesda, MD, USA, 2020. [Google Scholar]
- Mirati Therapeutics Inc. Adagrasib in Combination With TNO155 in Patients With Cancer (KRYSTAL 2); National Library of Medicine: Bethesda, MD, USA, 2020. [Google Scholar]
- Novartis Pharmaceuticals. Study of JDQ443 in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation; Novartis: Basel, Switzerland, 2021. [Google Scholar]
- Revolution Medicines, Inc. Dose Escalation of RMC-4630 Monotherapy in Relapsed/Refractory Solid Tumors; National Library of Medicine: Bethesda, MD, USA, 2018. [Google Scholar]
- Johnson, M.L.; Langdon, R.; Ellison, D.; Spira, A.; Amin, H.; Castine, M.; Daniel, D.; Larson, T.; Sohoni, S.; Chen, Y.C.; et al. EP08.02-111 RMC-4630, a SHP2 Inhibitor, in Combination with Sotorasib for Advanced KRASG12C NSCLC After Failure of Prior Standard Therapies: A Phase 2 Trial. J. Thorac. Oncol. 2022, 17, S454–S455. [Google Scholar] [CrossRef]
- Revolution Medicines, Inc. Dose-Escalation/Expansion of RMC-4630 and Cobimetinib in Relapsed/Refractory Solid Tumors and RMC-4630 and Osimertinib in EGFR Positive Locally Advanced/Metastatic NSCLC; Bladder Cancer Advocacy Network: Bethesda, MD, USA, 2019. [Google Scholar]
- The Netherlands Cancer Institute. Combination Therapy of RMC-4630 and LY3214996 in Metastatic KRAS Mutant Cancers; National Library of Medicine: Bethesda, MD, USA, 2022. [Google Scholar]
- Revolution Medicines, Inc. Combination Study of RMC-4630 and Sotorasib for NSCLC Subjects with KRASG12C Mutation After Failure of Prior Standard Therapies; National Library of Medicine: Bethesda, MD, USA, 2021. [Google Scholar]
- Falchook, G.; Li, B.T.; Marrone, K.A.; Bestvina, C.M.; Langer, C.J.; Krauss, J.C.; Strickler, J.H.; Meloni, A.; Dai, T.; Varrieur, T.; et al. OA03.03 Sotorasib in Combination with RMC-4630, a SHP2 Inhibitor, in KRAS p.G12C-Mutated NSCLC and Other Solid Tumors. J. Thorac. Oncol. 2022, 17, S8. [Google Scholar] [CrossRef]
- Safety and Efficacy Study of SAR442720 in Combination with Other Agents in Advanced Malignancies. 2023. Available online: https://clinicaltrials.gov/study/NCT04418661 (accessed on 16 June 2023).
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.-H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Pathak, M.K.; Yi, T.L. Sodium stibogluconate is a potent inhibitor of protein tyrosine phosphatases and augments cytokine responses in hemopoietic cell lines. J. Immunol. 2001, 167, 3391–3397. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.; Reuben, J.M.; Camacho, L.H.; Gao, H.; Lee, B.N.; Cohen, E.N.; Verschraegen, C.; Stephen, S.; Aaron, J.; Hong, D.; et al. Phase I Dose Escalation Study of Sodium Stibogluconate (SSG), a Protein Tyrosine Phosphatase Inhibitor, Combined with Interferon Alpha for Patients with Solid Tumors. J. Cancer 2011, 2, 81–89. [Google Scholar] [CrossRef]
- Jacobio Pharmaceuticals Co., Ltd. A First in Human, Dose Escalation Study of JAB-3068 (SHP2 Inhibitor) in Adult Patients with Advanced Solid Tumors; National Library of Medicine: Bethesda, MD, USA, 2018. [Google Scholar]
- Jacobio Pharmaceuticals Co., Ltd. A First-in-Human Study of JAB-3068 (SHP2 Inhibitor) in Adult Patients with Advanced Solid Tumors in China; National Library of Medicine: Bethesda, MD, USA, 2018. [Google Scholar]
- Jacobio Pharmaceuticals Co., Ltd. A Study of JAB-3312 in Adult Patients with Advanced Solid Tumors in China; National Library of Medicine: Bethesda, MD, USA, 2020. [Google Scholar]
- Jacobio Pharmaceuticals Co., Ltd. A First-in-Human, Phase 1 Study of JAB-3312 in Adult Patients with Advanced Solid Tumors; National Library of Medicine: Bethesda, MD, USA, 2019. [Google Scholar]
- Erasca, Inc. A Dose Escalation/Expansion Study of ERAS-601 in Patients with Advanced or Metastatic Solid Tumors; Erasca, Inc.: San Diego, CA, USA, 2020. [Google Scholar]
- Sun, Y.T.; Meyers, B.A.; Czako, B.; Leonard, P.; Mseeh, F.; Harris, A.L.; Wu, Q.; Johnson, S.; Parker, C.A.; Cross, J.B.; et al. Allosteric SHP2 Inhibitor, IACS-13909, Overcomes EGFR-Dependent and EGFR-Independent Resistance Mechanisms toward Osimertinib. Cancer Res. 2020, 80, 4840–4853. [Google Scholar] [CrossRef] [PubMed]
- Navire Pharma Inc., a.B.c. First-in-Human Study of the SHP2 Inhibitor BBP-398 in Patients with Advanced Solid Tumors; UCLA Health: Los Angeles, CA, USA, 2020. [Google Scholar]
- Shojaei, F.; Shojaei, F.; Ricono, J.M.; Fang, C.; Kabbinavar, F.; Goodenow, B.; Gillings, M. Abstract 1041: HBI-2376, HUYABIO clinical stage SHP2 inhibitor, possess robust in vitro potency and in vivo efficacy in several preclinical tumor models carrying KrasG12C or EGFR mutations. Cancer Res. 2022, 82, 1041. [Google Scholar] [CrossRef]
- Li, L.; Fu, B.; Han, H.; Sun, Z.; Zhao, X.; Jv, X.; Tong, J.; Zhao, J.; Zou, Z.; Chen, H.; et al. Abstract 5463: BPI-442096: A potent and selective inhibitor of SHP2 for the treatment of multiple cancers. Cancer Res. 2022, 82, 5463. [Google Scholar] [CrossRef]
- Nanjing Sanhome Pharmaceutical, Co., Ltd. A Study to Investigate Safety and Tolerability of SH3809 Tablet in Patients with Advanced Solid Tumors; National Library of Medicine: Bethesda, MD, USA, 2021. [Google Scholar]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Shi, Z.Q.; Yu, D.H.; Park, M.; Marshall, M.; Feng, G.S. Molecular mechanism for the Shp-2 tyrosine phosphatase function in promoting growth factor stimulation of Erk activity. Mol. Cell Biol. 2000, 20, 1526–1536. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Patel, H.; Nikanjam, M.; Fanta, P.T.; Hahn, M.E.; De, P.; Williams, C.; Guido, J.; et al. Molecular profiling of advanced malignancies guides first-line N-of-1 treatments in the I-PREDICT treatment-naïve study. Genome Med. 2021, 13, 155. [Google Scholar] [CrossRef]
- Adashek, J.J.; Subbiah, V.; Westphalen, C.B.; Naing, A.; Kato, S.; Kurzrock, R. Cancer: Slaying the nine-headed Hydra. Ann. Oncol. 2023, 34, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.R.; Bu, H.; Zhou, J.P.; Yang, C.Y.; Zhang, H.B. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J. Med. Chem. 2020, 63, 11368–11396. [Google Scholar] [CrossRef]
- Suzawa, K.; Offin, M.; Lu, D.; Kurzatkowski, C.; Vojnic, M.; Smith, R.S.; Sabari, J.K.; Tai, H.C.; Mattar, M.; Khodos, I.; et al. Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-mutant Non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, X.-Y.; Qiu, S.-J.; Yamato, I.; Sho, M.; Nakajima, Y.; Zhou, J.; Li, B.-Z.; Shi, Y.-H.; Xiao, Y.-S.; et al. Overexpression of PD-L1 Significantly Associates with Tumor Aggressiveness and Postoperative Recurrence in Human Hepatocellular Carcinoma. Clin. Cancer Res. 2009, 15, 971–979. [Google Scholar] [CrossRef]
- Bevins, N.J.; Okamura, R.; Montesion, M.; Adashek, J.J.; Goodman, A.M.; Kurzrock, R. Tumor Infiltrating Lymphocyte Expression of PD-1 Predicts Response to Anti-PD-1/PD-L1 Immunotherapy. J. Immunother. Precis. Oncol. 2022, 5, 90–97. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Brower, V. Anti-PD-L1 antibody active in metastatic bladder cancer. Lancet Oncol. 2015, 16, e11. [Google Scholar] [CrossRef]
- Barbee, M.S.; Ogunniyi, A.; Horvat, T.Z.; Dang, T.-O. Current Status and Future Directions of the Immune Checkpoint Inhibitors Ipilimumab, Pembrolizumab, and Nivolumab in Oncology. Ann. Pharmacother. 2015, 49, 907–937. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.O.N.; Tong, M.; Chung, K.P.S.; Zhou, L.; Che, N.; Tang, K.H.; Ding, J.; Lau, E.Y.T.; Ng, I.O.L.; Ma, S. Overriding Adaptive Resistance to Sorafenib Through Combination Therapy with Src Homology 2 Domain–Containing Phosphatase 2 Blockade in Hepatocellular Carcinoma. Hepatology 2020, 72, 155–168. [Google Scholar] [CrossRef]
- Hao, H.-X.; Wang, H.; Liu, C.; Kovats, S.; Velazquez, R.; Lu, H.; Pant, B.; Shirley, M.; Meyer, M.J.; Pu, M.; et al. Tumor Intrinsic Efficacy by SHP2 and RTK Inhibitors in KRAS-Mutant Cancers. Mol. Cancer Ther. 2019, 18, 2368–2380. [Google Scholar] [CrossRef]
- Yang, X.; Tang, C.; Luo, H.; Wang, H.; Zhou, X. Shp2 confers cisplatin resistance in small cell lung cancer via an AKT-mediated increase in CA916798. Oncotarget 2017, 8, 23664–23674. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kun, E.; Tsang, Y.T.M.; Ng, C.W.; Gershenson, D.M.; Wong, K.K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat. Rev. 2021, 92, 102137. [Google Scholar] [CrossRef] [PubMed]
- Torres-Ayuso, P.; Brognard, J. Shipping Out MEK Inhibitor Resistance with SHP2 Inhibitors. Cancer Discov. 2018, 8, 1210. [Google Scholar] [CrossRef]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef]
- Jiang, L.; Xu, W.; Chen, Y.; Zhang, Y. SHP2 inhibitor specifically suppresses the stemness of KRAS-mutant non-small cell lung cancer cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3231–3238. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; de Wit, N.; Gonçalves-Ribeiro, S.; et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef]
- Novartis Oncology Update. Available online: https://www.novartis.com/sites/novartis_com/files/2021-06-novartis-asco-call-presentation.pdf (accessed on 29 December 2022).
- Dardaei, L.; Wang, H.Q.; Singh, M.; Fordjour, P.; Shaw, K.X.; Yoda, S.; Kerr, G.; Yu, K.; Liang, J.; Cao, Y.; et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat. Med. 2018, 24, 512. [Google Scholar] [CrossRef]
- Kato, S.; Adashek, J.J.; Shaya, J.; Okamura, R.; Jimenez, R.E.; Lee, S.; Sicklick, J.K.; Kurzrock, R. Concomitant MEK and Cyclin Gene Alterations: Implications for Response to Targeted Therapeutics. Clin. Cancer Res. 2021, 27, 2792–2797. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Okamura, R.; Adashek, J.J.; Khalid, N.; Lee, S.; Nguyen, V.; Sicklick, J.K.; Kurzrock, R. Targeting G1/S phase cell-cycle genomic alterations and accompanying co-alterations with individualized CDK4/6 inhibitor–based regimens. JCI Insight 2021, 6, e142547. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Calizo, A.; Pollard, K.; Hirbe, A.C.; Pratilas, C.A. Abstract P125: Combined inhibition of SHP2 and CDK4/6 is active in NF1-associated malignant peripheral nerve sheath tumor. Mol. Cancer Ther. 2021, 20, P125. [Google Scholar] [CrossRef]
- Agianian, B.; Gavathiotis, E. Current Insights of BRAF Inhibitors in Cancer. J. Med. Chem. 2018, 61, 5775–5793. [Google Scholar] [CrossRef] [PubMed]
- Proietti, I.; Skroza, N.; Michelini, S.; Mambrin, A.; Balduzzi, V.; Bernardini, N.; Marchesiello, A.; Tolino, E.; Volpe, S.; Maddalena, P.; et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers 2020, 12, 1823. [Google Scholar] [CrossRef]
- Hertzman Johansson, C.; Egyhazi Brage, S. BRAF inhibitors in cancer therapy. Pharmacol. Ther. 2014, 142, 176–182. [Google Scholar] [CrossRef]
- Khan, S.; He, Y.; Zhang, X.; Yuan, Y.; Pu, S.; Kong, Q.; Zheng, G.; Zhou, D. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 2020, 39, 4909–4924. [Google Scholar] [CrossRef]
- Vemulapalli, V.; Donovan, K.A.; Seegar, T.C.; Rogers, J.M.; Bae, M.; Lumpkin, R.J.; Cao, R.; Henke, M.T.; Ray, S.S.; Fischer, E.S. Targeted degradation of the oncogenic phosphatase SHP2. Biochemistry 2021, 60, 2593–2609. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Brutkiewicz, R.R. Cell Signaling Pathways That Regulate Antigen Presentation. J. Immunol. 2016, 197, 2971–2979. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Name of Compound | Characteristics | Allosteric Site | Cell Line under Study | Target Mutation | Efficacy | Oral Bioavailability | Off-Target Inhibitory Effect | Combination |
|---|---|---|---|---|---|---|---|---|
| SHP099 [67,69,70,71,72,73,74] | Suppression of MAPK pathway activity in RTK-driven cancer cell lines and inhibition of malignant development in both in vitro and in vivo tumor models exhibited a synergistic effect on SHP2evoked illnesses or resistant malignancies caused by other mutations. | Tunnel allosteric site | Leukemia cell lines (inflammatory disease) | E69K mutation and leukemia-associated SHP2E69K mutant | Highly effective and selective | Orally accessible | No | BGJ398 (FGFR1 inhibitor) GSK1120212 (MEK inhibitor) Vemurafenib (BRAFV600E inhibitor) BVD523 (ERK inhibitor) Selumetinib (MEK inhibitor) |
| SHP389 [75,76] | Modulation of MAPK signaling in vivo and poor in vitro permeability | Tunnel allosteric site | Leukemia cell lines (inflammatory disease) | SHP2E76K mutant | Strong hERG selectivity | Limited oral bioavailability | - | - |
| SHP394 [15] | High lipophilic efficiency, improved potency, and enhanced pharmacokinetic properties SHP93, when administered orally to immunocompromised mice carrying subcutaneously implanted Detroit-562 tumor cells, demonstrated correlated and dose-dependent PK, PD, and effectiveness. | Tunnel allosteric site | Detroit-562 tumor cells, Caco-2 cells, KYSE520 cells | - | Potent, Selective, and Orally Efficacious and reduces tumor volume | Orally active | - | - |
| SHP836 [70] | In SHP2-WT, the published IC50 value for SHP836 is 5 µm, while the published IC50 value for SHP099 is 70 nm. | Tunnel allosteric site | - | - | SHP836 is much less effective than SHP099. | - | - | - |
| SHP244 [66,67] | Identified as a weak inhibitor of SHP2 with modest thermal stabilization of the enzyme | Latch allosteric site | JHH-7 and Hep3B cells | SHP2T253M/Q257L double mutant | good selectivity over the catalytic domain | - | - | RMC-4550 |
| Name of Compound | Company | Stage of Development | Diseases under Study | Combinations |
|---|---|---|---|---|
| SAR442720 (also known as RMC-4630) | Sanofi (Hongkong, China) (NCT04418661) | Phase 1 and 2 | Advanced Solid tumors and KRASG12C and KRAS mutated solid tumors | LY3214996 (ERK inhibitor) Sotorasib (KRASG12C inhibitor) Cobimetinib Osimertinib Adagrasib Pembrolizumab (PD-1 inhibitor) AMG510 (KRASG12C inhibitor) |
| BBP-398 (Formerly known as IACS-15509) | Navire Pharma Inc., a BridgeBio company (San Francisco, CA, USA) (NCT04528836) | Phase 1 and 2 | Solid tumors and NSCLC w/KRAS mutations | Nivolumab (PD-1 inhibitor) in NSCLC |
| RLY-1971 | Hoffmann-La Roche (Basel, Switzerland) (NCT04252339) | Phase 1 | Advanced solid tumors | Mono |
| TNO-155 | Novartis (Basel, Switzerland) (NCT04000529) (NCT04330664) (NCT04294160) | Phase 1 and 2 | Advanced solid tumors, EGFR-mutated NSCLC | EGF816 (nazartinib) (mutant-selective EGFR inhibitor) Spartalizumab Ribociclib JDQ443 + Tislelizumab (PD-1 inhibitor) Adagrasib Dabrafenib LTT462 |
| ERAS-601 | Erasca (San Diego, CA, USA) (NCT04670679) (NCT04959981) | Phase 1/1b | Advanced solid tumors and KRAS mutated NSCLC | Cetuximab Sotorasib |
| BPI-442096 | Betta Pharma (Hangzhou, China) (NCT05369312) | Phase 1 | Advanced solid tumors | Mono |
| ET0038 (development so far in China only) | Etern BioPharma (Shanghai, China) (Chinese) (NCT05354843) | Phase 1 | Advanced solid tumors | Mono |
| HS-10381 (development in China only) | Jiangsu Hansoh Pharmaceutical (Lianyungang, China) (Chinese) (NCT05378178) | Phase 1 | Advanced solid tumors | Mono |
| JAB-3312 | Jacobio Pharmaceuticals Co. (Beijing, China), (Chinese) (NCT05288205) | Phase 1 and 2 | Advanced solid tumors w/KRASG12C mutations | JAB-21822 (KRASG12C inhibitor) |
| JAB-3068 | Jacobio Pharmaceuticals Co., (Chinese) (NCT03565003) | Phase 1 and 2 | Advanced solid tumors: NSCLC, squamous esophageal, HNSCC | Mono |
| HBI-2376 | HUYABIO International, LLC. (San Diego, CA, USA) (NCT05163028) | Phase 1 | Advanced solid tumors/KRAS or EGFR Mutations | Mono |
| SH3809 | Nanjing Sanhome Pharmaceutical, Co., Ltd. (Nanjing, China) (NCT04843033) | Phase 1 | Advanced Solid Tumor | Mono |
| Sodium stibogluconate | M.D. Anderson Cancer Center/VioQuest Pharmaceuticals (Houston, TX, USA) (NCT00629200) | Phase 1 | Advanced Cancer | Mono |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tojjari, A.; Saeed, A.; Sadeghipour, A.; Kurzrock, R.; Cavalcante, L. Overcoming Immune Checkpoint Therapy Resistance with SHP2 Inhibition in Cancer and Immune Cells: A Review of the Literature and Novel Combinatorial Approaches. Cancers 2023, 15, 5384. https://doi.org/10.3390/cancers15225384
Tojjari A, Saeed A, Sadeghipour A, Kurzrock R, Cavalcante L. Overcoming Immune Checkpoint Therapy Resistance with SHP2 Inhibition in Cancer and Immune Cells: A Review of the Literature and Novel Combinatorial Approaches. Cancers. 2023; 15(22):5384. https://doi.org/10.3390/cancers15225384
Chicago/Turabian StyleTojjari, Alireza, Anwaar Saeed, Arezoo Sadeghipour, Razelle Kurzrock, and Ludimila Cavalcante. 2023. "Overcoming Immune Checkpoint Therapy Resistance with SHP2 Inhibition in Cancer and Immune Cells: A Review of the Literature and Novel Combinatorial Approaches" Cancers 15, no. 22: 5384. https://doi.org/10.3390/cancers15225384
APA StyleTojjari, A., Saeed, A., Sadeghipour, A., Kurzrock, R., & Cavalcante, L. (2023). Overcoming Immune Checkpoint Therapy Resistance with SHP2 Inhibition in Cancer and Immune Cells: A Review of the Literature and Novel Combinatorial Approaches. Cancers, 15(22), 5384. https://doi.org/10.3390/cancers15225384