
Tumor Necrosis Factor-Alpha and Adiponectin in Nonalcoholic Fatty Liver Disease-Associated Hepatocellular Carcinoma


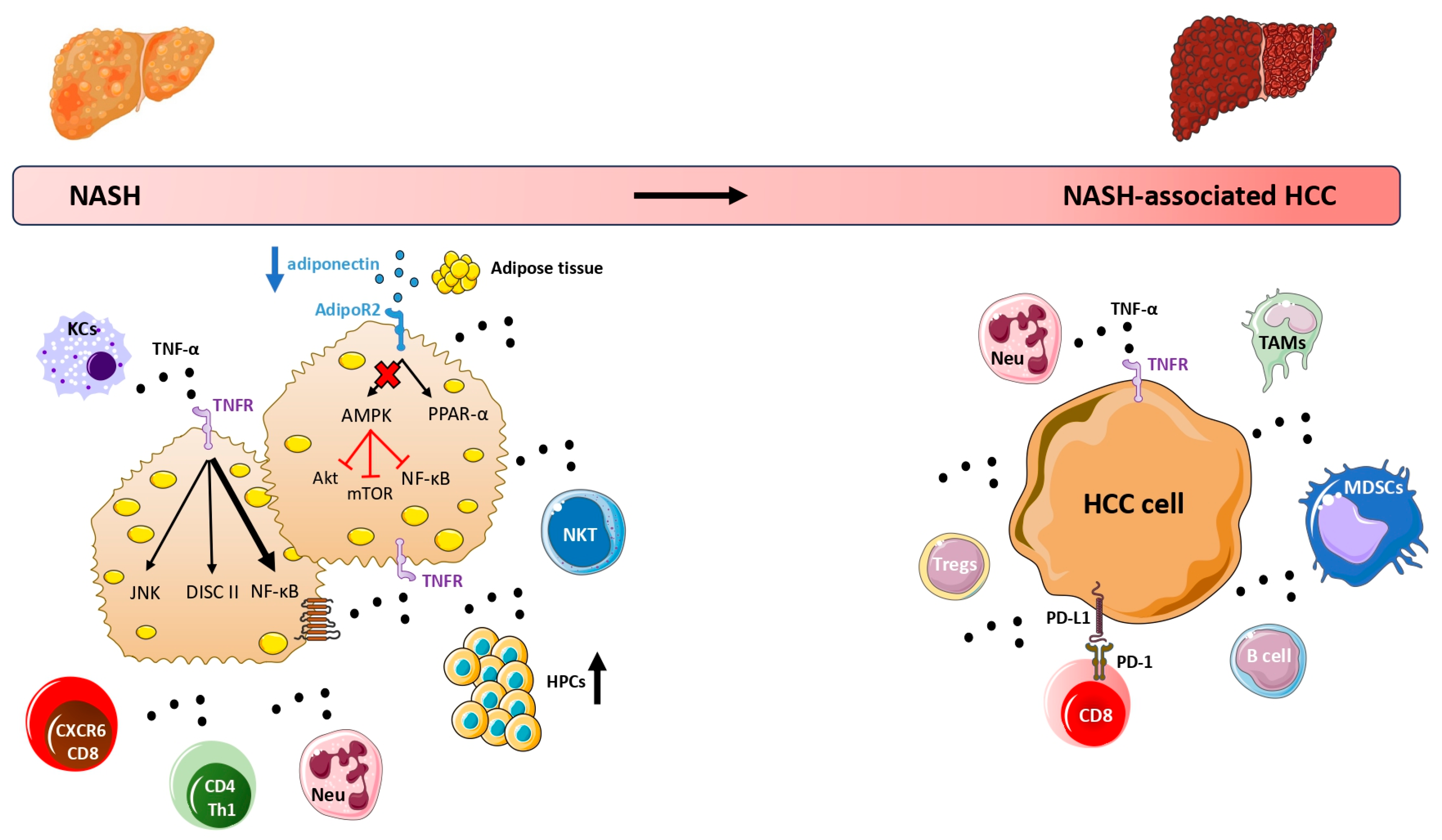
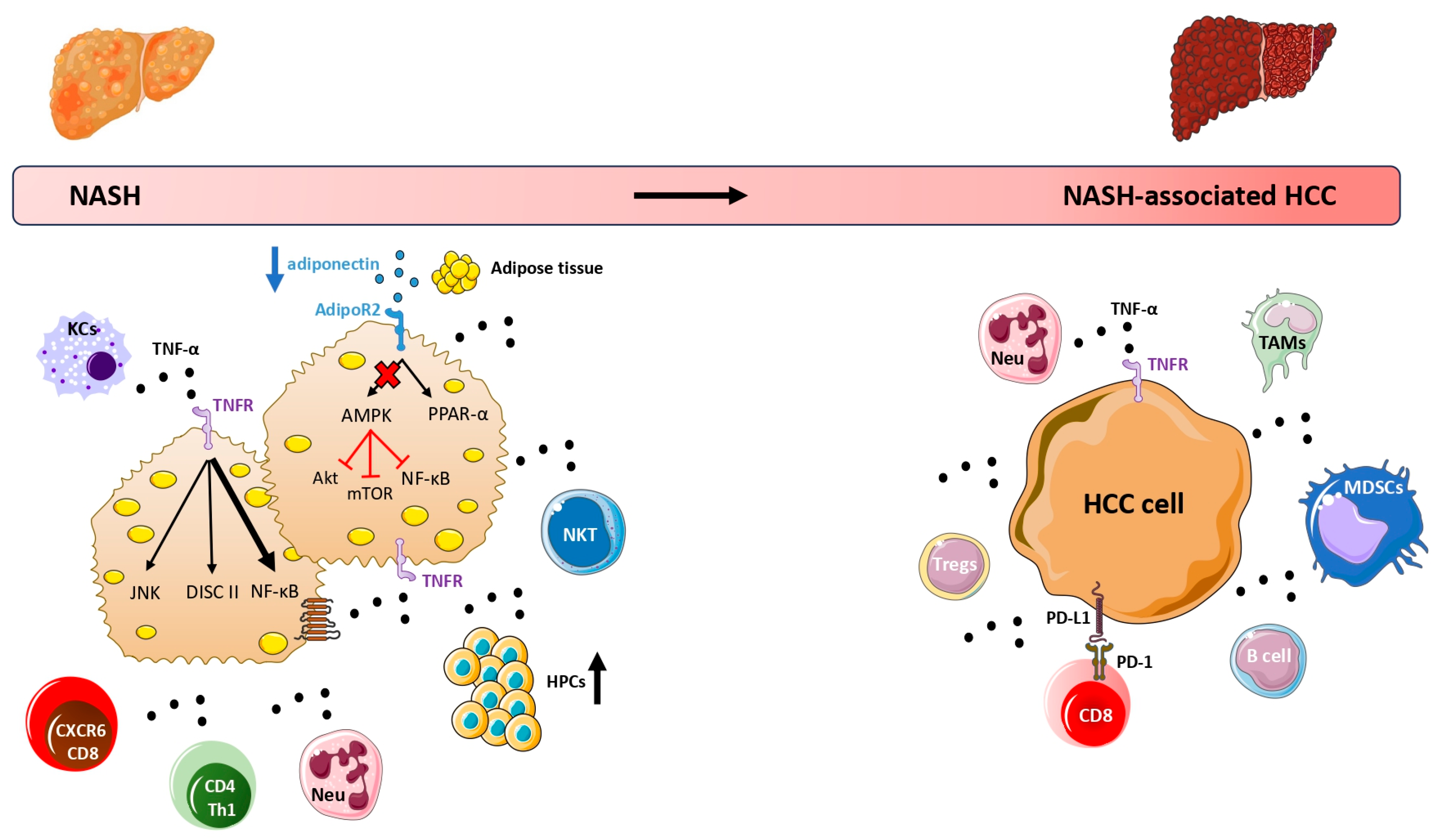{kind=link}
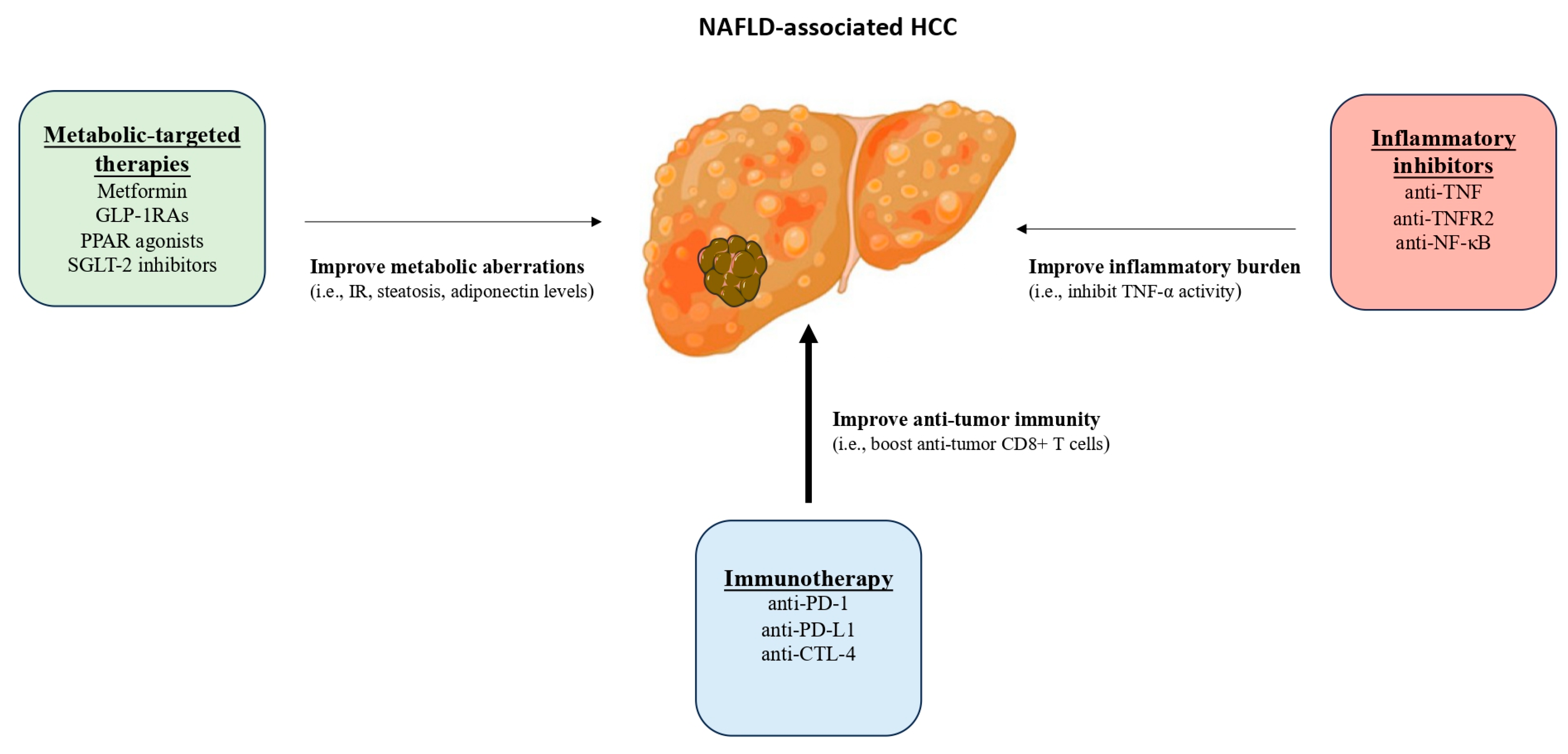
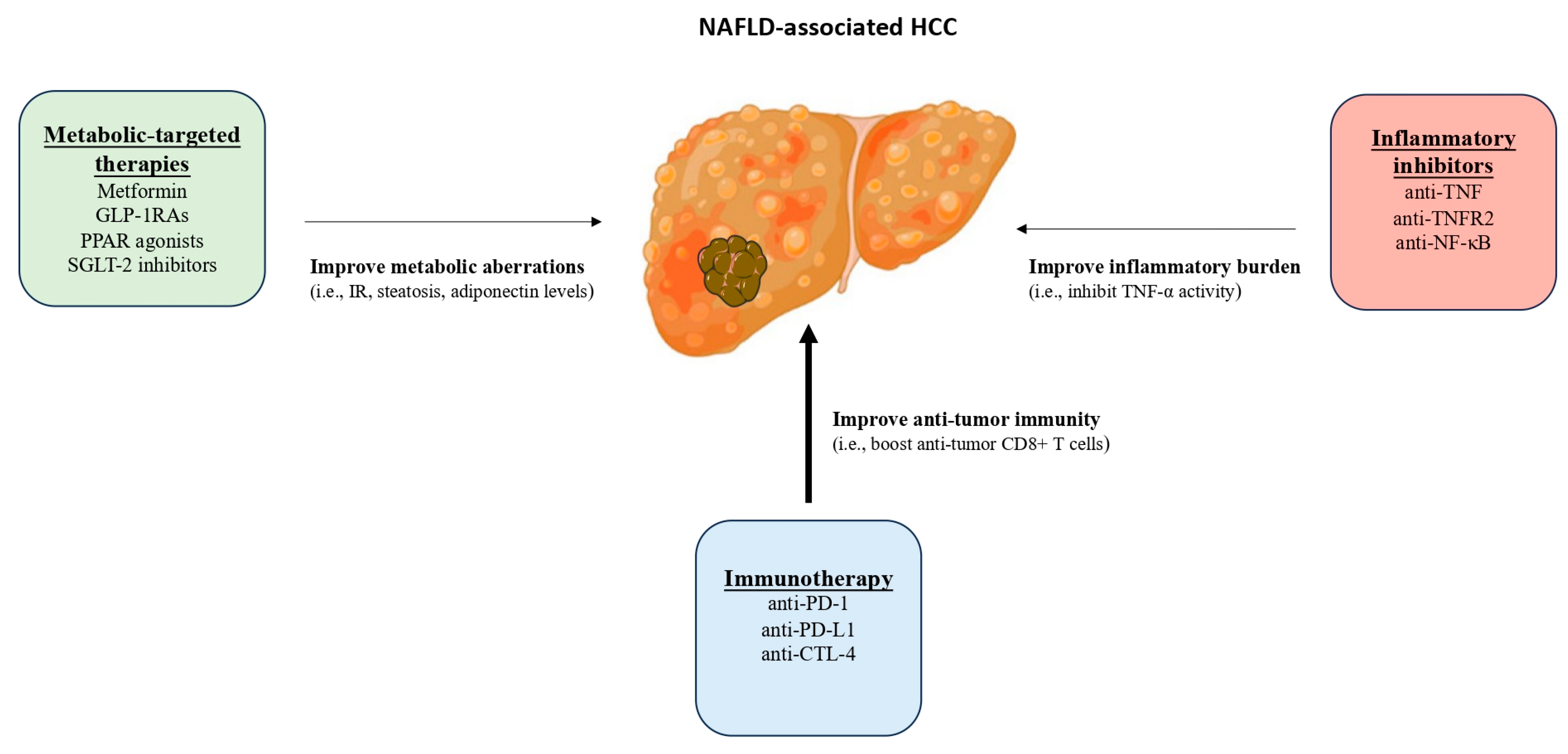{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. TNF-α and NAFLD-Associated HCC
2.1. Experimental Studies
2.2. Clinical Studies
3. Adiponectin and NAFLD-Associated HCC
3.1. Experimental Studies
3.2. Clinical Studies
4. Treatment Considerations for NAFLD-Associated HCC
5. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Henry, L.; Paik, J.; Younossi, Z.M. Review Article: The Epidemiologic Burden of Non-Alcoholic Fatty Liver Disease across the World. Aliment. Pharmacol. Ther. 2022, 56, 942–956. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Makri, E.; Goulas, A.; Polyzos, S.A. Epidemiology, Pathogenesis, Diagnosis and Emerging Treatment of Nonalcoholic Fatty Liver Disease. Arch. Med. Res. 2021, 52, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Chrysavgis, L.; Vachliotis, I.D.; Chartampilas, E.; Cholongitas, E. Nonalcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Insights in Epidemiology, Pathogenesis, Imaging, Prevention and Therapy. Semin. Cancer Biol. 2023, 93, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Pedica, F.; Colombo, M. Distinctive Features of Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease. Dig. Liver Dis. 2022, 54, 154–163. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Nonalcoholic Fatty Liver Disease: The Pathogenetic Roles of Insulin Resistance and Adipocytokines. Curr. Mol. Med. 2009, 9, 299–314. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipokines in Nonalcoholic Fatty Liver Disease. Metabolism 2016, 65, 1062–1079. [Google Scholar] [CrossRef]
- Vachliotis, I.D.; Polyzos, S.A. The Role of Tumor Necrosis Factor-Alpha in the Pathogenesis and Treatment of Nonalcoholic Fatty Liver Disease. Curr. Obes. Rep. 2023, 12, 191–206. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Y.; Liu, J. Tumor Necrosis Factor-α Signaling in Nonalcoholic Steatohepatitis and Targeted Therapies. J. Genet. Genomics 2021, 49, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, G.; Horst, A.K. TNF in the Liver: Targeting a Central Player in Inflammation. Semin. Immunopathol. 2022, 44, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Ramadori, P.; Kam, S.; Heikenwalder, M. T Cells: Friends and Foes in NASH Pathogenesis and Hepatocarcinogenesis. Hepatology 2022, 75, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and Nonalcoholic Fatty Liver Disease: From Pathophysiology to Therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Hüser, N.; Meiser, P.; Bayerl, F.; et al. Auto-Aggressive CXCR6+ CD8 T Cells Cause Liver Immune Pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH Limits Anti-Tumour Surveillance in Immunotherapy-Treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Yahoo, N.; Dudek, M.; Knolle, P.; Heikenwälder, M. Role of Immune Responses for Development of NAFLD-Associated Liver Cancer and Prospects for Therapeutic Modulation. J. Hepatol. 2023, 79, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Braunersreuther, V.; Viviani, G.L.; Mach, F.; Montecucco, F. Role of Cytokines and Chemokines in Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2012, 18, 727–735. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Yang, Y.M.; Kim, S.Y.; Seki, E. Inflammation and Liver Cancer: Molecular Mechanisms and Therapeutic Targets. Semin. Liver Dis. 2019, 39, 26–42. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor Necrosis Factor Signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Wullaert, A.; van Loo, G.; Heyninck, K.; Beyaert, R. Hepatic Tumor Necrosis Factor Signaling and Nuclear Factor-KappaB: Effects on Liver Homeostasis and Beyond. Endocr. Rev. 2007, 28, 365–386. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Sun, K.; Liu, W.; Sheng, D.; Zhao, S.; Gao, L.; Wei, L. Tumor Necrosis Factor-α Promotes Hepatocellular Carcinogenesis through the Activation of Hepatic Progenitor Cells. Cancer Lett. 2018, 434, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Gautheron, J.; Luedde, T. TNF-Dependent Signaling Pathways in Liver Cancer: Promising Targets for Therapeutic Strategies? Dig. Dis. 2012, 30, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Chrysavgis, L.; Giannakodimos, I.; Diamantopoulou, P.; Cholongitas, E. Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Clinical Challenges of an Intriguing Link. World J. Gastroenterol. 2022, 28, 310–331. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipose Tissue, Obesity and Non-Alcoholic Fatty Liver Disease. Minerva Endocrinol. 2017, 42, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Stergiopoulos, C. Adipocytokines in Insulin Resistance and Non-Alcoholic Fatty Liver Disease: The Two Sides of the Same Coin. Med. Hypotheses 2010, 74, 1089–1090. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The Immunology of Hepatocellular Carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Koo, S.-Y.; Park, E.-J.; Lee, C.-W. Immunological Distinctions between Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Exp. Mol. Med. 2020, 52, 1209–1219. [Google Scholar] [CrossRef]
- Zhao, X.; Rong, L.; Zhao, X.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P.; et al. TNF Signaling Drives Myeloid-Derived Suppressor Cell Accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef]
- Chen, Y.; Wen, H.; Zhou, C.; Su, Q.; Lin, Y.; Xie, Y.; Huang, Y.; Qiu, Q.; Lin, J.; Huang, X.; et al. TNF-α Derived from M2 Tumor-Associated Macrophages Promotes Epithelial-Mesenchymal Transition and Cancer Stemness through the Wnt/β-Catenin Pathway in SMMC-7721 Hepatocellular Carcinoma Cells. Exp. Cell Res. 2019, 378, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.; Bridle, K.R.; Crawford, D.H.G.; Jayachandran, A. TNF-α-mediated Epithelial-to-mesenchymal Transition Regulates Expression of Immune Checkpoint Molecules in Hepatocellular Carcinoma. Mol. Med. Rep. 2020, 21, 1849–1860. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The Role of PD-1/PD-L1 and Application of Immune-Checkpoint Inhibitors in Human Cancers. Front. Immunol. 2022, 13, 964442. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, C.; Du, J.-X.; Zhao, J.; Shi, M.-T.; Jin, M.-W.; Liu, H. Adipocytes Promote Tumor Progression and Induce PD-L1 Expression via TNF-α/IL-6 Signaling. Cancer Cell Int. 2020, 20, 179. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of Inflammatory Cytokines with Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 880298. [Google Scholar] [CrossRef]
- Potoupni, V.; Georgiadou, M.; Chatzigriva, E.; Polychronidou, G.; Markou, E.; Zapantis Gakis, C.; Filimidou, I.; Karagianni, M.; Anastasilakis, D.; Evripidou, K.; et al. Circulating Tumor Necrosis Factor-α Levels in Non-Alcoholic Fatty Liver Disease: A Systematic Review and a Meta-Analysis. J. Gastroenterol. Hepatol. 2021, 36, 3002–3014. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and Cancer Risk: Emerging Biological Mechanisms and Perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and Genetic Obesity Promote Liver Inflammation and Tumorigenesis by Enhancing IL-6 and TNF Expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Kumar, D.P.; Santhekadur, P.K.; Seneshaw, M.; Mirshahi, F.; Uram-Tuculescu, C.; Sanyal, A.J. A Regulatory Role of Apoptosis Antagonizing Transcription Factor in the Pathogenesis of Nonalcoholic Fatty Liver Disease and Hepatocellular Carcinoma. Hepatology 2019, 69, 1520–1534. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Lo, G.-H.; Lai, K.-H.; Cheng, J.-S.; Lin, C.-K.; Hsu, P.-I. Increased Serum Concentrations of Tumor Necrosis Factor-Alpha Are Associated with Disease Progression and Malnutrition in Hepatocellular Carcinoma. J. Chin. Med. Assoc. 2003, 66, 593–598. [Google Scholar] [PubMed]
- Okuda, K.; Ohtsuki, T.; Obata, H.; Tomimatsu, M.; Okazaki, N.; Hasegawa, H.; Nakajima, Y.; Ohnishi, K. Natural History of Hepatocellular Carcinoma and Prognosis in Relation to Treatment. Study of 850 Patients. Cancer 1985, 56, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Sobin, L.H.; Fleming, I.D. TMN Classification of Malignant Tumours. Cancer 1997, 80, 1803–1804. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.; Hu, X.; Liu, S.; He, B. Prognostic and Therapeutic Values of Tumor Necrosis Factor-Alpha in Hepatocellular Carcinoma. Med. Sci. Monit. 2016, 22, 3694–3704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, J.; Li, H.; Zhang, S.; Hu, W.; Wu, L.; Han, B. High TNF-α and/or P38MAPK Expression Predicts a Favourable Prognosis in Patients with T1N0M0 Hepatocellular Carcinoma: An Immunohistochemical Study. Oncol. Lett. 2019, 17, 4948–4956. [Google Scholar] [PubMed]
- Llovet, J.M.; Brú, C.; Bruix, J. Prognosis of Hepatocellular Carcinoma: The BCLC Staging Classification. Semin. Liver Dis. 1999, 19, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Zhang, M.; Hu, J.; Li, Z.; Han, B. The High Expression of TNF-α and NF-κB in Tumor Microenvironment Predicts Good Prognosis of Patients with BCLC-0-B Hepatocellular Carcinoma. Transl. Cancer Res. 2019, 8, 532–541. [Google Scholar] [CrossRef]
- Wungu, C.D.K.; Ariyanto, F.C.; Prabowo, G.I.; Soetjipto; Handajani, R. Association between Five Types of Tumor Necrosis Factor-α Gene Polymorphism and Hepatocellular Carcinoma Risk: A Meta-Analysis. BMC Cancer 2020, 20, 1134. [Google Scholar] [CrossRef]
- Kartikasari, A.E.R.; Cassar, E.; Razqan, M.A.M.; Szydzik, C.; Huertas, C.S.; Mitchell, A.; Plebanski, M. Elevation of Circulating TNF Receptor 2 in Cancer: A Systematic Meta-Analysis for Its Potential as a Diagnostic Cancer Biomarker. Front. Immunol. 2022, 13, 918254. [Google Scholar] [CrossRef]
- Yang, Y.; Islam, M.S.; Hu, Y.; Chen, X. TNFR2: Role in Cancer Immunology and Immunotherapy. ImmunoTargets Ther. 2021, 10, 103–122. [Google Scholar] [CrossRef]
- Vanamee, É.S.; Faustman, D.L. TNFR2: A Novel Target for Cancer Immunotherapy. Trends Mol. Med. 2017, 23, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A. Adiponectin in Health and Disease: Current Evidence and Therapeutic Perspectives. Curr. Med. Chem. 2012, 19, 5425–5426. [Google Scholar] [CrossRef]
- Duan, X.-F.; Tang, P.; Li, Q.; Yu, Z.-T. Obesity, Adipokines and Hepatocellular Carcinoma. Int. J. Cancer 2013, 133, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Tsiaousi, E. The Role of Adiponectin in the Pathogenesis and Treatment of Non-Alcoholic Fatty Liver Disease. Diabetes Obes. Metab. 2010, 12, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Wieser, V.; Moschen, A.R.; Tilg, H. Adipocytokines and Hepatocellular Carcinoma. Dig. Dis. 2012, 30, 508–513. [Google Scholar] [CrossRef]
- Saxena, N.K.; Fu, P.P.; Nagalingam, A.; Wang, J.; Handy, J.; Cohen, C.; Tighiouart, M.; Sharma, D.; Anania, F.A. Adiponectin Modulates C-Jun N-Terminal Kinase and Mammalian Target of Rapamycin and Inhibits Hepatocellular Carcinoma. Gastroenterology 2010, 139, 1762–1773. [Google Scholar] [CrossRef] [PubMed]
- Heiker, J.T.; Kosel, D.; Beck-Sickinger, A.G. Molecular Mecha- Nisms of Signal Transduction via Adiponectin and Adiponectin Receptors. Biol. Chem. 2010, 391, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Marques, V.; Arella, F.; Afonso, M.B.; Santos, A.A.; Rodrigues, C.M.P. Decoding the Role of Leptin and Adiponectin in Obesity-Related Gastrointestinal Cancer. Clin. Sci. 2023, 137, 1095–1114. [Google Scholar] [CrossRef]
- Kamada, Y.; Matsumoto, H.; Tamura, S.; Fukushima, J.; Kiso, S.; Fukui, K.; Igura, T.; Maeda, N.; Kihara, S.; Funahashi, T.; et al. Hypoadiponectinemia Accelerates Hepatic Tumor Formation in a Nonalcoholic Steatohepatitis Mouse Model. J. Hepatol. 2007, 47, 556–564. [Google Scholar] [CrossRef]
- Asano, T.; Watanabe, K.; Kubota, N.; Gunji, T.; Omata, M.; Kadowaki, T.; Ohnishi, S. Adiponectin Knockout Mice on High Fat Diet Develop Fibrosing Steatohepatitis. J. Gastroenterol. Hepatol. 2009, 24, 1669–1676. [Google Scholar] [CrossRef]
- Nazmy, E.A.; El-Khouly, O.A.; Zaki, M.M.A.; Elsherbiny, N.M.; Said, E.; Al-Gayyar, M.M.H.; Salem, H.A. Targeting P53/TRAIL/Caspase-8 Signaling by Adiponectin Reverses Thioacetamide-Induced Hepatocellular Carcinoma in Rats. Environ. Toxicol. Pharmacol. 2019, 72, 103240. [Google Scholar] [CrossRef] [PubMed]
- Al-Gayyar, M.M.H.; Abbas, A.; Hamdan, A.M. Chemopreventive and Hepatoprotective Roles of Adiponectin (SULF2 Inhibitor) in Hepatocelluar Carcinoma. Biol. Chem. 2016, 397, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. The Multi-Hit Process and the Antagonistic Roles of Tumor Necrosis Factor-Alpha and Adiponectin in Non Alcoholic Fatty Liver Disease. Hippokratia 2009, 13, 127. [Google Scholar] [PubMed]
- Kucukoglu, O.; Sowa, J.-P.; Mazzolini, G.D.; Syn, W.-K.; Canbay, A. Hepatokines and Adipokines in NASH-Related Hepatocellular Carcinoma. J. Hepatol. 2021, 74, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Vachher, M.; Bansal, S.; Kumar, B.; Yadav, S.; Arora, T.; Wali, N.M.; Burman, A. Contribution of Organokines in the Development of NAFLD/NASH Associated Hepatocellular Carcinoma. J. Cell. Biochem. 2022, 123, 1553–1584. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum Total Adiponectin in Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Metabolism 2011, 60, 313–326. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Nonlinear Distribution of Adiponectin in Patients with Nonalcoholic Fatty Liver Disease Limits Its Use in Linear Regression Analysis. J. Clin. Gastroenterol. 2010, 44, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, G.P.; Armandi, A.; Rosso, C.; Gaia, S.; Aneli, S.; Rolle, E.; Abate, M.L.; Olivero, A.; Nicolosi, A.; Guariglia, M.; et al. Biomarkers of Oncogenesis, Adipose Tissue Dysfunction and Systemic Inflammation for the Detection of Hepatocellular Carcinoma in Patients with Nonalcoholic Fatty Liver Disease. Cancers 2021, 13, 2305. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J. Serum Alpha-Fetoprotein in Patients with Nonalcoholic Fatty Liver Disease. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2411–2412. [Google Scholar]
- Wei, T.; Ye, P.; Peng, X.; Wu, L.-L.; Yu, G.-Y. Circulating Adiponectin Levels in Various Malignancies: An Updated Meta-Analysis of 107 Studies. Oncotarget 2016, 7, 48671–48691. [Google Scholar] [CrossRef]
- Jiang, H.; Hu, D.; Wang, J.; Zhang, B.; He, C.; Ning, J. Adiponectin and the Risk of Gastrointestinal Cancers in East Asians: Mendelian Randomization Analysis. Cancer Med. 2022, 11, 2397–2404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yuan, Q.; Li, M.; Chai, D.; Deng, W.; Wang, W. The Association of Leptin and Adiponectin with Hepatocellular Carcinoma Risk and Prognosis: A Combination of Traditional, Survival, and Dose-Response Meta-Analysis. BMC Cancer 2020, 20, 1167. [Google Scholar] [CrossRef] [PubMed]
- Sadik, N.A.E.-H.; Ahmed, A.; Ahmed, S. The Significance of Serum Levels of Adiponectin, Leptin, and Hyaluronic Acid in Hepatocellular Carcinoma of Cirrhotic and Noncirrhotic Patients. Hum. Exp. Toxicol. 2012, 31, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Siegel, A.B.; Goyal, A.; Salomao, M.; Wang, S.; Lee, V.; Hsu, C.; Rodriguez, R.; Hershman, D.L.; Brown, R.S., Jr.; Neugut, A.I.; et al. Serum Adiponectin Is Associated with Worsened Overall Survival in a Prospective Cohort of Hepatocellular Carcinoma Patients. Oncology 2015, 88, 57–68. [Google Scholar] [CrossRef]
- Shin, E.; Yu, Y.-D.; Kim, D.-S.; Won, N.H. Adiponectin Receptor Expression Predicts Favorable Prognosis in Cases of Hepatocellular Carcinoma. Pathol. Oncol. Res. 2014, 20, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-N.; Yang, S.-F.; Tsai, H.-H.; Lee, K.-T.; Yeh, Y.-T. Increased Adiponectin Associated with Poor Survival in Hepatocellular Carcinoma. J. Gastroenterol. 2014, 49, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Ramai, D.; Singh, J.; Lester, J.; Khan, S.R.; Chandan, S.; Tartaglia, N.; Ambrosi, A.; Serviddio, G.; Facciorusso, A. Systematic Review with Meta-Analysis: Bariatric Surgery Reduces the Incidence of Hepatocellular Carcinoma. Aliment. Pharmacol. Ther. 2021, 53, 977–984. [Google Scholar] [CrossRef]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC Strategy for Prognosis Prediction and Treatment Recommendation: The 2022 Update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Vogel, A.; Martinelli, E.; ESMO Guidelines Committee. Electronic address: Clinicalguidelines@esmo.org; ESMO Guidelines Committee Updated Treatment Recommendations for Hepatocellular Carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef]
- Bruix, J.; da Fonseca, L.G.; Reig, M. Insights into the Success and Failure of Systemic Therapy for Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 617–630. [Google Scholar] [CrossRef]
- Pinter, M.; Pinato, D.J.; Ramadori, P.; Heikenwalder, M. NASH and Hepatocellular Carcinoma: Immunology and Immunotherapy. Clin. Cancer Res. 2023, 29, 513–520. [Google Scholar] [CrossRef]
- Zuazo, M.; Gato-Cañas, M.; Llorente, N.; Ibañez-Vea, M.; Arasanz, H.; Kochan, G.; Escors, D. Molecular Mechanisms of Programmed Cell Death-1 Dependent T Cell Suppression: Relevance for Immunotherapy. Ann. Transl. Med. 2017, 5, 385. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.K.; Puigvehí, M.; Castet, F.; Lourdusamy, V.; Montal, R.; Tabrizian, P.; Buckstein, M.; Kim, E.; Villanueva, A.; Schwartz, M.; et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-Analysis of Randomized Controlled Trials (2002–2020). Gastroenterology 2021, 161, 879–898. [Google Scholar] [CrossRef]
- Ho, W.J.; Danilova, L.; Lim, S.J.; Verma, R.; Xavier, S.; Leatherman, J.M.; Sztein, M.B.; Fertig, E.J.; Wang, H.; Jaffee, E.; et al. Viral Status, Immune Microenvironment and Immunological Response to Checkpoint Inhibitors in Hepatocellular Carcinoma. J. Immunother. Cancer 2020, 8, e000394. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Dong, Z.; Chen, Z.; Hong, J.; Yan, L.; Li, H.; Yao, S.; Yan, Y.; Yang, Y.; Yang, C.; et al. Viral Status and Efficacy of Immunotherapy in Hepatocellular Carcinoma: A Systematic Review with Meta-Analysis. Front. Immunol. 2021, 12, 733530. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Kazakos, E.; Polyzos, S.A.; Papaefthymiou, A.; Zavos, C.; Tzitiridou-Chatzopoulou, M.; Chatzopoulos, D.; Vardaka, E.; Gatopoulou, A.; Kyrailidi, F.; et al. Potential Impact of Trained Innate Immunity on the Pathophysiology of Metabolic Dysfunction-Associated Fatty Liver Disease. Clin. Immunol. 2023, 256, 109776. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Kazakos, E.; Kyrailidi, F.; Polyzos, S.A.; Zavos, C.; Arapoglou, S.; Boziki, M.; Mouratidou, M.C.; Tzitiridou-Chatzopoulou, M.; Chatzopoulos, D.; et al. Innate Immunity and Nonalcoholic Fatty Liver Disease. Ann. Gastroenterol. 2023, 36, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Deretzi, G. Nonalcoholic Fatty Liver Disease: Multimodal Treatment Options for a Pathogenetically Multiple-Hit Disease. J. Clin. Gastroenterol. 2012, 46, 272–284. [Google Scholar] [CrossRef]
- Makri, E.S.; Makri, E.; Polyzos, S.A. Combination Therapies for Nonalcoholic Fatty Liver Disease. J. Pers. Med. 2022, 12, 1166. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Xiao, H. Metformin Actions on the Liver: Protection Mechanisms Emerging in Hepatocytes and Immune Cells against NASH-Related HCC. Int. J. Mol. Sci. 2021, 22, 5016. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-R.; Lu, Z.-H.; Su, Y.; Zhao, N.; Dong, C.-L.; Sun, L.; Zhao, S.-F.; Li, Y. Relationship of Serum Adiponectin Levels and Metformin Therapy in Patients with Type 2 Diabetes. Horm. Metab. Res. 2016, 48, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Pinyopornpanish, K.; Leerapun, A.; Pinyopornpanish, K.; Chattipakorn, N. Effects of Metformin on Hepatic Steatosis in Adults with Nonalcoholic Fatty Liver Disease and Diabetes: Insights from the Cellular to Patient Levels. Gut Liver 2021, 15, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Matafome, P.; Louro, T.; Rodrigues, L.; Crisóstomo, J.; Nunes, E.; Amaral, C.; Monteiro, P.; Cipriano, A.; Seiça, R. Metformin and Atorvastatin Combination Further Protect the Liver in Type 2 Diabetes with Hyperlipidaemia. Diabetes. Metab. Res. Rev. 2011, 27, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Rongjiong, Z.; Kahaer, M.; Chunhui, J.; Wulasihan, M. Therapeutic Efficacy of Liraglutide versus Metformin in Modulating the Gut Microbiota for Treating Type 2 Diabetes Mellitus Complicated with Nonalcoholic Fatty Liver Disease. Front. Microbiol. 2023, 14, 1088187. [Google Scholar] [CrossRef] [PubMed]
- Wabitsch, S.; McCallen, J.D.; Kamenyeva, O.; Ruf, B.; McVey, J.C.; Kabat, J.; Walz, J.S.; Rotman, Y.; Bauer, K.C.; Craig, A.J.; et al. Metformin Treatment Rescues CD8+ T-Cell Response to Immune Checkpoint Inhibitor Therapy in Mice with NAFLD. J. Hepatol. 2022, 77, 748–760. [Google Scholar] [CrossRef]
- Lujambio, A.; Sarobe, P. Metformin Keeps CD8+ T Cells Active and Moving in NASH-HCC Immunotherapy. J. Hepatol. 2022, 77, 593–595. [Google Scholar] [CrossRef]
- Lin, H.; Yiu, D.C.-Y.; Chin, S.; Liu, K.; Yip, T.C.-F. Metformin in Patients with Hepatocellular Carcinoma Receiving Immunotherapy. J. Hepatol. 2023, 78, e180–e182. [Google Scholar] [CrossRef]
- Casadei Gardini, A.; Faloppi, L.; De Matteis, S.; Foschi, F.G.; Silvestris, N.; Tovoli, F.; Palmieri, V.; Marisi, G.; Brunetti, O.; Vespasiani-Gentilucci, U.; et al. Metformin and Insulin Impact on Clinical Outcome in Patients with Advanced Hepatocellular Carcinoma Receiving Sorafenib: Validation Study and Biological Rationale. Eur. J. Cancer 2017, 86, 106–114. [Google Scholar] [CrossRef]
- Abd El-Fattah, E.E.; Zakaria, A.Y. Metformin Modulate Immune Fitness in Hepatocellular Carcinoma: Molecular and Cellular Approach. Int. Immunopharmacol. 2022, 109, 108889. [Google Scholar] [CrossRef] [PubMed]
- Athyros, V.G.; Polyzos, S.A.; Kountouras, J.; Katsiki, N.; Anagnostis, P.; Doumas, M.; Mantzoros, C.S. Non-Alcoholic Fatty Liver Disease Treatment in Patients with Type 2 Diabetes Mellitus; New Kids on the Block. Curr. Vasc. Pharmacol. 2020, 18, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liang, H.; Huang, L.; Zhou, H.; Wang, Z. Liraglutide Enhances the Effect of Checkpoint Blockade through the Inhibition of Neutrophil Extracellular Traps in Murine Lung and Liver Cancers. FEBS Open Bio 2022. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Maleki, M.; Atkin, S.L.; Jamialahmadi, T.; Sahebkar, A. Impact of Incretin-Based Therapies on Adipokines and Adiponectin. J. Diabetes Res. 2021, 2021, 3331865. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide Safety and Efficacy in Patients with Non-Alcoholic Steatohepatitis (LEAN): A Multicentre, Double-Blind, Randomised, Placebo-Controlled Phase 2 Study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Goulis, D.G.; Giouleme, O.; Germanidis, G.S.; Goulas, A. Anti-Obesity Medications for the Management of Nonalcoholic Fatty Liver Disease. Curr. Obes. Rep. 2022, 11, 166–179. [Google Scholar] [CrossRef]
- Kojima, M.; Takahashi, H.; Kuwashiro, T.; Tanaka, K.; Mori, H.; Ozaki, I.; Kitajima, Y.; Matsuda, Y.; Ashida, K.; Eguchi, Y.; et al. Glucagon-like Peptide-1 Receptor Agonist Prevented the Progression of Hepatocellular Carcinoma in a Mouse Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 5722. [Google Scholar] [CrossRef]
- Zhou, M.; Mok, M.T.; Sun, H.; Chan, A.W.; Huang, Y.; Cheng, A.S. The Antidiabetic Drug Exenatide, a Glucagon-like Peptide-1 Receptor Agonist, Counteracts Hepatocarcinogenesis through CAMP-PKA-EGFR-STAT3 Axis. Oncogene 2017, 36, 4135–4149. [Google Scholar] [CrossRef]
- van Dalem, J.; Driessen, J.H.M.; Burden, A.M.; Stehouwer, C.D.A.; Klungel, O.H.; de Vries, F.; Brouwers, M.C.G.J. Thiazolidinediones and Glucagon-like Peptide-1 Receptor Agonists and the Risk of Nonalcoholic Fatty Liver Disease: A Cohort Study. Hepatology 2021, 74, 2467–2477. [Google Scholar] [CrossRef]
- Giri, S.R.; Bhoi, B.; Trivedi, C.; Rath, A.; Rathod, R.; Sharma, A.; Ranvir, R.; Kadam, S.; Ingale, K.; Patel, H.; et al. Saroglitazar Suppresses the Hepatocellular Carcinoma Induced by Intraperitoneal Injection of Diethylnitrosamine in C57BL/6 Mice Fed on Choline Deficient, l-Amino Acid- Defined, High-Fat Diet. BMC Cancer 2023, 23, 59. [Google Scholar] [CrossRef]
- Jojima, T.; Wakamatsu, S.; Kase, M.; Iijima, T.; Maejima, Y.; Shimomura, K.; Kogai, T.; Tomaru, T.; Usui, I.; Aso, Y. The SGLT2 Inhibitor Canagliflozin Prevents Carcinogenesis in a Mouse Model of Diabetes and Non-Alcoholic Steatohepatitis-Related Hepatocarcinogenesis: Association with SGLT2 Expression in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 5237. [Google Scholar] [CrossRef]
- Akbari, R.; Behdarvand, T.; Afarin, R.; Yaghooti, H.; Jalali, M.T.; Mohammadtaghvaei, N. Saroglitazar Improved Hepatic Steatosis and Fibrosis by Modulating Inflammatory Cytokines and Adiponectin in an Animal Model of Non-Alcoholic Steatohepatitis. BMC Pharmacol. Toxicol. 2021, 22, 53. [Google Scholar] [CrossRef]
- Nakano, S.; Katsuno, K.; Isaji, M.; Nagasawa, T.; Buehrer, B.; Walker, S.; Wilkison, W.O.; Cheatham, B. Remogliflozin Etabonate Improves Fatty Liver Disease in Diet-Induced Obese Male Mice. J. Clin. Exp. Hepatol. 2015, 5, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Tobita, H.; Sato, S.; Miyake, T.; Ishihara, S.; Kinoshita, Y. Effects of Dapagliflozin on Body Composition and Liver Tests in Patients with Nonalcoholic Steatohepatitis Associated with Type 2 Diabetes Mellitus: A Prospective, Open-Label, Uncontrolled Study. Curr. Ther. Res. Clin. Exp. 2017, 87, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kang, E.S.; Boutari, C.; Rhee, E.-J.; Mantzoros, C.S. Current and Emerging Pharmacological Options for the Treatment of Nonalcoholic Steatohepatitis. Metabolism 2020, 111, 154203. [Google Scholar] [CrossRef] [PubMed]
- Makri, E.S.; Goulas, A.; Polyzos, S.A. Sodium-Glucose Co-Transsporter 2 Inhibitors in Nonalcoholic Fatty Liver Disease. Eur. J. Pharmacol. 2021, 907, 174272. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Adiponectin as a Potential Therapeutic Agent for Nonalcoholic Steatohepatitis. Hepatol. Res. 2010, 40, 446–447. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Adiponectin in Non-Alcoholic Fatty Liver Disease Treatment: Therapeutic Perspectives and Unresolved Dilemmas. Int. J. Clin. Pract. 2011, 65, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.-I.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A Small-Molecule AdipoR Agonist for Type 2 Diabetes and Short Life in Obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef]
- Postow, M.A.; Hellmann, M.D. Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 1165. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ruiz, E.; Minute, L.; Otano, I.; Alvarez, M.; Ochoa, M.C.; Belsue, V.; de Andrea, C.; Rodriguez-Ruiz, M.E.; Perez-Gracia, J.L.; Marquez-Rodas, I.; et al. Prophylactic TNF Blockade Uncouples Efficacy and Toxicity in Dual CTLA-4 and PD-1 Immunotherapy. Nature 2019, 569, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jian, Y.-B. Antitumor Necrosis Factor-α Antibodies as a Novel therapy for Hepatocellular Carcinoma. Exp. Ther. Med. 2018, 16, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Bagalagel, A.; Diri, R.; Noor, A.; Almasri, D.; Bakhsh, H.; Kutbi, H.I.; Al-Gayyar, M.M. Evaluating the Anticancer Activity of Blocking TNF Type 1 Receptors in Thioacetamide-Induced Hepatocellular Carcinoma in a Rat Model. Cureus 2022, 14, e32519. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF-α Is a Potential Therapeutic Target to Overcome Sorafenib Resistance in Hepatocellular Carcinoma. eBioMedicine 2019, 40, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, X.; Bai, X.; Liang, T. Targeting TNFR2: A Novel Breakthrough in the Treatment of Cancer. Front. Oncol. 2022, 12, 862154. [Google Scholar] [CrossRef]
- Pocino, K.; Stefanile, A.; Basile, V.; Napodano, C.; D’Ambrosio, F.; Di Santo, R.; Callà, C.A.M.; Gulli, F.; Saporito, R.; Ciasca, G.; et al. Cytokines and Hepatocellular Carcinoma: Biomarkers of a Deadly Embrace. J. Pers. Med. 2022, 13, 13010005. [Google Scholar] [CrossRef]
- Rico Montanari, N.; Anugwom, C.M.; Boonstra, A.; Debes, J.D. The Role of Cytokines in the Different Stages of Hepatocellular Carcinoma. Cancers 2021, 13, 13194876. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vachliotis, I.D.; Valsamidis, I.; Polyzos, S.A. Tumor Necrosis Factor-Alpha and Adiponectin in Nonalcoholic Fatty Liver Disease-Associated Hepatocellular Carcinoma. Cancers 2023, 15, 5306. https://doi.org/10.3390/cancers15215306
Vachliotis ID, Valsamidis I, Polyzos SA. Tumor Necrosis Factor-Alpha and Adiponectin in Nonalcoholic Fatty Liver Disease-Associated Hepatocellular Carcinoma. Cancers. 2023; 15(21):5306. https://doi.org/10.3390/cancers15215306
Chicago/Turabian StyleVachliotis, Ilias D., Ioannis Valsamidis, and Stergios A. Polyzos. 2023. "Tumor Necrosis Factor-Alpha and Adiponectin in Nonalcoholic Fatty Liver Disease-Associated Hepatocellular Carcinoma" Cancers 15, no. 21: 5306. https://doi.org/10.3390/cancers15215306
APA StyleVachliotis, I. D., Valsamidis, I., & Polyzos, S. A. (2023). Tumor Necrosis Factor-Alpha and Adiponectin in Nonalcoholic Fatty Liver Disease-Associated Hepatocellular Carcinoma. Cancers, 15(21), 5306. https://doi.org/10.3390/cancers15215306