

Integrated Network Analysis of microRNAs, mRNAs, and Proteins Reveals the Regulatory Interaction between hsa-mir-200b and CFL2 Associated with Advanced Stage and Poor Prognosis in Patients with Intestinal Gastric Cancer

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Acquisition of Gastric Cancer Datasets, Patient Samples, and Study Design
2.2. Differential Expression Analysis of DEGs and DEMs
2.3. Prediction of microRNA and mRNA Interactions
2.4. Proteomic Data for IGC Patients
2.5. In Silico Analysis and Integration Network of Differentially Expressed microRNAs, Genes, and Proteins
2.6. ROC Curves and Survival Analysis of DEM and DEP Hubs
3. Results
3.1. Identification of DEGs and DEMs in IGC Patients
3.2. Construction of an Integrated Regulatory Network in IGC
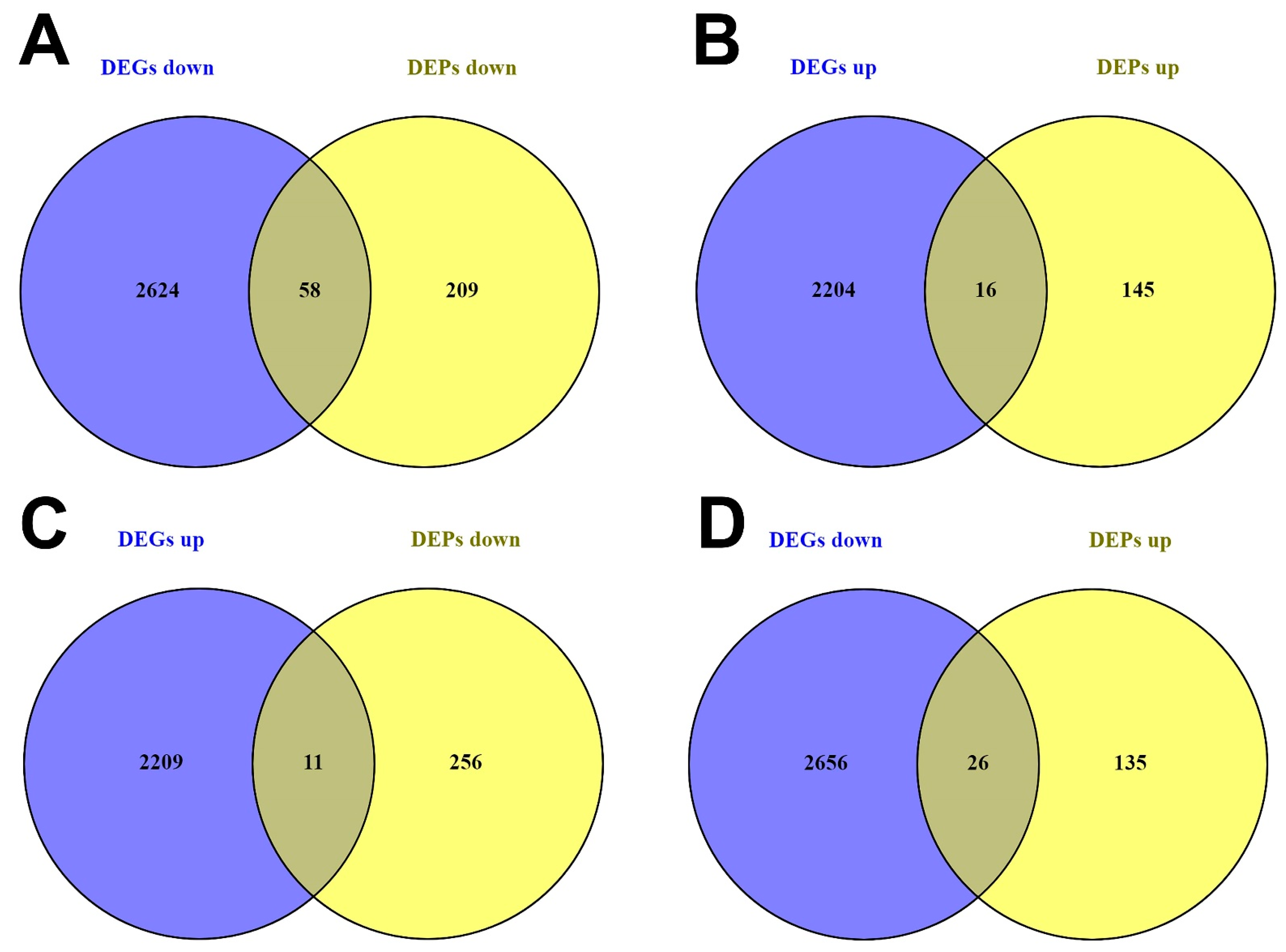
3.2.1. DEG and DEP Selection
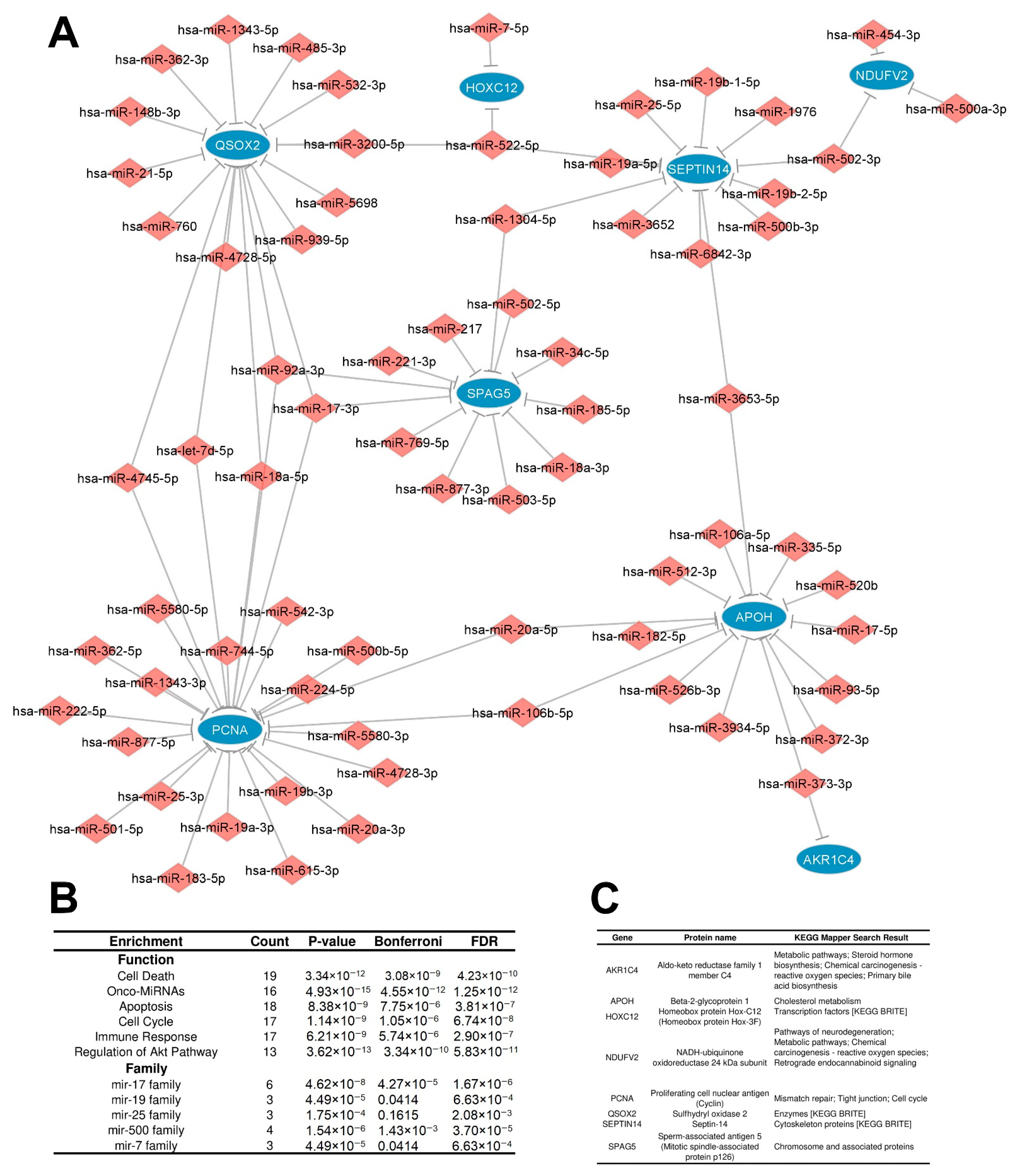
3.2.2. Distinctly Altered Regulatory Circuits Involving DEMs, DEGs, and DEPs Are Related to IGC
3.3. Functional Enrichment Analysis of Regulatory Circuits
3.4. Identification of Key Regulators
3.5. Impact of Hubs in IGC Carcinogenesis
3.5.1. Diagnostic Value of Key Hubs in IGC
3.5.2. Hub Expression Levels Are Related to Clinicopathological Characteristics and Prognosis in IGC Patients
3.6. Upregulation of hsa-mir-200b Represses Expression of CFL2 and Impacts Prognosis of IGC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Nagini, S. Carcinoma of the stomach: A review of epidemiology, pathogenesis, molecular genetics and chemoprevention. World J. Gastrointest. Oncol. 2012, 4, 156. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.K.; Raj, S.S.; Shankar, E.M.; Ganapathy, E.; Ebrahim, A.S.; Farooq, S.M. Gastric Carcinoma: A Review on Epidemiology, Current Surgical and Chemotherapeutic Options. In Gastric Carcinoma—New Insights into Current Management; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef]
- Karimi, P.; Islami, F.; Anandasabapathy, S.; Freedman, N.D.; Kamangar, F. Gastric Cancer: Descriptive Epidemiology, Risk Factors, Screening, and Prevention. Cancer Epidemiol. Biomark. Prev. 2014, 23, 700–713. [Google Scholar] [CrossRef]
- Yong, X.; Tang, B.; Li, B.-S.; Xie, R.; Hu, C.-J.; Luo, G.; Qin, Y.; Dong, H.; Yang, S.-M. Helicobacter pylori virulence factor CagA promotes tumorigenesis of gastric cancer via multiple signaling pathways. Cell Commun. Signal. CCS 2015, 13, 30. [Google Scholar] [CrossRef]
- Laurén, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Bottai, G.; Diao, L.; Baggerly, K.; Paladini, L.; Győrffy, B.; Raschioni, C.; Pusztai, L.; Calin, G.; Santarpia, L. Integrated MicroRNA–mRNA Profiling Identifies Oncostatin M as a Marker of Mesenchymal-Like ER-Negative/HER2-Negative Breast Cancer. Int. J. Mol. Sci. 2017, 18, 194. [Google Scholar] [CrossRef]
- Li, X.; Yu, X.; He, Y.; Meng, Y.; Liang, J.; Huang, L.; Du, H.; Wang, X.; Liu, W. Integrated Analysis of MicroRNA (miRNA) and mRNA Profiles Reveals Reduced Correlation between MicroRNA and Target Gene in Cancer. BioMed. Res. Int. 2018, 2018, 1972606. [Google Scholar] [CrossRef]
- Hu, S.; Liao, Y.; Zheng, J.; Gou, L.; Regmi, A.; Zafar, M.I.; Chen, L. In Silico Integration Approach Reveals Key MicroRNAs and Their Target Genes in Follicular Thyroid Carcinoma. BioMed Res. Int. 2019, 2019, 2725192. [Google Scholar] [CrossRef]
- Xiao, Y. Construction of a circRNA-miRNA-mRNA network to explore the pathogenesis and treatment of pancreatic ductal adenocarcinoma. J. Cell. Biochem. 2019, 121, 394–406. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Y.; Xiao, F.; Zhang, J.; Wang, Y.; Yao, Y.; Jiao, Y. Comprehensive Analysis of a circRNA-miRNA-mRNA Network to Reveal Potential Inflammation-Related Targets for Gastric Adenocarcinoma. Mediat. Inflamm. 2020, 2020, 9435608. [Google Scholar] [CrossRef]
- Tian, Y.; Xing, Y.; Zhang, Z.; Peng, R.; Zhang, L.; Sun, Y. Bioinformatics Analysis of Key Genes and circRNA-miRNA-mRNA Regulatory Network in Gastric Cancer. BioMed Res. Int. 2020, 2020, 2862701. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Jia, X.; Wu, Q.; Shi, L.; Ma, Z.; Ba, N.; Zhao, H.; Xia, X.; Zhang, Z. Esomeprazole affects the proliferation, metastasis, apoptosis and chemosensitivity of gastric cancer cells by regulating lncRNA/circRNA-miRNA-mRNA ceRNA networks. PubMed 2020, 20, 329. [Google Scholar] [CrossRef]
- Zhu, T.; Lou, Q.; Shi, Z.; Chen, G. Identification of Key miRNA-Gene Pairs in Gastric Cancer through Integrated Analysis of mRNA and miRNA Microarray. Am. J. Transl. Res. 2021, 13, 253–269. Available online: https://pubmed.ncbi.nlm.nih.gov/33527022/ (accessed on 10 September 2023). [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Elkan-Miller, T.; Ulitsky, I.; Hertzano, R.; Rudnicki, A.; Dror, A.A.; Lenz, D.R.; Elkon, R.; Irmler, M.; Beckers, J.; Shamir, R.; et al. Proteomics, and MicroRNA Analyses Reveals Novel MicroRNA Regulation of Targets in the Mammalian Inner Ear. PLoS ONE 2011, 6, e18195. [Google Scholar] [CrossRef]
- Li, Z.; Qin, T.; Wang, K.; Hackenberg, M.; Yan, J.; Gao, Y.; Yu, L.-R.; Shi, L.; Su, Z.; Chen, T. Integrated microRNA; mRNA, and protein expression profiling reveals microRNA regulatory networks in rat kidney treated with a carcinogenic dose of aristolochic acid. BMC Genom. 2015, 16, 365. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Zhang, J.; Liang, X.; Zhang, X.; Wang, T.; Yin, S. Integrated Analysis of Transcriptomic, miRNA and Proteomic Changes of a Novel Hybrid Yellow Catfish Uncovers Key Roles for miRNAs in Heterosis. Mol. Cell. Proteom. 2019, 18, 1437–1453. [Google Scholar] [CrossRef]
- Seo, J.; Jin, D.; Choi, C.-H.; Lee, H. Integration of MicroRNA, mRNA, and Protein Expression Data for the Identification of Cancer-Related MicroRNAs. PLoS ONE 2017, 12, e0168412. [Google Scholar] [CrossRef]
- Santos, E.C.; Gomes, R.B.; Fernandes, P.V.; Ferreira, M.A.; Abdelhay, E.S.F.W. The protein-protein interaction network of intestinal gastric cancer patients reveals hub proteins with potential prognostic value. Cancer Biomark. 2021, 33, 83–96. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2015, 44, e71. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The multiMiR R package and database: Integration of microRNA–target interactions along with their disease and drug associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Lu, M.; Shi, B.; Wang, J.; Cao, Q.; Cui, Q. TAM: A method for enrichment and depletion analysis of a microRNA category in a list of microRNAs. BMC Bioinform. 2010, 11, 419. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, X.; Wan, Y.; Zhang, S.; Zhao, Y.; Fan, R.; Cui, Q.; Zhou, Y. TAM 2.0: Tool for MicroRNA set analysis. Nucleic Acids Res. 2018, 46, W180–W185. [Google Scholar] [CrossRef]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef]
- Krapivsky, P.L.; Rodgers, G.J.; Redner, S. Degree Distributions of Growing Networks. Phys. Rev. Lett. 2001, 86, 5401–5404. [Google Scholar] [CrossRef] [PubMed]
- López-Ratón, M.; Rodríguez-Álvarez, M.X.; Suárez, C.C.; Sampedro, F.G. OptimalCutpoints: AnRPackage for Selecting Optimal Cutpoints in Diagnostic Tests. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef]
- Metz, C.E. Basic principles of ROC analysis. Semin. Nucl. Med. 1978, 8, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Safari, S.; Baratloo, A.; Elfil, M.; Negida, A. Evidence Based Emergency Medicine; Part 5 Receiver Operating Curve and Area under the Curve. Emergency 2016, 4, 111–113. Available online: https://pubmed.ncbi.nlm.nih.gov/27274525/ (accessed on 10 September 2023).
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Concepcion, C.P.; Bonetti, C.; Ventura, A. The MicroRNA-17-92 Family of MicroRNA Clusters in Development and Disease. Cancer J. 2012, 18, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Sárközy, M.; Kahán, Z.; Csont, T. A myriad of roles of miR-25 in health and disease. Oncotarget 2018, 9, 21580–21612. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, J. Why Do Hubs Tend to Be Essential in Protein Networks? PLoS Genet. 2006, 2, e88. [Google Scholar] [CrossRef]
- Albert, R.; Jeong, H.; Barabási, A.-L. Error and attack tolerance of complex networks. Nature 2000, 406, 378–382. [Google Scholar] [CrossRef]
- Marques, M.; Evangelista, A.; Macedo, T.; Vieira, R.; Scapulatempo-Vieira, C.; Reis, R.; Carvalho, A.; Silva, I. Expression of tumor suppressors miR-195 and let-7a as potential biomarkers of invasive breast cancer. Clinics 2018, 73, e184. [Google Scholar] [CrossRef]
- Li, L.; Feng, T.; Zhang, W.; Gao, S.; Wang, R.; Lv, W.; Zhu, T.; Yu, H.; Qian, B. MicroRNA Biomarker hsa-miR-195-5p for Detecting the Risk of Lung Cancer. Int. J. Genom. 2020, 2020, 7415909. [Google Scholar] [CrossRef]
- Zhao, D.-L.; Wu, Q.-L. Effect of inhibition to Yes-related proteins-mediated Wnt/β-catenin signaling pathway through miR-195-5p on apoptosis of gastric cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6486–6496. [Google Scholar] [CrossRef]
- Wang, F.; Ruan, L.; Yang, J.; Zhao, Q.; Wei, W. TRIM14 promotes the migration and invasion of gastric cancer by regulating epithelial-to-mesenchymal transition via activation of AKT signaling regulated by miR-195-5p. Oncol. Rep. 2018, 40, 3273–3284. [Google Scholar] [CrossRef]
- Bi, J.; Guo, A.; Lai, Y.R.; Li, B.; Zhong, J.M.; Wu, H.; Xie, Z.; He, Y.; Lv, Z.; Lau, S.H.; et al. Overexpression of clusterin correlates with tumor progression, metastasis in gastric cancer: A study on tissue microarrays. Neoplasma 2010, 57, 191–197. [Google Scholar] [CrossRef]
- Nie, H.; Mu, J.; Wang, J.; Li, Y. miR-195-5p regulates multi-drug resistance of gastric cancer cells via targeting ZNF139. Oncol. Rep. 2018, 40, 1370–1378. [Google Scholar] [CrossRef]
- Zhao, C.; Li, Y.; Chen, G.; Wang, F.; Shen, Z.; Zhou, R. Overexpression of miR-15b-5p promotes gastric cancer metastasis by regulating PAQR3. Oncol. Rep. 2017, 38, 352–358. [Google Scholar] [CrossRef]
- Yuan, C.; Zhang, Y.; Tu, W.; Guo, Y. Integrated miRNA profiling and bioinformatics analyses reveal upregulated miRNAs in gastric cancer. Oncol. Lett. 2019, 18, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Gilles, M.-E.; Slack, F.J. Let-7 microRNA as a potential therapeutic target with implications for immunotherapy. Expert Opin. Ther. Targets 2018, 22, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jiao, X.; Pestell, T.G.; Fan, C.; Qin, S.; Mirabelli, E.; Ren, H.; Pestell, R.G. MicroRNAs and cancer stem cells: The sword and the shield. Oncogene 2013, 33, 4967–4977. [Google Scholar] [CrossRef]
- Sun, X.; Liu, J.; Xu, C.; Tang, S.-C.; Ren, H. The insights of Let-7 miRNAs in oncogenesis and stem cell potency. J. Cell. Mol. Med. 2016, 20, 1779–1788. [Google Scholar] [CrossRef]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Liu, H.; Wang, R.; Huang, M.; Wu, Q.; Wang, Y.; Zhang, W.; Liu, Y. Hsa-let-7d-5p Promotes Gastric Cancer Progression by Targeting PRDM5. J. Oncol. 2022, 2022, 2700651. [Google Scholar] [CrossRef] [PubMed]
- Xiao-lin, W.; Chang, H.; Gao, G.; Su, B.; Deng, Q.; Zhou, H.; Wang, Q.; Lin, Y.; Ding, Y. Silencing of PRDM5 increases cell proliferation and inhibits cell apoptosis in glioma. Int. J. Neurosci. 2020, 131, 144–153. [Google Scholar] [CrossRef]
- Takeno, A.; Takemasa, I.; Doki, Y.; Yamasaki, M.; Miyata, H.; Takiguchi, S.; Fujiwara, Y.; Matsubara, K.; Monden, M. Integrative approach for differentially overexpressed genes in gastric cancer by combining large-scale gene expression profiling and network analysis. Br. J. Cancer 2008, 99, 1307–1315. [Google Scholar] [CrossRef]
- Aziz, S.; Rasheed, F.; Zahra, R.; König, S. Gastric Cancer Pre-Stage Detection and Early Diagnosis of Gastritis Using Serum Protein Signatures. Molecules 2022, 27, 2857. [Google Scholar] [CrossRef]
- Xu, T.; Xie, M.; Jing, X.; Jiang, H.; Wu, X.; Wang, X.; Shu, Y. Loss of miR-26b-5p promotes gastric cancer progression via miR-26b-5p-PDE4B/CDK8-STAT3 feedback loop. J. Transl. Med. 2023, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Trümbach, D.; Prakash, N. The conserved miR-8/miR-200 microRNA family and their role in invertebrate and vertebrate neurogenesis. Cell Tissue Res. 2014, 359, 161–177. [Google Scholar] [CrossRef]
- Davalos, V.; Moutinho, C.; Villanueva, A.; Boque, R.; Silva, P.; Carneiro, F.; Esteller, M. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 2012, 31, 2062–2074. [Google Scholar] [CrossRef]
- Pui-Wah, C.; Shu-Wing, N. The Functions of MicroRNA-200 Family in Ovarian Cancer: Beyond Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2017, 18, 1207. [Google Scholar] [CrossRef]
- Nam, E.J.; Yoon, H.; Kim, S.W.; Kim, H.; Kim, Y.T.; Kim, J.H.; Kim, J.W.; Kim, S. MicroRNA Expression Profiles in Serous Ovarian Carcinoma. Clin. Cancer Res. 2008, 14, 2690–2695. [Google Scholar] [CrossRef]
- Elson-Schwab, I.; Lorentzen, A.; Marshall, C.J. MicroRNA-200 Family Members Differentially Regulate Morphological Plasticity and Mode of Melanoma Cell Invasion. PLoS ONE 2010, 5, e13176. [Google Scholar] [CrossRef]
- Moh-Moh-Aung, A.; Fujisawa, M.; Ito, S.; Katayama, H.; Ohara, T.; Ota, Y.; Yoshimura, T.; Matsukawa, A. Decreased miR-200b-3p in cancer cells leads to angiogenesis in HCC by enhancing endothelial ERG expression. Sci. Rep. 2020, 10, 10418. [Google Scholar] [CrossRef]
- Tang, H.; Deng, M.; Tang, Y.; Xie, X.; Guo, J.; Kong, Y.; Ye, F.; Su, Q.; Xie, X. miR-200b and miR-200c as Prognostic Factors and Mediators of Gastric Cancer Cell Progression. Clin. Cancer Res. 2013, 19, 5602–5612. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Guo, F.; Huo, B.; Lv, Y.; Wang, Y.; Liu, W. Expression and clinical significance of the microRNA-200 family in gastric cancer. Oncol. Lett. 2015, 9, 2317–2324. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Xie, F.; Xu, D.; Xu, W.; Ge, X.; Lv, S.; Li, S. MiR-200b Suppresses Gastric Cancer Cell Migration and Invasion by Inhibiting NRG1 through ERBB2/ERBB3 Signaling. J. Oncol. 2021, 2021, 4470778. [Google Scholar] [CrossRef]
- Li, D.; Li, Q. MicroRNA-200b-3p restrains gastric cancer cell proliferation, migration, and invasion via C-X-C motif chemokine ligand 12/CXC chemokine receptor 7 axis. Bioengineered 2022, 13, 6509–6520. [Google Scholar] [CrossRef]
- Minn, Y.-K.; Lee, D.H.; Hyung, W.J.; Kim, J.E.; Choi, J.; Yang, S.H.; Song, H.T.; Lim, B.J.; Kim, S.H. MicroRNA-200 family members and ZEB2 are associated with brain metastasis in gastric adenocarcinoma. Int. J. Oncol. 2014, 45, 2403–2410. [Google Scholar] [CrossRef]
- Valladares-Ayerbes, M.; Reboredo, M.; Medina-Villaamil, V.; Iglesias-Díaz, P.; Lorenzo-Patiño, M.J.; Haz, M.; Santamarina, I.; Blanco, M.; Fernandez-Tajes, J.; Quindós, M.; et al. Circulating miR-200c as a diagnostic and prognostic biomarker for gastric cancer. J. Transl. Med. 2012, 10, 186. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, Z.; Zhang, H.; Zhu, M.; Zhu, W.; Zhou, X.; Liu, P. Prognostic value of candidate microRNAs in gastric cancer: A validation study. Cancer Biomark. 2017, 18, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Kanellos, G.; Frame, M.C. Cellular functions of the ADF/cofilin family at a glance. J. Cell Sci. 2016, 129, 3211–3218. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; Magalhaes, M.A.O.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef]
- Agrawal, P.B.; Greenleaf, R.S.; Tomczak, K.K.; Lehtokari, V.-L.; Wallgren-Pettersson, C.; Wallefeld, W.; Laing, N.G.; Darras, B.T.; Maciver, S.K.; Dormitzer, P.R.; et al. Nemaline Myopathy with Minicores Caused by Mutation of the CFL2 Gene Encoding the Skeletal Muscle Actin–Binding Protein, Cofilin-2. Am. J. Hum. Genet. 2007, 80, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Ockeloen, C.W.; Gilhuis, H.J.; Pfundt, R.; Kamsteeg, E.J.; Agrawal, P.B.; Beggs, A.H.; Hama-Amin, A.D.; Diekstra, A.; Knoers, N.V.A.M.; Lammens, M.; et al. Congenital myopathy caused by a novel missense mutation in the CFL2 gene. Neuromuscul. Disord. 2012, 22, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.B.; Joshi, M.; Savic, T.; Chen, Z.; Beggs, A.H. Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout demonstrates requirement of cofilin-2 for muscle maintenance. Hum. Mol. Genet. 2012, 21, 2341–2356. [Google Scholar] [CrossRef]
- Kuramitsu, Y. Differential expression of up-regulated cofilin-1 and down-regulated cofilin-2 characteristic of pancreatic cancer tissues. Oncol. Rep. 2011, 26, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Wilson, J.M.; Harvel, N.; Liu, J.; Pei, L.; Huang, S.; Hawthorn, L.; Shi, H. A systematic evaluation of miRNA:mRNA interactions involved in the migration and invasion of breast cancer cells. J. Transl. Med. 2013, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, Z.; Li, S.; Wang, L. Circ_0006404 Accelerates Prostate Cancer Progression Through Regulating miR-1299/CFL2 Signaling. OncoTargets Ther. 2021, 14, 83–95. [Google Scholar] [CrossRef]
- Qi, M.; Yu, B.; Yu, H.; Fan, L. Integrated analysis of a ceRNA network reveals potential prognostic lncRNAs in gastric cancer. Cancer Med. 2020, 9, 1798–1817. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, M.; Hu, X.; She, J.; Sun, R.; Qin, S.; Li, D. miRNA-194 predicts favorable prognosis in gastric cancer and inhibits gastric cancer cell growth by targeting CCND1. FEBS Open Bio. 2021, 11, 1814–1826. [Google Scholar] [CrossRef]
- Huang, Y.-K. Circulating microRNAs and long non-coding RNAs in gastric cancer diagnosis: An update and review. World J. Gastroenterol. 2015, 21, 9863. [Google Scholar] [CrossRef]
- Yuan, K.; Li, B.; Yuan, Y.; Tan, M.; Tan, J.; Dai, W.; Feng, W.; Zuo, J. Deregulation of MicroRNA-375 Inhibits Proliferation and Migration in Gastric Cancer in Association with Autophagy-Mediated AKT/mTOR Signaling Pathways. Technol. Cancer Res. Treat. 2018, 17, 153303381880649. [Google Scholar] [CrossRef]
- Bian, Y.; Guo, J.; Qiao, L.; Sun, X. miR-3189-3p Mimics Enhance the Effects of S100A4 siRNA on the Inhibition of Proliferation and Migration of Gastric Cancer Cells by Targeting CFL2. Int. J. Mol. Sci. 2018, 19, 236. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Features | Cohort | |
|---|---|---|
| State | Primary tumor | 180 |
| Normal tissue | 18 | |
| Gender | Male | 119 |
| Female | 61 | |
| Ethnicity | White | 100 |
| Asian | 51 | |
| Black or African American | 4 | |
| Not reported | 25 | |
| Major pathologic TNM stages | Stage I | 31 |
| Stage II | 62 | |
| Stage III | 62 | |
| Stage IV | 12 | |
| Pathologic T stage | T1 | 11 |
| T2 | 50 | |
| T3 | 69 | |
| T4 | 44 | |
| Pathologic N stage | N0 | 65 |
| N1 | 43 | |
| N2 | 33 | |
| N3 | 29 | |
| Pathologic M stage | M0 | 166 |
| M1 | 6 | |
| Molecular subtype | Chromosomal instability | 108 |
| Microsatellite instability | 44 | |
| Genomically stable | 14 | |
| Epstein–Barr virus (EBV)+ | 14 | |
| Hub | Score (DEGs/DEPs) | Number of Regulators | Regulatory Circuit | Expression in IGC DEG/DEP |
|---|---|---|---|---|
| CDKN1A | 102.0 | 102 | RRC1 | Down/Down |
| PPP1CB | 29.0 | 29 | RRC1 | Down/Down |
| CKB | 27.0 | 27 | RRC1 | Down/Down |
| TTC9 | 18.0 | 18 | RRC1 | Down/Down |
| CFL2 | 47.0 | 47 | RRC1 | Down/Down |
| PCNA | 25.0 | 25 | RRC2 | Up/Down |
| QSOX2 | 17.0 | 17 | RRC2 | Up/Down |
| APOH | 14.0 | 14 | RRC2 | Up/Down |
| SPAG5 | 12.0 | 12 | RRC2 | Up/Down |
| SEPTIN14 | 12.0 | 12 | RRC2 | Up/Down |
| SNRPD1 | 19.0 | 19 | IRC | Up/Up |
| BID | 12.0 | 12 | IRC | Up/Up |
| UBE2C | 11.0 | 11 | IRC | Up/Up |
| HSPE1 | 10.0 | 10 | IRC | Up/Up |
| OLFM4 | 5.0 | 5 | IRC | Up/Up |
| Hub | Number of Targets | % of DEGs/DEPs Targeted in the Circuit | Regulatory Circuit | DEM Expression in IGC |
|---|---|---|---|---|
| hsa-miR-210-3p | 14 | 30.4 | RRC1 | Up |
| hsa-miR-200b-3p | 9 | 19.6 | RRC1 | Up |
| hsa-miR-335-5p | 7 | 15.2 | RRC1 | Up |
| hsa-miR-20a-5p | 7 | 15.2 | RRC1 | Up |
| hsa-miR-148b-3p | 7 | 15.2 | RRC1 | Up |
| hsa-miR-17-5p | 6 | 13.0 | RRC1 | Up |
| hsa-miR-15b-5p | 6 | 13.0 | RRC1 | Up |
| hsa-miR-15a-5p | 6 | 13.0 | RRC1 | Up |
| hsa-miR-106b-5p | 6 | 13.0 | RRC1 | Up |
| hsa-miR-92a-3p | 6 | 13.0 | RRC1 | Up |
| hsa-miR-17-3p | 3 | 37.5 | RRC2 | Up |
| hsa-miR-522-5p | 3 | 37.5 | RRC2 | Up |
| hsa-miR-92a-3p | 3 | 37.5 | RRC2 | Up |
| hsa-let-7d-5p | 2 | 25.0 | RRC2 | Up |
| hsa-miR-18a-5p | 2 | 25.0 | RRC2 | Up |
| hsa-miR-3653-5p | 2 | 25.0 | RRC2 | Up |
| hsa-miR-373-3p | 2 | 25.0 | RRC2 | Up |
| hsa-miR-4745-5p | 2 | 25.0 | RRC2 | Up |
| hsa-miR-20a-5p | 2 | 25.0 | RRC2 | Up |
| hsa-miR-106b-5p | 2 | 25.0 | RRC2 | Up |
| hsa-miR-1304-5p | 2 | 25.0 | RRC2 | Up |
| hsa-miR-502-3p | 2 | 25.0 | RRC2 | Up |
| hsa-miR-23b-3p | 4 | 33.3 | IRC | Down |
| hsa-miR-1-3p | 4 | 33.3 | IRC | Down |
| hsa-let-7b-5p | 3 | 25.0 | IRC | Down |
| hsa-miR-195-5p | 3 | 25.0 | IRC | Down |
| hsa-miR-26b-5p | 3 | 25.0 | IRC | Down |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
dos Santos, E.C.; Rohan, P.; Binato, R.; Abdelhay, E. Integrated Network Analysis of microRNAs, mRNAs, and Proteins Reveals the Regulatory Interaction between hsa-mir-200b and CFL2 Associated with Advanced Stage and Poor Prognosis in Patients with Intestinal Gastric Cancer. Cancers 2023, 15, 5374. https://doi.org/10.3390/cancers15225374
dos Santos EC, Rohan P, Binato R, Abdelhay E. Integrated Network Analysis of microRNAs, mRNAs, and Proteins Reveals the Regulatory Interaction between hsa-mir-200b and CFL2 Associated with Advanced Stage and Poor Prognosis in Patients with Intestinal Gastric Cancer. Cancers. 2023; 15(22):5374. https://doi.org/10.3390/cancers15225374
Chicago/Turabian Styledos Santos, Everton Cruz, Paulo Rohan, Renata Binato, and Eliana Abdelhay. 2023. "Integrated Network Analysis of microRNAs, mRNAs, and Proteins Reveals the Regulatory Interaction between hsa-mir-200b and CFL2 Associated with Advanced Stage and Poor Prognosis in Patients with Intestinal Gastric Cancer" Cancers 15, no. 22: 5374. https://doi.org/10.3390/cancers15225374
APA Styledos Santos, E. C., Rohan, P., Binato, R., & Abdelhay, E. (2023). Integrated Network Analysis of microRNAs, mRNAs, and Proteins Reveals the Regulatory Interaction between hsa-mir-200b and CFL2 Associated with Advanced Stage and Poor Prognosis in Patients with Intestinal Gastric Cancer. Cancers, 15(22), 5374. https://doi.org/10.3390/cancers15225374