
Multigene Profiling of Circulating Tumor Cells in Esophageal Squamous Cell Carcinoma Identifies Prognostic Cancer Driver Genes Associated with Epithelial-Mesenchymal-Transition Progression and Chemoresistance
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
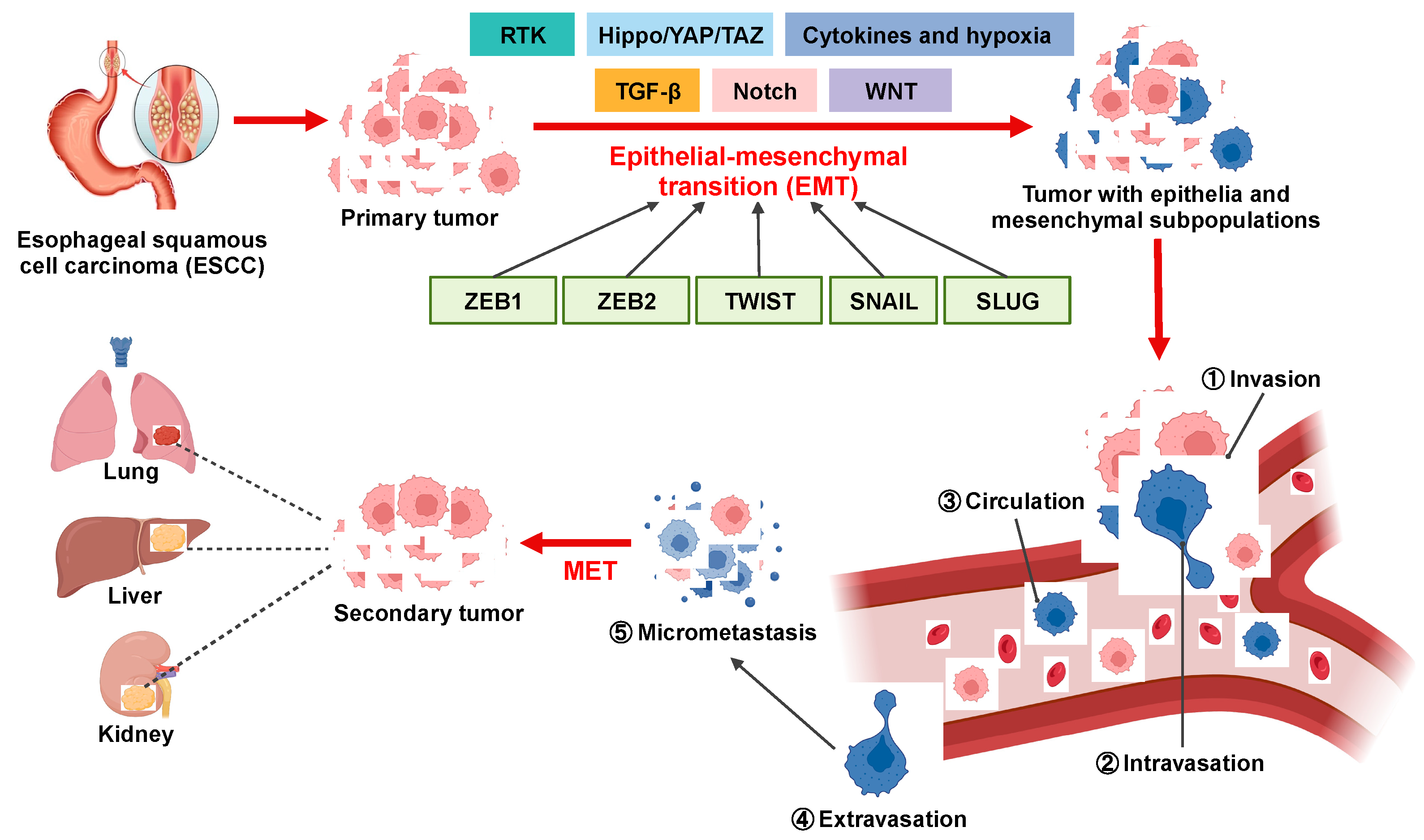
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Spike-in Experiments
2.3. Patient and Sample Collection
2.4. CTC Enrichment, RNA Extraction and RT-qPCR
2.5. Lentivirus Preparation and Transfection
2.6. RNA Sequencing and Transcriptomic Analysis
2.7. Western Blot Analysis
2.8. Wound Healing Assay
2.9. Matrigel-Coated Transwell Assay
2.10. Colony Formation Assay
2.11. MTT Assay Following Cytotoxic Drug Treatments
2.12. In Vivo Tumorigenicity and In Vivo Metastasis Assay
2.13. Statistics
3. Results
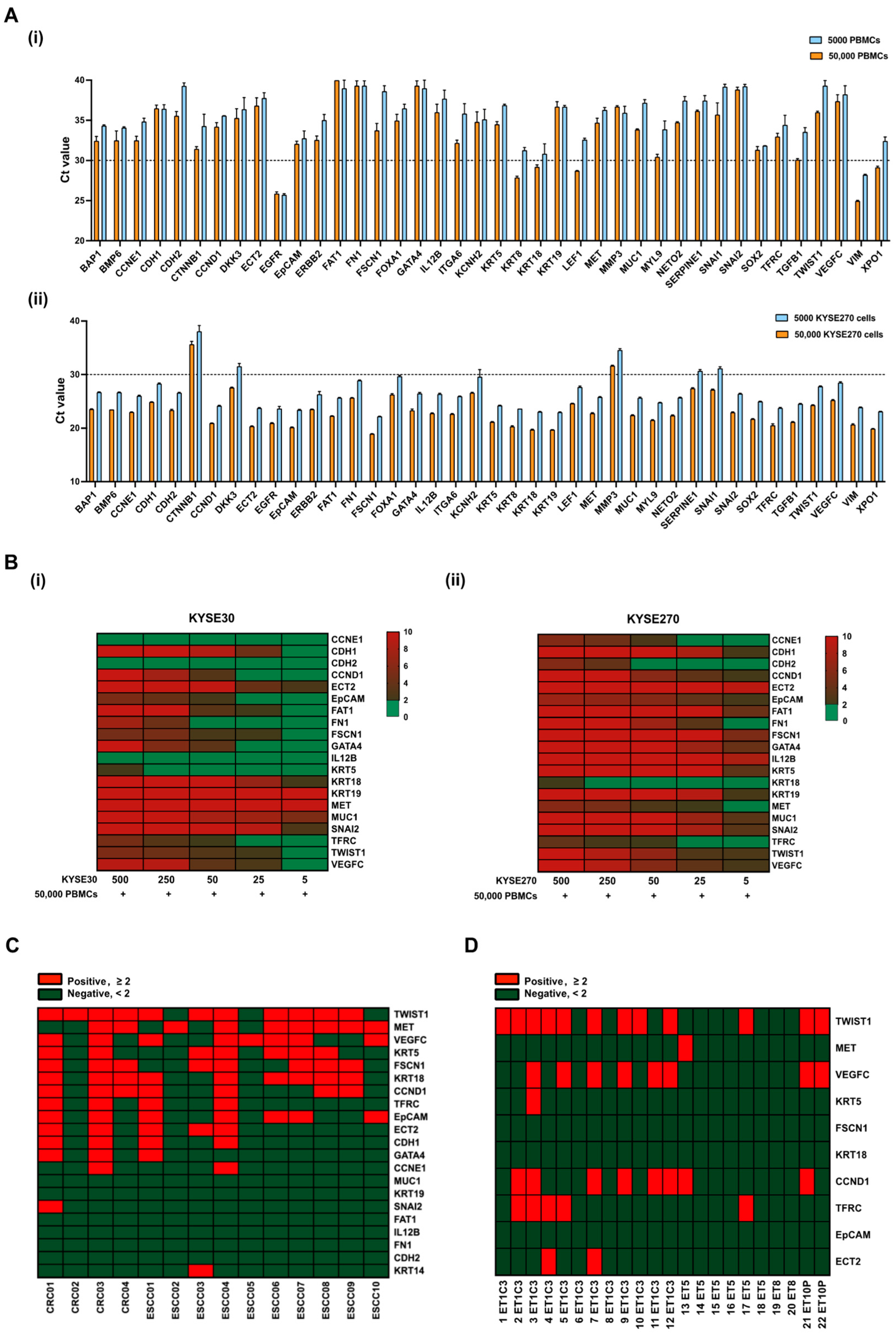
3.1. Establishment of Gene Panels for CTC Detection in ESCC Patients
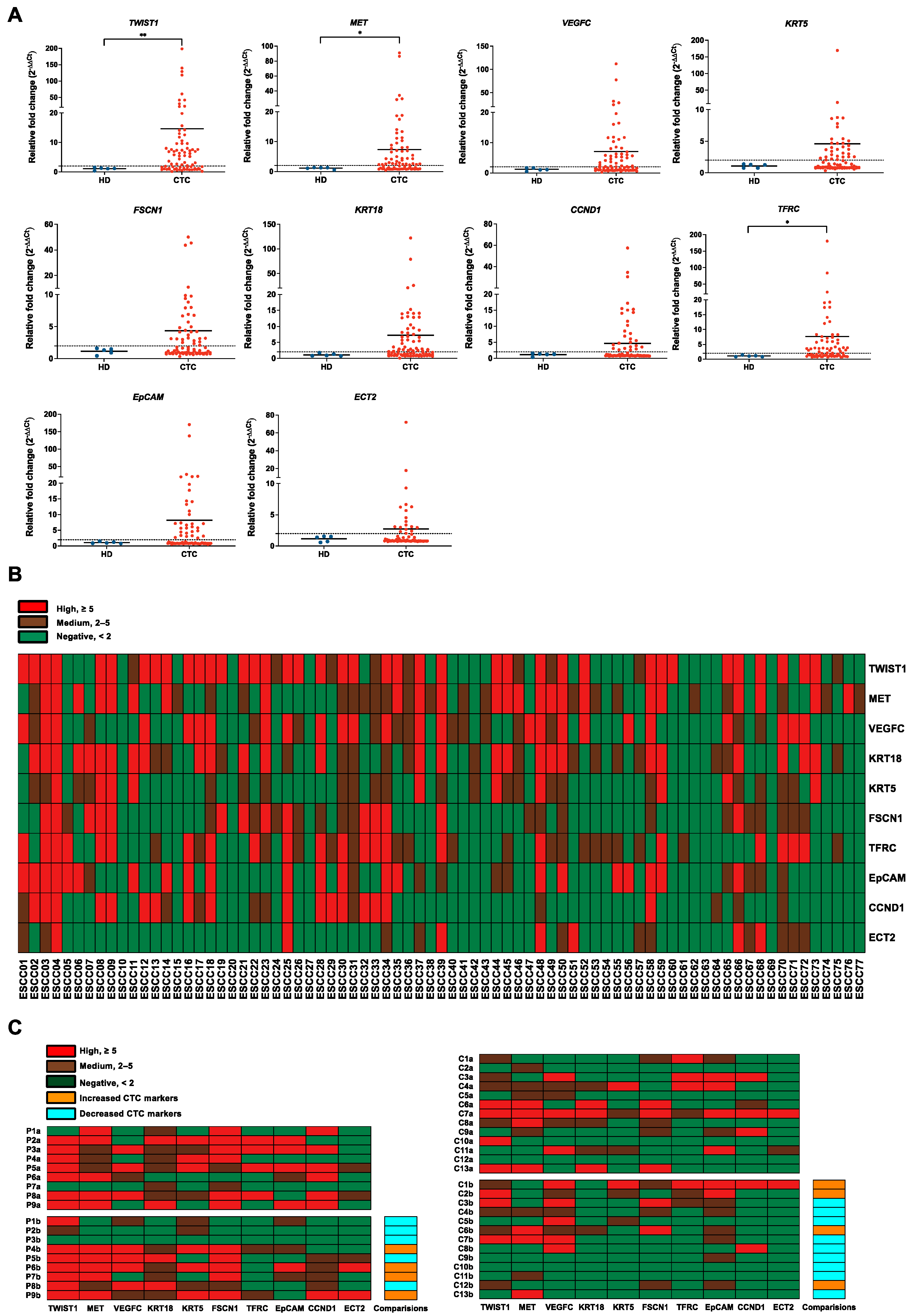
3.2. Evaluation of Gene Panels for Characterizing CTCs in ESCC Patients
3.3. Correlations of CTC Marker Expression with Clinicopathological Features in ESCC Patients
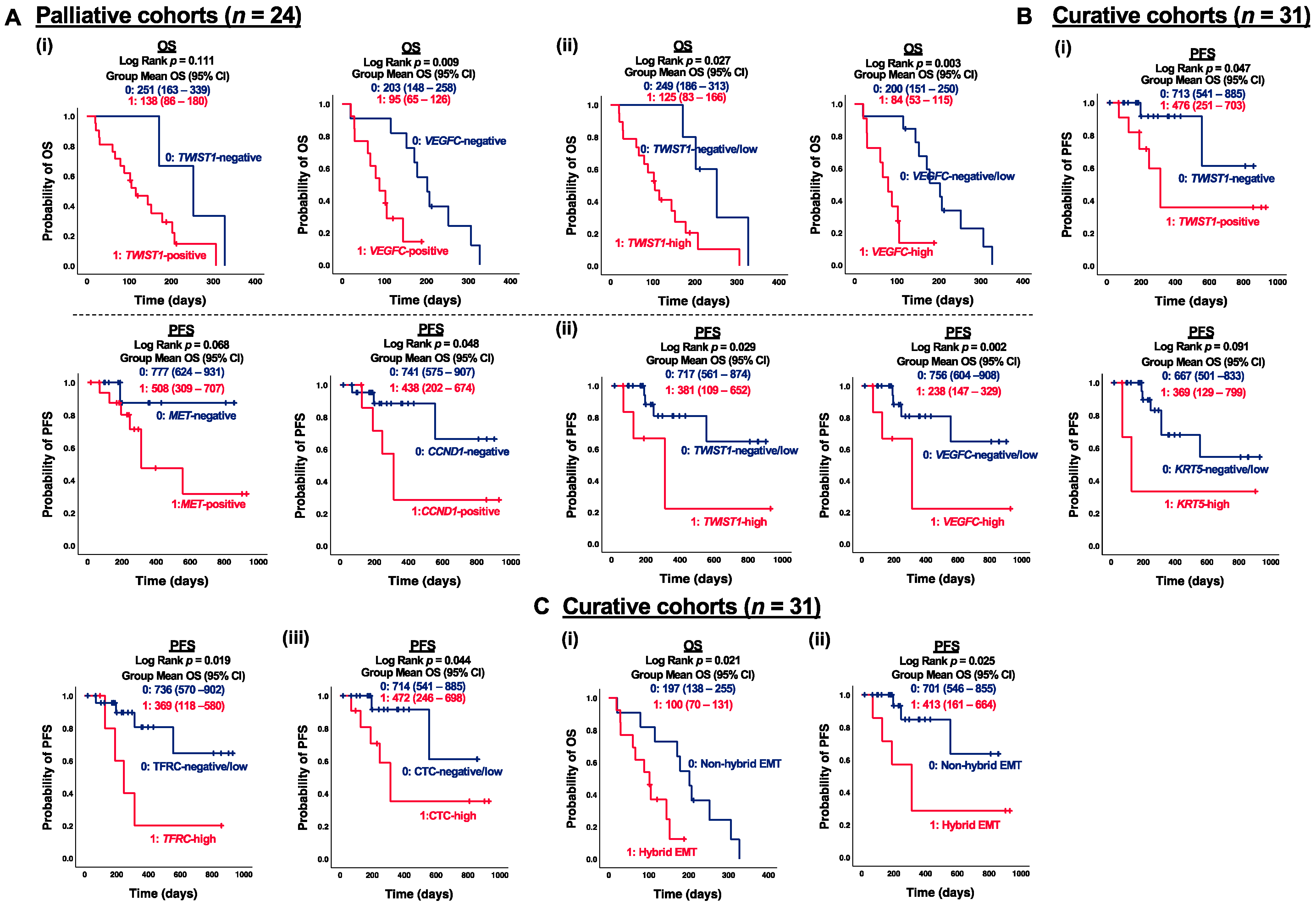
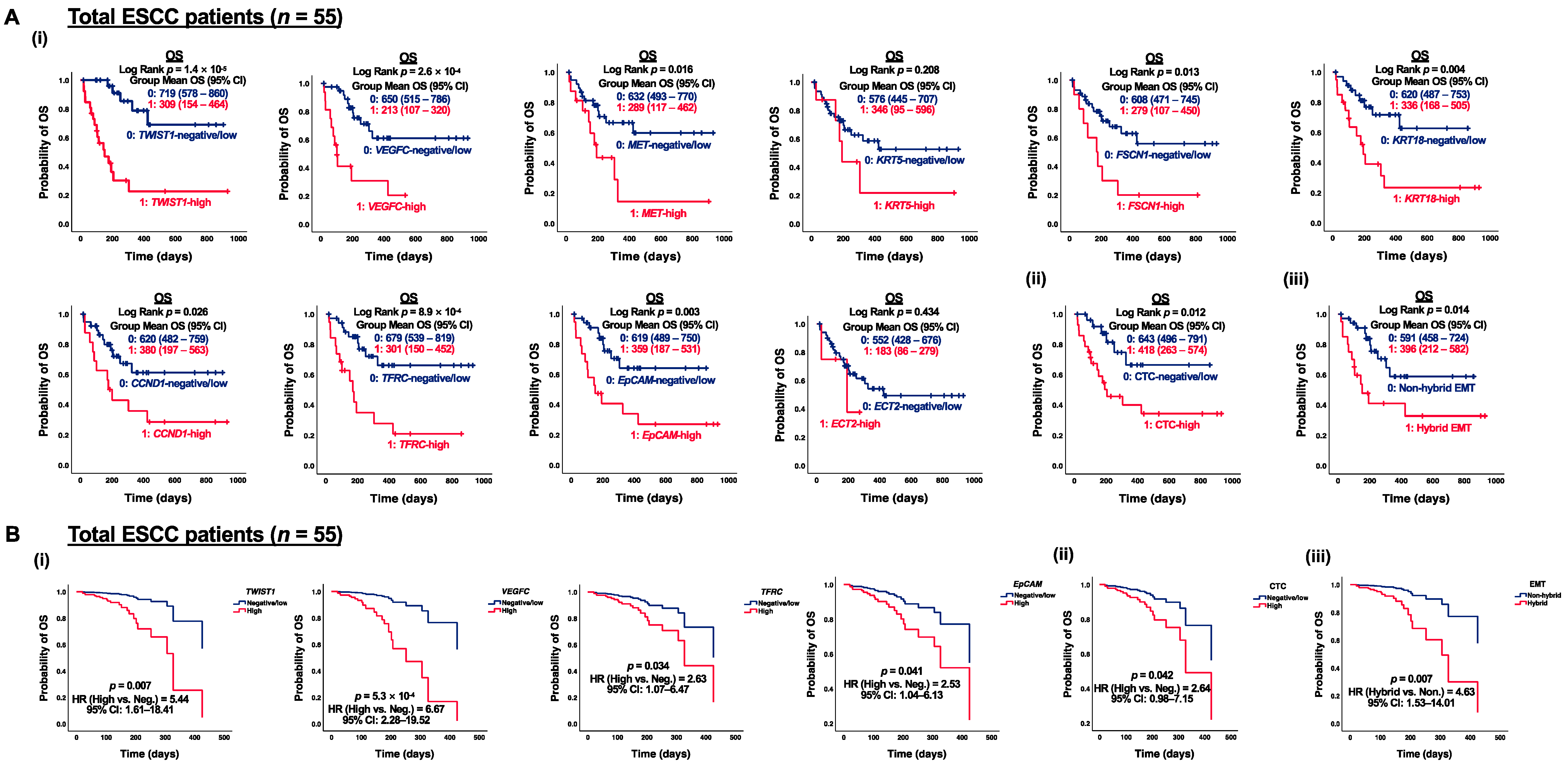
3.4. Correlation of CTC Marker Expression at Baseline with PFS or OS in ESCC Patients
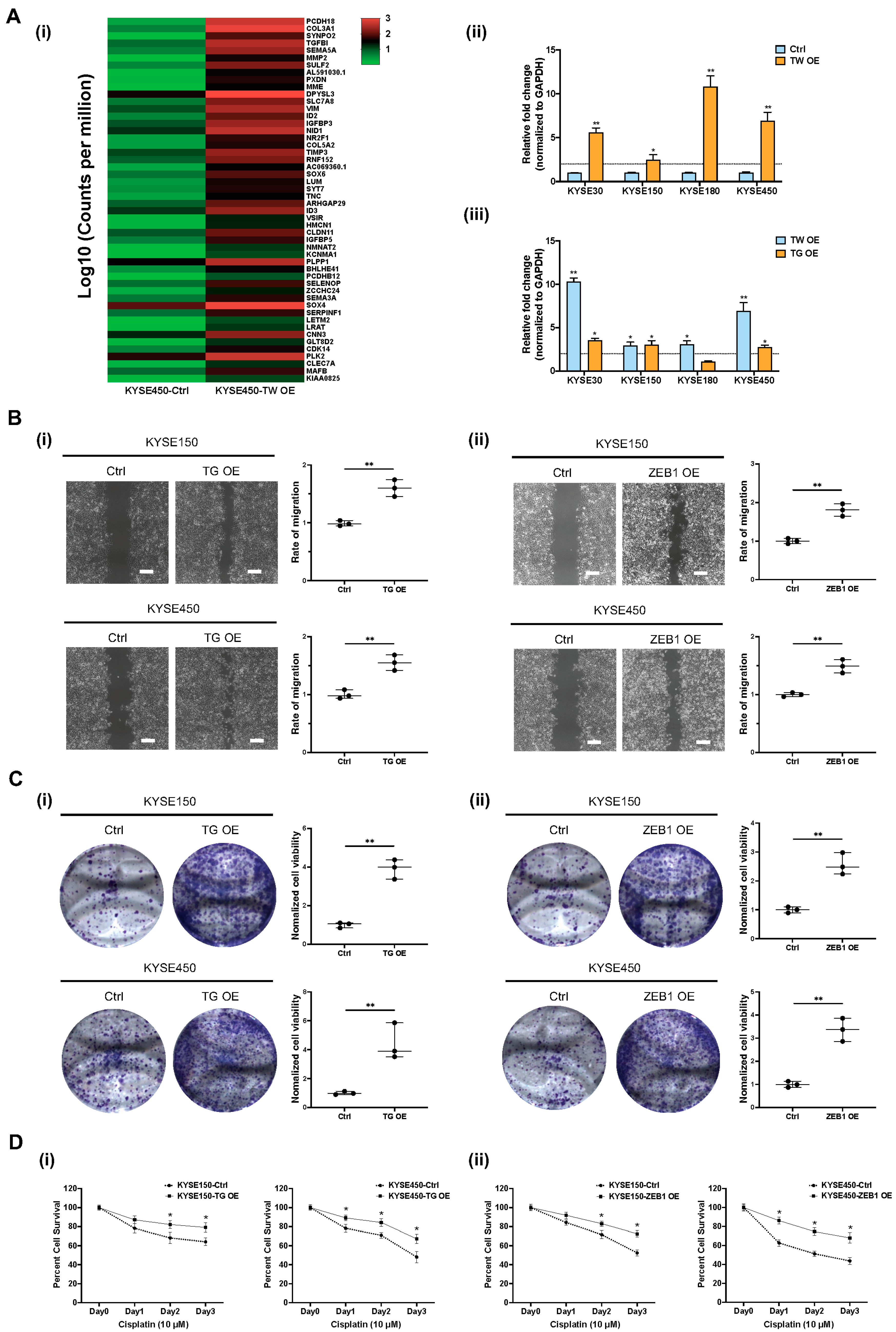
3.5. Functional Role of TWIST1-TGFBI-ZEB1 Signaling in ESCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Soerjomataram, I.; Rumgay, H.; Coleman, H.G.; Thrift, A.P.; Vignat, J.; Laversanne, M.; Ferlay, J.; Arnold, M. The Global Landscape of Esophageal Squamous Cell Carcinoma and Esophageal Adenocarcinoma Incidence and Mortality in 2020 and Projections to 2040: New Estimates from GLOBOCAN 2020. Gastroenterology 2022, 163, 649–658.e642. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A. Update on metastatic gastric and esophageal cancers. J. Clin. Oncol. 2015, 33, 1760–1769. [Google Scholar] [CrossRef]
- Thrumurthy, S.G.; Chaudry, M.A.; Thrumurthy, S.S.D.; Mughal, M. Oesophageal cancer: Risks, prevention, and diagnosis. BMJ 2019, 366, l4373. [Google Scholar] [CrossRef] [PubMed]
- Enzinger, P.C.; Mayer, R.J. Esophageal cancer. N. Engl. J. Med. 2003, 349, 2241–2252. [Google Scholar] [CrossRef]
- Joosse, S.A.; Gorges, T.M.; Pantel, K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Ilie, M.; Hofman, V.; Long-Mira, E.; Selva, E.; Vignaud, J.M.; Padovani, B.; Mouroux, J.; Marquette, C.H.; Hofman, P. “Sentinel” circulating tumor cells allow early diagnosis of lung cancer in patients with chronic obstructive pulmonary disease. PLoS ONE 2014, 9, e111597. [Google Scholar] [CrossRef]
- Ujiie, D.; Matsumoto, T.; Endo, E.; Okayama, H.; Fujita, S.; Kanke, Y.; Watanabe, Y.; Hanayama, H.; Hayase, S.; Saze, Z.; et al. Circulating tumor cells after neoadjuvant chemotherapy are related with recurrence in esophageal squamous cell carcinoma. Esophagus 2021, 18, 566–573. [Google Scholar] [CrossRef]
- Ko, J.M.Y.; Lam, K.O.; Kwong, D.L.W.; Wong, I.Y.; Chan, F.S.; Wong, C.L.; Chan, K.K.; Law, T.T.; Chiu, K.W.H.; Lam, C.C.S.; et al. Circulating Tumor Cell Enumeration for Serial Monitoring of Treatment Outcomes for Locally Advanced Esophageal Squamous Cell Carcinoma. Cancers 2023, 15, 832. [Google Scholar] [CrossRef]
- Matsushita, D.; Uenosono, Y.; Arigami, T.; Yanagita, S.; Nishizono, Y.; Hagihara, T.; Hirata, M.; Haraguchi, N.; Arima, H.; Kijima, Y.; et al. Clinical Significance of Circulating Tumor Cells in Peripheral Blood of Patients with Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2015, 22, 3674–3680. [Google Scholar] [CrossRef] [PubMed]
- Su, P.J.; Wu, M.H.; Wang, H.M.; Lee, C.L.; Huang, W.K.; Wu, C.E.; Chang, H.K.; Chao, Y.K.; Tseng, C.K.; Chiu, T.K.; et al. Circulating Tumour Cells as an Independent Prognostic Factor in Patients with Advanced Oesophageal Squamous Cell Carcinoma Undergoing Chemoradiotherapy. Sci. Rep. 2016, 6, 31423. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Lim, M.; Park, J.; Oh, J.M.; Kim, H.; Jeong, H.; Lee, S.J.; Park, H.C.; Jung, S.; Kim, B.C.; et al. FAST: Size-Selective, Clog-Free Isolation of Rare Cancer Cells from Whole Blood at a Liquid-Liquid Interface. Anal. Chem. 2017, 89, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Hofman, V.; Ilie, M.I.; Long, E.; Selva, E.; Bonnetaud, C.; Molina, T.; Venissac, N.; Mouroux, J.; Vielh, P.; Hofman, P. Detection of circulating tumor cells as a prognostic factor in patients undergoing radical surgery for non-small-cell lung carcinoma: Comparison of the efficacy of the CellSearch Assay and the isolation by size of epithelial tumor cell method. Int. J. Cancer 2011, 129, 1651–1660. [Google Scholar] [CrossRef]
- Ko, J.M.Y.; Ng, H.Y.; Lam, K.O.; Chiu, K.W.H.; Kwong, D.L.W.; Lo, A.W.I.; Wong, J.C.; Lin, R.C.W.; Fong, H.C.H.; Li, J.Y.K.; et al. Liquid Biopsy Serial Monitoring of Treatment Responses and Relapse in Advanced Esophageal Squamous Cell Carcinoma. Cancers 2020, 12, 1352. [Google Scholar] [CrossRef]
- Zhang, X.; Peng, L.; Luo, Y.; Zhang, S.; Pu, Y.; Chen, Y.; Guo, W.; Yao, J.; Shao, M.; Fan, W.; et al. Dissecting esophageal squamous-cell carcinoma ecosystem by single-cell transcriptomic analysis. Nat. Commun. 2021, 12, 5291. [Google Scholar] [CrossRef] [PubMed]
- Warkiani, M.E.; Khoo, B.L.; Wu, L.; Tay, A.K.; Bhagat, A.A.; Han, J.; Lim, C.T. Ultra-fast, label-free isolation of circulating tumor cells from blood using spiral microfluidics. Nat. Protoc. 2016, 11, 134–148. [Google Scholar] [CrossRef]
- Lei, L.C.; Yu, V.Z.; Ko, J.M.Y.; Ning, L.; Lung, M.L. FANCD2 Confers a Malignant Phenotype in Esophageal Squamous Cell Carcinoma by Regulating Cell Cycle Progression. Cancers 2020, 12, 2545. [Google Scholar] [CrossRef]
- Christ, A.; Gunther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Bassler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e114. [Google Scholar] [CrossRef]
- Yu, V.Z.; So, S.S.; Lung, B.C.C.; Hou, G.Z.; Wong, C.W.Y.; Chung, M.K.Y.; Wong, Y.H.; Chan, K.K.; Chan, S.Y.; Law, T.T.; et al. ΔNp63-restricted viral mimicry response impedes cancer cell viability and promotes antitumor immunity in esophageal squamous cell carcinoma. 2023; Submitted. [Google Scholar]
- Sun, Q.; Wang, Y.; Officer, A.; Pecknold, B.; Lee, G.; Harismendy, O.; Desgrosellier, J.S. Stem-like breast cancer cells in the activated state resist genetic stress via TGFBI-ZEB1. NPJ Breast Cancer 2022, 8, 5. [Google Scholar] [CrossRef]
- Zavridou, M.; Mastoraki, S.; Strati, A.; Koutsodontis, G.; Klinakis, A.; Psyrri, A.; Lianidou, E. Direct comparison of size-dependent versus EpCAM-dependent CTC enrichment at the gene expression and DNA methylation level in head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 6551. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, M.; Zhao, J.; Ju, H. Circulating tumor cells in perioperative esophageal cancer patients: Quantitative assay system and potential clinical utility. Clin. Cancer Res. 2007, 13, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Yie, S.M.; Wu, S.M.; Chen, S.; Lou, B.; He, X.; Ye, S.R.; Xie, K.; Rao, L.; Gao, E.; et al. Detection of survivin-expressing circulating cancer cells in the peripheral blood of patients with esophageal squamous cell carcinoma and its clinical significance. Clin. Exp. Metastasis 2009, 26, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Yano, M.; Motoori, M.; Kishi, K.; Miyashiro, I.; Shingai, T.; Gotoh, K.; Noura, S.; Takahashi, H.; Ohue, M.; et al. CEA-antigen and SCC-antigen mRNA expression in peripheral blood predict hematogenous recurrence after resection in patients with esophageal cancer. Ann. Surg. Oncol. 2010, 17, 2779–2786. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.D.; Yuan, X.; Xue, J.J.; Wang, R.; Zhang, Z.R.; Tong, J.D. Clinical significance of carcinoembryonic antigen-, cytokeratin 19-, or survivin-positive circulating tumor cells in the peripheral blood of esophageal squamous cell carcinoma patients treated with radiotherapy. Dis. Esophagus 2012, 25, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Markou, A.; Lazaridou, M.; Paraskevopoulos, P.; Chen, S.; Świerczewska, M.; Budna, J.; Kuske, A.; Gorges, T.M.; Joosse, S.A.; Kroneis, T.; et al. Multiplex Gene Expression Profiling of In Vivo Isolated Circulating Tumor Cells in High-Risk Prostate Cancer Patients. Clin. Chem. 2018, 64, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.H.; Wang, Z.R.; Chen, C.L.; Di, L.; Bi, Z.F.; Li, Z.H.; Liu, Y.M. Molecular detection of epithelial-mesenchymal transition markers in circulating tumor cells from pancreatic cancer patients: Potential role in clinical practice. World J. Gastroenterol. 2019, 25, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Aktas, B.; Tewes, M.; Fehm, T.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009, 11, R46. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, T.; Peng, X.; Liu, J.; Liu, F.; Wu, S.; Liu, S.; Dong, Y.; Xie, S.; Ma, S. Mesenchymal phenotype of circulating tumor cells is associated with distant metastasis in breast cancer patients. Cancer Manag. Res. 2017, 9, 691–700. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Papadaki, M.A.; Mala, A.; Merodoulaki, A.C.; Vassilakopoulou, M.; Mavroudis, D.; Agelaki, S. Investigating the Role of CTCs with Stem/EMT-like Features in Metastatic Breast Cancer Patients Treated with Eribulin Mesylate. Cancers 2022, 14, 3903. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Chen, K.; Che, J.; Hang, J.; Li, H. Detection of Epithelial-Mesenchymal Transition Status of Circulating Tumor Cells in Patients with Esophageal Squamous Carcinoma. BioMed Res. Int. 2018, 2018, 7610154. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.; Liu, W.; Huang, L.; Wang, Y.; Li, D.; Wang, G.; Zhao, Z.; Chi, X.; Xue, Y.; et al. Long Noncoding RNA VESTAR Regulates Lymphangiogenesis and Lymph Node Metastasis of Esophageal Squamous Cell Carcinoma by Enhancing VEGFC mRNA Stability. Cancer Res. 2021, 81, 3187–3199. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Yeow, W.S.; Zou, C.; Wassell, R.; Wang, C.; Pestell, R.G.; Quong, J.N.; Quong, A.A. Cyclin D1/cyclin-dependent kinase 4 interacts with filamin A and affects the migration and invasion potential of breast cancer cells. Cancer Res. 2010, 70, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Xu, J.D.; Wang, W.J.; Cao, X.X.; Chen, Q.; Tang, F.; Chen, Z.Q.; Liu, X.P.; Xu, Z.D. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer Res. 2009, 15, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef]
- Roberts, C.M.; Tran, M.A.; Pitruzzello, M.C.; Wen, W.; Loeza, J.; Dellinger, T.H.; Mor, G.; Glackin, C.A. TWIST1 drives cisplatin resistance and cell survival in an ovarian cancer model, via upregulation of GAS6, L1CAM, and Akt signalling. Sci. Rep. 2016, 6, 37652. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Kim, H.S.; Yoon, H. Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression. Int. J. Mol. Sci. 2020, 21, 7613. [Google Scholar] [CrossRef]
- Wu, J.; Liao, Q.; He, H.; Zhong, D.; Yin, K. TWIST interacts with β-catenin signaling on osteosarcoma cell survival against cisplatin. Mol. Carcinog. 2014, 53, 440–446. [Google Scholar] [CrossRef]
- Bildsoe, H.; Fan, X.; Wilkie, E.E.; Ashoti, A.; Jones, V.J.; Power, M.; Qin, J.; Wang, J.; Tam, P.P.L.; Loebel, D.A.F. Transcriptional targets of TWIST1 in the cranial mesoderm regulate cell-matrix interactions and mesenchyme maintenance. Dev. Biol. 2016, 418, 189–203. [Google Scholar] [CrossRef]
- Thapa, N.; Lee, B.H.; Kim, I.S. TGFBIp/betaig-h3 protein: A versatile matrix molecule induced by TGF-beta. Int. J. Biochem. Cell Biol. 2007, 39, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Lecker, L.S.M.; Berlato, C.; Maniati, E.; Delaine-Smith, R.; Pearce, O.M.T.; Heath, O.; Nichols, S.J.; Trevisan, C.; Novak, M.; McDermott, J.; et al. TGFBI Production by Macrophages Contributes to an Immunosuppressive Microenvironment in Ovarian Cancer. Cancer Res. 2021, 81, 5706–5719. [Google Scholar] [CrossRef] [PubMed]
- Chiavarina, B.; Costanza, B.; Ronca, R.; Blomme, A.; Rezzola, S.; Chiodelli, P.; Giguelay, A.; Belthier, G.; Doumont, G.; Van Simaeys, G.; et al. Metastatic colorectal cancer cells maintain the TGFβ program and use TGFBI to fuel angiogenesis. Theranostics 2021, 11, 1626–1640. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Parameters | Patients (n = 55) | CTC-Positive b | TWIST1 c | MET | VEGFC | KRT5 | FSCN1 | KRT18 | CCND1 | TFRC | EpCAM | ECT2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (within group %) | 46 (83.6) | 33 (60.0) | 34 (61.8) | 25 (45.5) | 25 (45.5) | 23 (41.8) | 33 (60.0) | 22 (40.0) | 29 (52.7) | 25 (45.5) | 14 (25.5) | |
| Median age (range) | 68 ± (33–82) | |||||||||||
| ≤68 | 28 (50.9) | 24 (85.7) | 19 (67.9) | 19(67.9) | 13 (46.4) | 16 (57.1) | 11 (39.3) | 19 (67.9) | 15 (53.6) | 14 (50.0) | 12 (42.9 | 7 (25.0) |
| >68 | 27 (49.1) | 22 (81.5) | 14 (51.9) | 15 (55.6) | 12 (44.4) | 9 (33.3) | 12 (44.4) | 14 (51.9) | 7 (25.9) | 15 (55.6) | 13 (48.1) | 7 (25.9) |
| Gender | ||||||||||||
| Female | 6 (10.9) | 5 (83.3) | 5 (83.3) | 4 (66.7) | 2 (33.3) | 4 (66.7) | 3 (50.0) | 5 (83.3) | 2 (33.3) | 3 (50.0) | 1 (16.7) | 1 (16.7) |
| Male | 49 (89.1) | 41 (83.7) | 28 (57.1) | 30 (61.2) | 23 (46.9) | 21 (42.9) | 20 (40.8) | 28 (57.1) | 20 (40.8) | 26 (53.3) | 24 (49.0) | 13 (26.5) |
| Tumor location | ||||||||||||
| Lower | 25 (45.5) | 19 (76.0) | 15 (60.0) | 15 (60.0) | 10 (40.0) | 13 (52.0) | 15 (40.0) | 16 (64.0) | 8 (32.0) | 11 (44.0) | 10 (40.0) | 4 (16.0) |
| Upper/Middle | 30 (54.5) | 27 (90.0) | 18 (60.0) | 19 (63.3) | 15 (50.0) | 12 (40.0) | 13 (43.3) | 17 (56.7) | 14 (46.7) | 18 (60.0) | 15 (50.0) | 10 (33.3) |
| TNM stage a | p d < 0.001 | p = 0.001 | p = 0.026 | |||||||||
| 0–II | 22 (40.0) | 17 (81.0) | 6 (27.3) | 10 (45.5) | 9 (40.9) | 4 (18.2) | 6 (27.3) | 9 (40.9) | 6 (27.3) | 11 (50.0) | 9 (40.9) | 5 (22.7) |
| III–IV | 33 (60.0) | 29 (85.3) | 27 (81.8) | 24 (72.7) | 16 (48.5) | 21 (63.6) | 17 (51.5) | 24 (72.7) | 16 (48.5) | 18 (54.5) | 16 (48.5) | 9 (27.3) |
| Tumor size | p = 0.031 | p = 0.033 | ||||||||||
| T0–T2 | 10 (18.2) | 5 (55.6) | 3 (30.0) | 3 (30.0) | 2 (20.0) | 4 (40.0) | 3 (30.0) | 5 (50.0) | 2 (20.0) | 3 (30.0) | 2 (20.0) | 0 (0.00) |
| T3–T4 | 45 (81.8) | 41 (89.1) | 30 (66.7) | 31 (68.9) | 23 (51.1) | 21 (46.7) | 20 (44.4) | 28 (62.2) | 20 (44.4) | 26 (57.8) | 23 (51.1) | 14 (31.1) |
| Node stage | p = 0.006 | p = 0.001 | p = 0.029 | |||||||||
| N0–N1 | 27 (49.1) | 22 (84.6) | 11 (40.7) | 13 (48.1) | 11 (40.7) | 6 (22.2) | 7 (25.9) | 14 (51.9) | 9 (33.3) | 13 (48.1) | 13 (48.1) | 5 (18.5) |
| N2–N3 | 28 (50.9) | 24 (82.8) | 22 (78.6) | 21 (75.0) | 14 (50.0) | 19 (67.9) | 16 (57.1) | 19 (67.9) | 13 (46.4) | 16 (57.1) | 12 (42.9) | 19 (32.1) |
| Distant Metastasis | p = 0.004 | p < 0.001 | p = 0.002 | p = 0.029 | ||||||||
| NO | 41 (74.5) | 32 (78.0) | 20 (48.8) | 22 (53.7) | 18 (43.9) | 13 (31.7) | 12 (29.3) | 21 (51.2) | 13 (31.7) | 19 (46.3) | 16 (39.0) | 8 (19.5) |
| YES | 14 (25.5) | 14 (100.0) | 13 (92.9) | 12 (85.7) | 7 (50.0) | 12 (85.7) | 11 (78.6) | 12 (85.7) | 9 (64.3) | 10 (71.4) | 9 (64.3) | 6 (42.9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Z.; Ko, J.M.-Y.; Yu, V.Z.; Lam, K.-O.; Kwong, D.L.-W.; Wong, I.Y.-H.; Chan, F.S.-Y.; Wong, C.L.-Y.; Chan, K.-K.; Law, T.-T.; et al. Multigene Profiling of Circulating Tumor Cells in Esophageal Squamous Cell Carcinoma Identifies Prognostic Cancer Driver Genes Associated with Epithelial-Mesenchymal-Transition Progression and Chemoresistance. Cancers 2023, 15, 5329. https://doi.org/10.3390/cancers15225329
Tan Z, Ko JM-Y, Yu VZ, Lam K-O, Kwong DL-W, Wong IY-H, Chan FS-Y, Wong CL-Y, Chan K-K, Law T-T, et al. Multigene Profiling of Circulating Tumor Cells in Esophageal Squamous Cell Carcinoma Identifies Prognostic Cancer Driver Genes Associated with Epithelial-Mesenchymal-Transition Progression and Chemoresistance. Cancers. 2023; 15(22):5329. https://doi.org/10.3390/cancers15225329
Chicago/Turabian StyleTan, Zhen, Josephine Mun-Yee Ko, Valen Zhuoyou Yu, Ka-On Lam, Dora Lai-Wan Kwong, Ian Yu-Hong Wong, Fion Siu-Yin Chan, Claudia Lai-Yin Wong, Kwan-Kit Chan, Tsz-Ting Law, and et al. 2023. "Multigene Profiling of Circulating Tumor Cells in Esophageal Squamous Cell Carcinoma Identifies Prognostic Cancer Driver Genes Associated with Epithelial-Mesenchymal-Transition Progression and Chemoresistance" Cancers 15, no. 22: 5329. https://doi.org/10.3390/cancers15225329
APA StyleTan, Z., Ko, J. M.-Y., Yu, V. Z., Lam, K.-O., Kwong, D. L.-W., Wong, I. Y.-H., Chan, F. S.-Y., Wong, C. L.-Y., Chan, K.-K., Law, T.-T., Choy, F. S.-F., Ng, H.-Y., Law, S. Y.-K., & Lung, M. L. (2023). Multigene Profiling of Circulating Tumor Cells in Esophageal Squamous Cell Carcinoma Identifies Prognostic Cancer Driver Genes Associated with Epithelial-Mesenchymal-Transition Progression and Chemoresistance. Cancers, 15(22), 5329. https://doi.org/10.3390/cancers15225329