
Correlation Analyses between Histological Staging and Molecular Alterations in Tumor-Derived and Cell-Free DNA of Early-Stage Primary Cutaneous Melanoma


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples and Study Design
2.2. Histopathological and Immunohistochemical Analyses
2.3. DNA Isolation
2.4. StripAssay
2.5. Digital PCR Reactions
2.6. Statistical Analysis
3. Results
3.1. Patients Clinicopathological Characteristics
3.2. Histological Features and Molecular Findings
3.3. Diagnostical Characterization of the dPCR Analyses
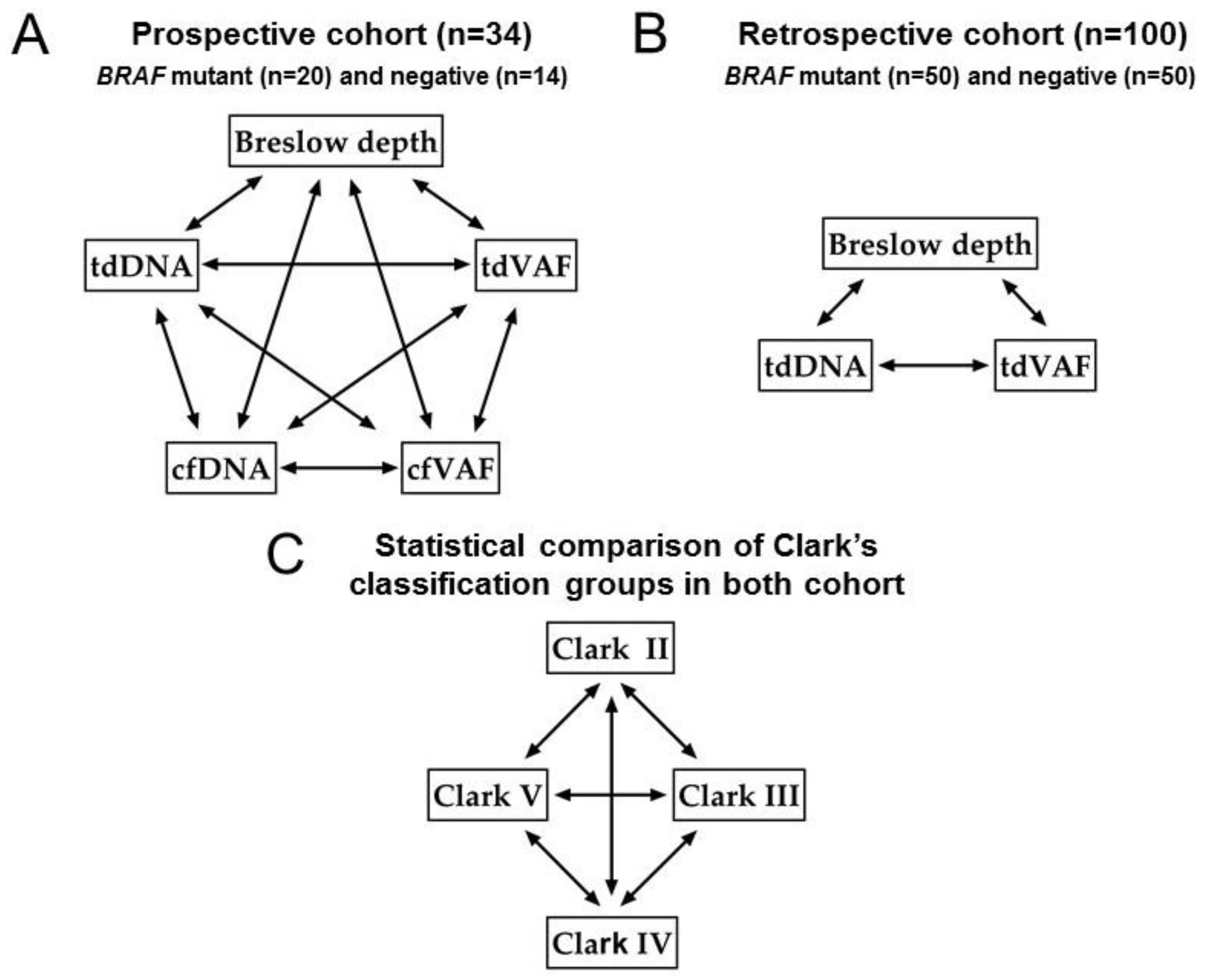
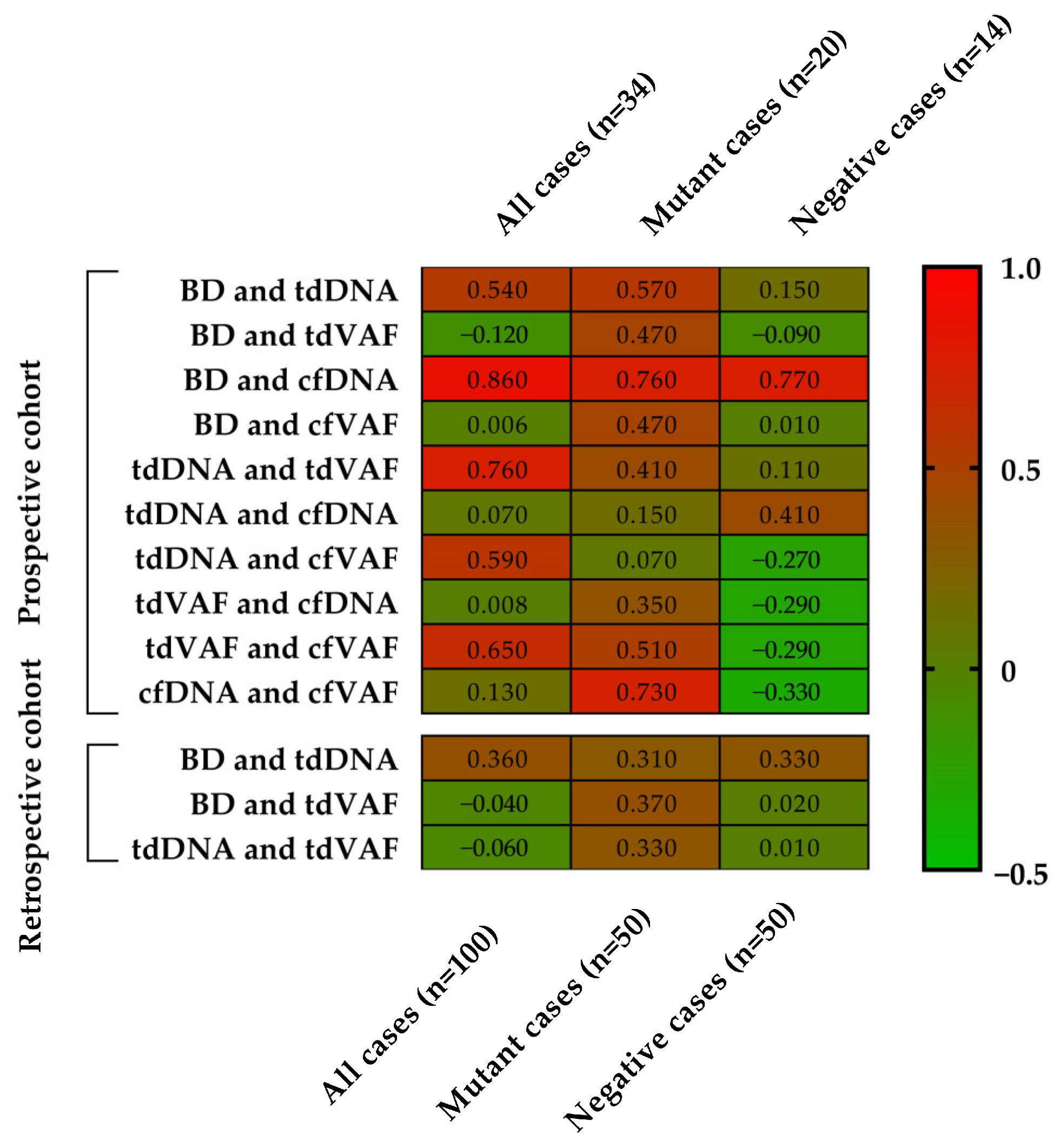
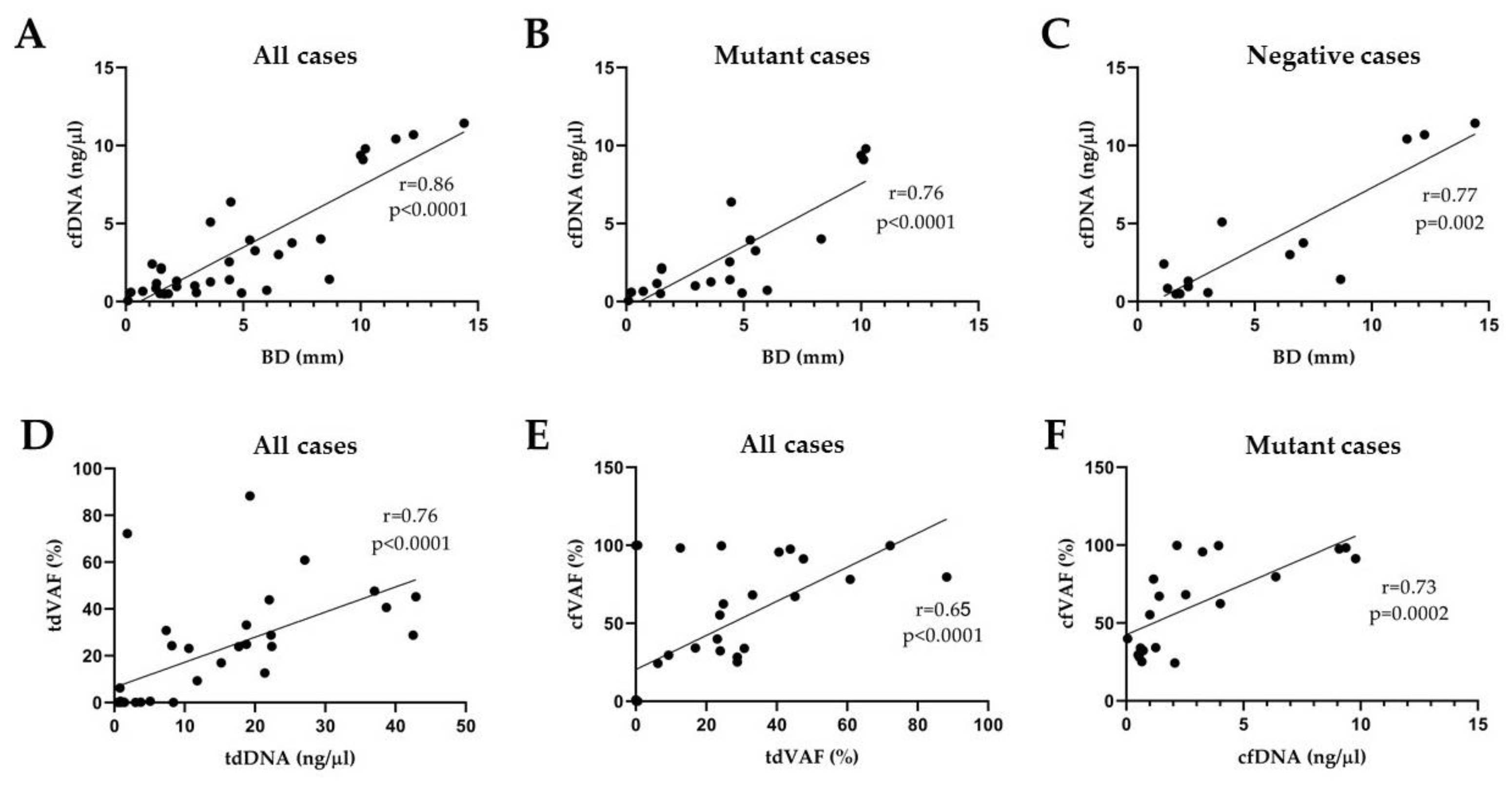
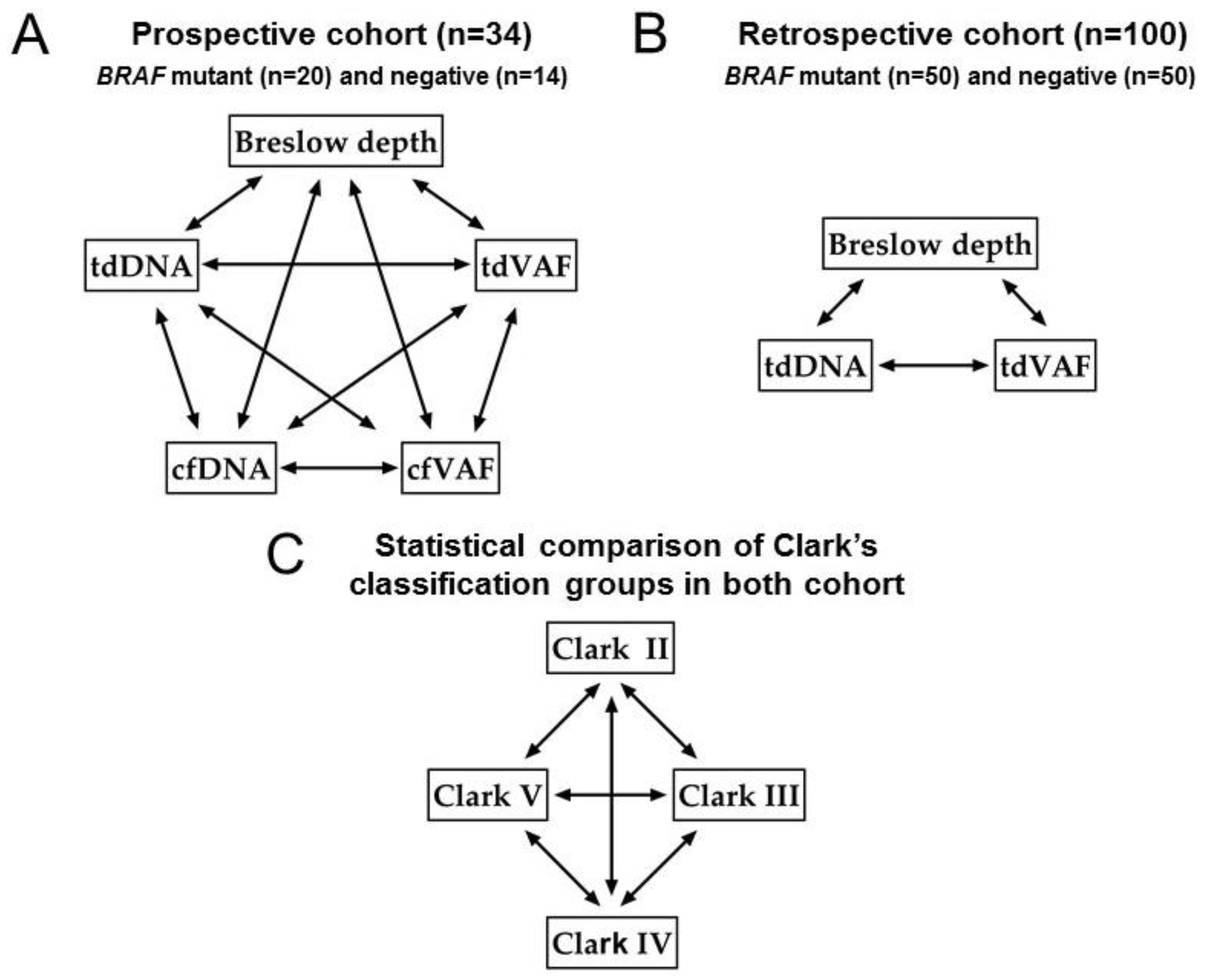
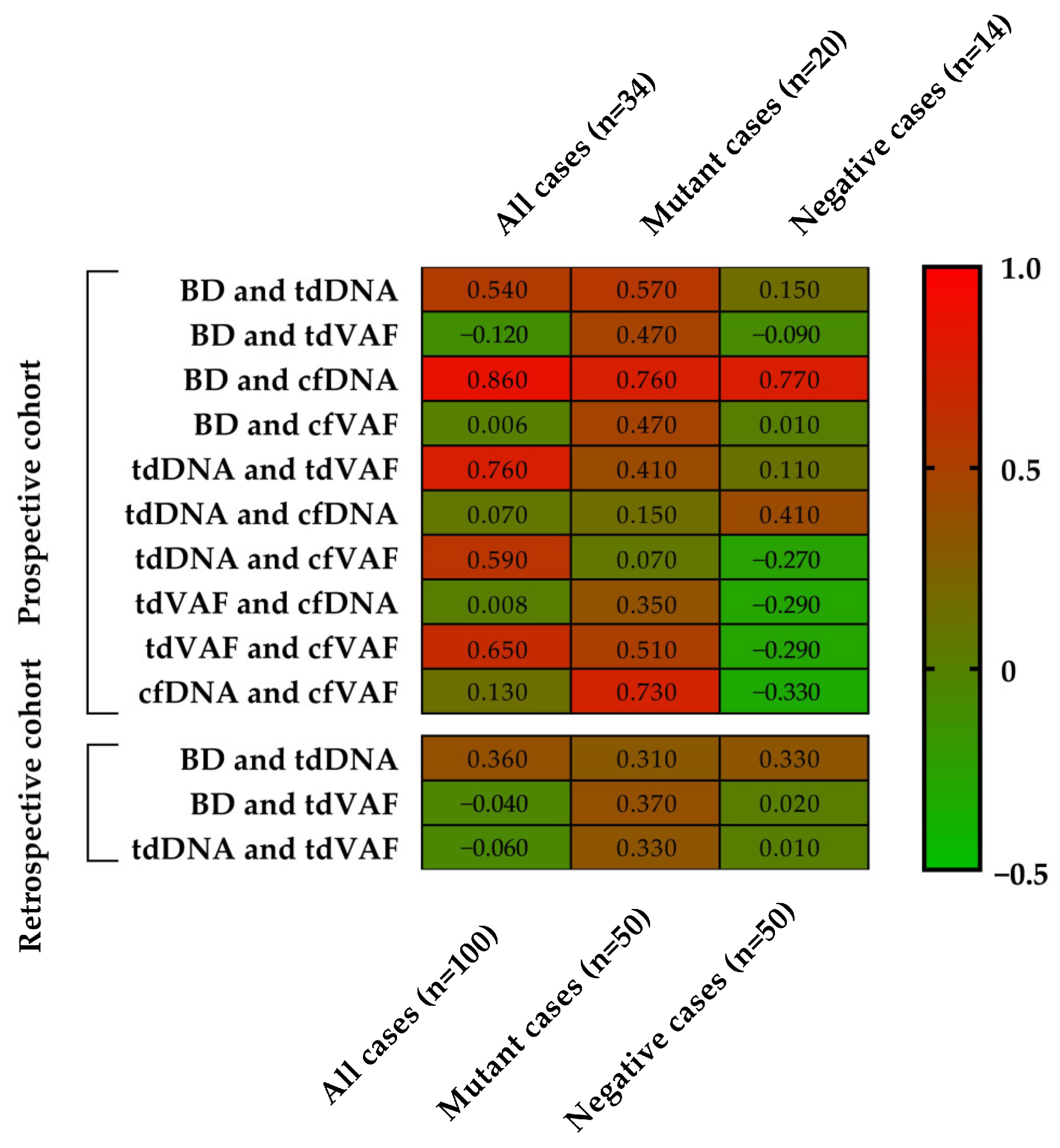
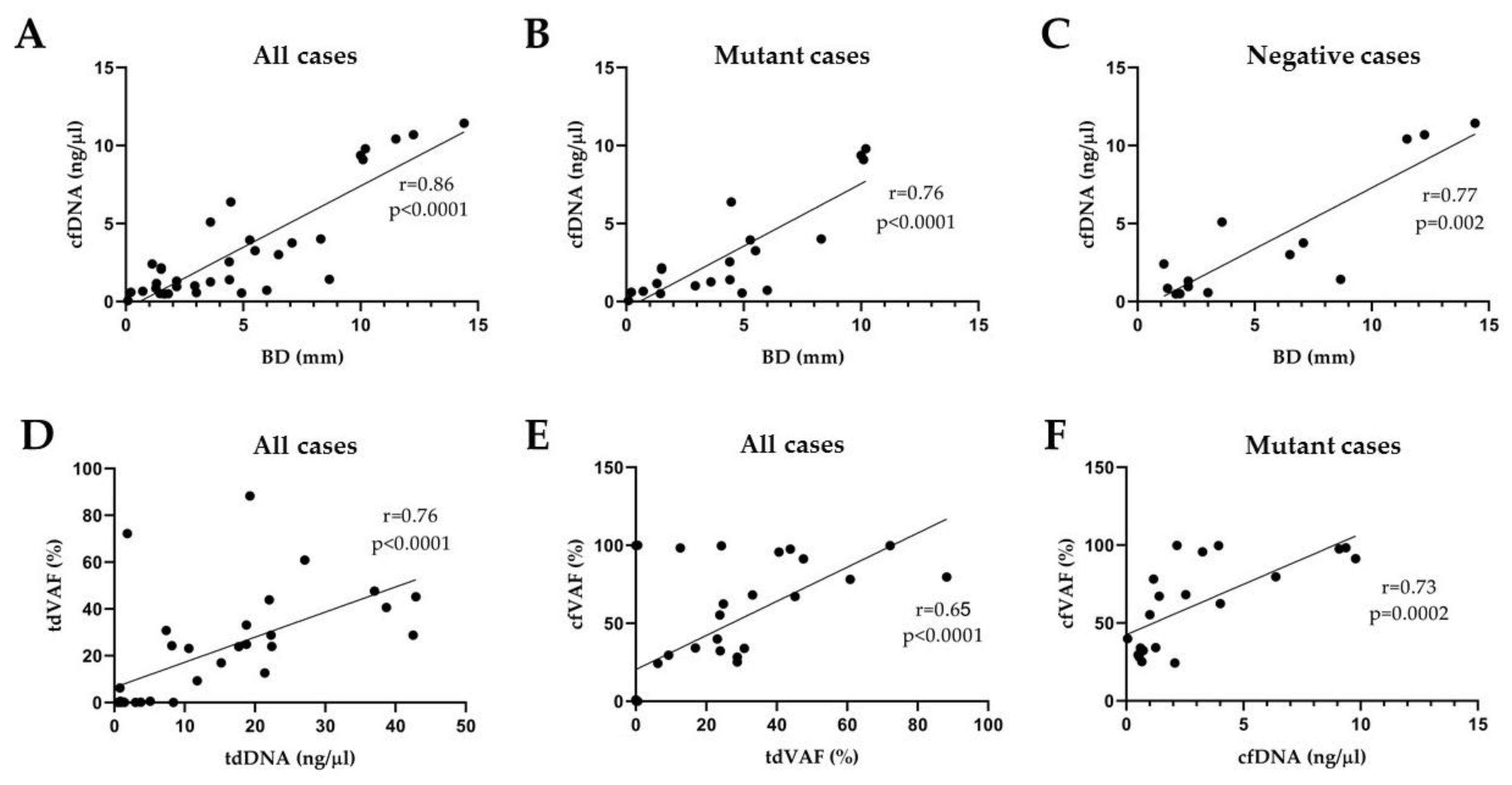
3.4. Correlation Analyses
3.5. Statistical Comparison of Clark’s Classification Groups
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abbas, O.; Miller, D.D.; Bhawan, J. Cutaneous Malignant Melanoma: Update on Diagnostic and Prognostic Biomarkers. Am. J. Dermatopathol. 2014, 36, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef]
- Thanh, D.N.H.; Prasath, V.B.S.; Hieu, L.M.; Hien, N.N. Melanoma Skin Cancer Detection Method Based on Adaptive Principal Curvature, Colour Normalisation and Feature Extraction with the ABCD Rule. J. Digit. Imaging 2020, 33, 574–585. [Google Scholar] [CrossRef]
- Pavri, S.N.; Clune, J.; Ariyan, S.; Narayan, D. Malignant Melanoma: Beyond the Basics. Plast. Reconstr. Surg. 2016, 138, 330e. [Google Scholar] [CrossRef]
- Takahashi, J.; Nagasawa, S. Immunostimulatory Effects of Radiotherapy for Local and Systemic Control of Melanoma: A Review. Int. J. Mol. Sci. 2020, 21, 9324. [Google Scholar] [CrossRef]
- Seedor, R.S.; Orloff, M. Treatment of Metastatic Melanoma in the Elderly. Curr. Oncol. Rep. 2022, 24, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Dhomen, N.; Marais, R. BRAF Signaling and Targeted Therapies in Melanoma. Hematol. Oncol. Clin. N. Am. 2009, 23, 529–545. [Google Scholar] [CrossRef]
- Siroy, A.E.; Boland, G.M.; Milton, D.R.; Roszik, J.; Frankian, S.; Malke, J.; Haydu, L.; Prieto, V.G.; Tetzlaff, M.; Ivan, D.; et al. Beyond BRAFV600: Clinical Mutation Panel Testing by next-Generation Sequencing in Advanced Melanoma. J. Investig. Dermatol. 2015, 135, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.-J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The Role of BRAF V600 Mutation in Melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Frisone, D.; Friedlaender, A.; Malapelle, U.; Banna, G.; Addeo, A. A BRAF New World. Crit. Rev. Oncol. Hematol. 2020, 152, 103008. [Google Scholar] [CrossRef]
- Tímár, J.; Ladányi, A. Molecular Pathology of Skin Melanoma: Epidemiology, Differential Diagnostics, Prognosis and Therapy Prediction. Int. J. Mol. Sci. 2022, 23, 5384. [Google Scholar] [CrossRef]
- Daum, G.; Eisenmann-Tappe, I.; Fries, H.-W.; Troppmair, J.; Rapp, U.R. The Ins and Outs of Raf Kinases. Trends Biochem. Sci. 1994, 19, 474–480. [Google Scholar] [CrossRef]
- Cutler, R.E.; Stephens, R.M.; Saracino, M.R.; Morrison, D.K. Autoregulation of the Raf-1 Serine/Threonine Kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 9214–9219. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Garnett, M.J.; Marais, R. Guilty as Charged: B-RAF Is a Human Oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for Carcinogenesis and Molecular Therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef]
- Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid Biopsies Come of Age: Towards Implementation of Circulating Tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating Liquid Biopsies into the Management of Cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Gaiser, M.R.; von Bubnoff, N.; Gebhardt, C.; Utikal, J.S. Liquid Biopsy to Monitor Melanoma Patients. J. Dtsch. Dermatol. Ges. 2018, 16, 405–414. [Google Scholar] [CrossRef]
- Nikanjam, M.; Kato, S.; Kurzrock, R. Liquid Biopsy: Current Technology and Clinical Applications. J. Hematol. Oncol. 2022, 15, 131. [Google Scholar] [CrossRef]
- Kamińska, P.; Buszka, K.; Zabel, M.; Nowicki, M.; Alix-Panabières, C.; Budna-Tukan, J. Liquid Biopsy in Melanoma: Significance in Diagnostics, Prediction and Treatment Monitoring. Int. J. Mol. Sci. 2021, 22, 9714. [Google Scholar] [CrossRef] [PubMed]
- Váraljai, R.; Elouali, S.; Lueong, S.S.; Wistuba-Hamprecht, K.; Seremet, T.; Siveke, J.T.; Becker, J.C.; Sucker, A.; Paschen, A.; Horn, P.A.; et al. The Predictive and Prognostic Significance of Cell-Free DNA Concentration in Melanoma. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Dube, S.; Qin, J.; Ramakrishnan, R. Mathematical Analysis of Copy Number Variation in a DNA Sample Using Digital PCR on a Nanofluidic Device. PLoS ONE 2008, 3, e2876. [Google Scholar] [CrossRef]
- Quan, P.-L.; Sauzade, M.; Brouzes, E. DPCR: A Technology Review. Sensors 2018, 18, 1271. [Google Scholar] [CrossRef]
- Mao, X.; Liu, C.; Tong, H.; Chen, Y.; Liu, K. Principles of Digital PCR and Its Applications in Current Obstetrical and Gynecological Diseases. Am. J. Transl. Res. 2019, 11, 7209–7222. [Google Scholar]
- Whale, A.S.; Cowen, S.; Foy, C.A.; Huggett, J.F. Methods for Applying Accurate Digital PCR Analysis on Low Copy DNA Samples. PLoS ONE 2013, 8, e58177. [Google Scholar] [CrossRef]
- Thierry, A.R. A Targeted Q-PCR-Based Method for Point Mutation Testing by Analyzing Circulating DNA for Cancer Management Care. In Clinical Applications of PCR; Luthra, R., Singh, R.R., Patel, K.P., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 1–16. ISBN 978-1-4939-3360-0. [Google Scholar]
- Büttner, P.; Garbe, C.; Bertz, J.; Burg, G.; d’Hoedt, B.; Drepper, H.; Guggenmoos-Holzmann, I.; Lechner, W.; Lippold, A.; Orfanos, C.E. Primary Cutaneous Melanoma. Optimized Cutoff Points of Tumor Thickness and Importance of Clark’s Level for Prognostic Classification. Cancer 1995, 75, 2499–2506. [Google Scholar] [CrossRef] [PubMed]
- Bunnell, A.M.; Nedrud, S.M.; Fernandes, R.P. Classification and Staging of Melanoma in the Head and Neck. Oral Maxillofac. Surg. Clin. N. Am. 2022, 34, 221–234. [Google Scholar] [CrossRef]
- Puckett, Y.; Wilson, A.M.; Farci, F.; Thevenin, C. Melanoma Pathology. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Sefrioui, D.; Sarafan-Vasseur, N.; Beaussire, L.; Baretti, M.; Gangloff, A.; Blanchard, F.; Clatot, F.; Sabourin, J.-C.; Sesboüé, R.; Frebourg, T.; et al. Clinical Value of Chip-Based Digital-PCR Platform for the Detection of Circulating DNA in Metastatic Colorectal Cancer. Dig. Liver Dis. 2015, 47, 884–890. [Google Scholar] [CrossRef] [PubMed]
- García-Foncillas, J.; Tabernero, J.; Élez, E.; Aranda, E.; Benavides, M.; Camps, C.; Jantus-Lewintre, E.; López, R.; Muinelo-Romay, L.; Montagut, C.; et al. Prospective Multicenter Real-World RAS Mutation Comparison between OncoBEAM-Based Liquid Biopsy and Tissue Analysis in Metastatic Colorectal Cancer. Br. J. Cancer 2018, 119, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus Tissue Biopsy for Detecting Acquired Resistance and Tumor Heterogeneity in Gastrointestinal Cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Esagian, S.M.; Grigoriadou, G.Ι.; Nikas, I.P.; Boikou, V.; Sadow, P.M.; Won, J.-K.; Economopoulos, K.P. Comparison of Liquid-Based to Tissue-Based Biopsy Analysis by Targeted next Generation Sequencing in Advanced Non-Small Cell Lung Cancer: A Comprehensive Systematic Review. J. Cancer Res. Clin. Oncol. 2020, 146, 2051–2066. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Lee, J.H.; Rizos, H. Monitoring Melanoma Using Circulating Free DNA. Am. J. Clin. Dermatol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Ricciardi, E.; Giordani, E.; Ziccheddu, G.; Falcone, I.; Giacomini, P.; Fanciulli, M.; Russillo, M.; Cerro, M.; Ciliberto, G.; Morrone, A.; et al. Metastatic Melanoma: Liquid Biopsy as a New Precision Medicine Approach. Int. J. Mol. Sci. 2023, 24, 4014. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Prospective Cohort | Retrospective Cohort | |||||||
|---|---|---|---|---|---|---|---|---|
| Clark II (n = 4) | Clark III (n = 14) | Clark IV (n = 10) | Clark V (n = 6) | Clark II (n = 9) | Clark III (n = 41) | Clark IV (n = 37) | Clark V (n = 13) | |
| Age (year) | 54 (42–66) | 59 (42–78) | 59 (37–89) | 72 (36–100) | 62 (35–82) | 62 (29–85) | 66 (28–83) | 66 (50–88) |
| Breslow depth (mm) | 0.2 (0.1–0.2) | 3.5 (0.7–10.1) | 5.4 (1.5–11.5) | 9.1 (2.2–14.4) | 1.1 (0.3–2.2) | 2.7 (0.1–9.2) | 5.1 (1.1–15.3) | 9.4 (1.8–35) |
| tdDNA concentration (ng/µL) | 9 (7.4–10.6) | 14.4 (0.5–42.9) | 15 (0.4–38.7) | 6.2 (0.7–21.4) | 11.1 (0.4–26.3) | 19.5 (1.2–53) | 17.7 (1.6–46.8) | 19.8 (3.4–49.7) |
| all tdVAF of BRAF p.V600E (%) | 26.9 (23.1–30.8) | 24.6 (0–72.2) | 22.7 (0–88.3) | 2.1 (0–12.6) | 4.5 (0–14) | 18.5 (0–80.5) | 15.7 (0–82.7) | 12.3 (0–51.6) |
| mutant tdVAF of BRAF p.V600E (%) | 26.9 (23.1–30.8) | 35.6 (6.2–72.2) | 37.8 (9.3–88.3) | 12.6 | 7.2 (0–14) | 31.5 (0–80.5) | 34.1 (6.2–82.7) | 31.8 (0–51.6) |
| negative tdVAF of BRAF p.V600E (%) | – | 0.24 (0–0.6) | 0.1 (0–0.2) | 0 (0–0.1) | 0.1 (0–0.2) | 0.1 (0–0.6) | 1.6 (0–22) | 0.2 (0–0.6) |
| cfDNA concentration (ng/µL) | 0.3 (0.1–0.6) | 2.3 (0.5–9.1) | 3.7 (0.5–10.4) | 6.5 (1.4–11.4) | - | |||
| all cfVAF of BRAF p.V600E (%) | 37 (33.9–39.9) | 44.1 (0–99.8) | 36.4 (0–95.7) | 16.4 (0–98.3) | - | |||
| mutant cfVAF of BRAF p.V600E (%) | 37 (33.9–39.9) | 64.2 (24.3–99.8) | 60.4 (29.5–95.7) | 98.3 | - | |||
| negative cfVAF of BRAF p.V600E (%) | – | 0 (0–0.1) | 0.4 (0–0.9) | 0 (0–0.1) | - | |||
| Prospective Cohort | Retrospective Cohort | |||||
|---|---|---|---|---|---|---|
| All (n = 34) | Mutant Cases (n = 20) | Negative Cases (n = 14) | All (n = 100) | Mutant Cases (n = 50) | Negative Cases (n = 50) | |
| Age (year) | 61 (36–100) | 55 (36–78) | 71 (45–100) | 64 (28–88) | 60 (35–82) | 67 (28–88) |
| Breslow depth (mm) | 4.83 (0.09–14.41) | 4.35 (0.09–10.2) | 5.51 (1.12–14.41) | 4.27 (0–35) | 4.21 (0.1–35) | 4.33 (0–15.1) |
| tdDNA concentration (ng/µL) | 18.75 (0.48–59.43) | 20.34 (0.8–42.9) | 2.06 (0.48–59.43) | 17.39 (0.19–53) | 14.03 (0.58–45.4) | 20.75 (0.19–53) |
| tdVAF of BRAF p.V600E (%) | 20.2 (0–88.25) | 34.26 (6.23–88.25) | 0.11 (0–0.55) | 15.33 (0–82.74) | 31.16 (0–82.74) | 0.74 (0–21.96) |
| cfDNA concentration (ng/µL) | 3.33 (0.05–11.43) | 3.02 (0.05–9.79) | 3.78 (0.48–11.43) | - | ||
| cfVAF of BRAF p.V600E (%) | 36.55 (0–99.8) | 62.05 (24.27–99.8) | 0.12 (0–0.93) | - | ||
| All Cases (n = 134) | Prospective Cases (n = 34) | Retrospective Cases (n = 100) | |
|---|---|---|---|
| Sensitivity (%) | 98.6 | 100 | 99 |
| Specificity (%) | 97 | 100 | 98 |
| Positive predictive value (%) | 97.2 | 100 | 98 |
| Negative predictive value (%) | 98.5 | 100 | 99 |
| Prospective Cohort (n = 34) | Retrospective Cohort (n = 100) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| BD | tdDNA | tdVAF | cfDNA | cfVAF | BD | tdDNA | tdVAF | ||
| All cases | Clark II vs. III | 0.0131 | n.s. | n.s. | n.s. | n.s. | 0.0022 | n.s. | n.s. |
| Clark II vs. IV | 0.0303 | n.s. | n.s. | n.s. | n.s. | <0.0001 | n.s. | n.s. | |
| Clark II vs. V | n.s. | n.s. | 0.0357 | n.s. | n.s. | <0.0001 | n.s. | n.s. | |
| Clark III vs. IV | n.s. | n.s. | n.s. | n.s. | n.s. | <0.0001 | n.s. | n.s. | |
| Clark III vs. Clark V | 0.0063 | n.s. | 0.0048 | 0.017 | n.s. | <0.0001 | n.s. | n.s. | |
| Clark IV vs. Clark V | n.s. | n.s. | n.s. | n.s. | n.s. | 0.0168 | n.s. | n.s. | |
| Mutant cases | Clark II vs. III | 0.0256 | n.s. | n.s. | 0.0256 | n.s. | 0.001 | n.s. | 0.0127 |
| Clark II vs. IV | n.s. | n.s. | n.s. | n.s. | n.s. | <0.0001 | n.s. | 0.0019 | |
| Clark II vs. V | n.s. | n.s. | n.s. | n.s. | n.s. | 0.0079 | n.s. | n.s. | |
| Clark III vs. IV | n.s. | n.s. | n.s. | 0.0002 | n.s. | n.s. | n.s. | n.s. | |
| Clark III vs. Clark V | n.s. | n.s. | n.s. | n.s. | n.s. | <0.0001 | n.s. | n.s. | |
| Clark IV vs. Clark V | n.s. | n.s. | n.s. | n.s. | n.s. | 0.0039 | n.s. | n.s. | |
| Negative cases | Clark II vs. III | - | - | - | - | - | n.s. | n.s. | n.s. |
| Clark II vs. IV | - | - | - | - | - | 0.0069 | n.s. | n.s. | |
| Clark II vs. V | - | - | - | - | - | 0.0485 | n.s. | n.s. | |
| Clark III vs. IV | n.s. | n.s. | n.s. | n.s. | n.s. | <0.0001 | n.s. | n.s. | |
| Clark III vs. Clark V | 0.0238 | n.s. | n.s. | n.s. | n.s. | 0.0018 | n.s. | n.s. | |
| Clark IV vs. Clark V | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Csoma, S.L.; Madarász, K.; Chang Chien, Y.C.; Emri, G.; Bedekovics, J.; Méhes, G.; Mokánszki, A. Correlation Analyses between Histological Staging and Molecular Alterations in Tumor-Derived and Cell-Free DNA of Early-Stage Primary Cutaneous Melanoma. Cancers 2023, 15, 5141. https://doi.org/10.3390/cancers15215141
Csoma SL, Madarász K, Chang Chien YC, Emri G, Bedekovics J, Méhes G, Mokánszki A. Correlation Analyses between Histological Staging and Molecular Alterations in Tumor-Derived and Cell-Free DNA of Early-Stage Primary Cutaneous Melanoma. Cancers. 2023; 15(21):5141. https://doi.org/10.3390/cancers15215141
Chicago/Turabian StyleCsoma, Szilvia Lilla, Kristóf Madarász, Yi Che Chang Chien, Gabriella Emri, Judit Bedekovics, Gábor Méhes, and Attila Mokánszki. 2023. "Correlation Analyses between Histological Staging and Molecular Alterations in Tumor-Derived and Cell-Free DNA of Early-Stage Primary Cutaneous Melanoma" Cancers 15, no. 21: 5141. https://doi.org/10.3390/cancers15215141
APA StyleCsoma, S. L., Madarász, K., Chang Chien, Y. C., Emri, G., Bedekovics, J., Méhes, G., & Mokánszki, A. (2023). Correlation Analyses between Histological Staging and Molecular Alterations in Tumor-Derived and Cell-Free DNA of Early-Stage Primary Cutaneous Melanoma. Cancers, 15(21), 5141. https://doi.org/10.3390/cancers15215141