
Auranofin Induces Lethality Driven by Reactive Oxygen Species in High-Grade Serous Ovarian Cancer Cells


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Cell Lines
2.2. Cellular Vitality
2.3. Cellular Viability
2.4. Clonogenic Survival
2.5. Cell Cycle Distribution
2.6. Protein Lysate Preparation and Western Blot Analysis
2.7. Detection of DNA Damage
2.8. Detection of Annexin-V Binding
2.9. Measurement of Caspase-3/7 Activation
2.10. Treatment with a Caspase Inhibitor
2.11. Detection of Mitochondrial Membrane Depolarization
2.12. Assessment of Intracellular ROS Levels
2.13. Drug Interaction between Auranofin and L-BSO
2.14. Measurement of TrxR Activity
2.15. In Vitro Analysis of Total GSH
2.16. Statistics
3. Results
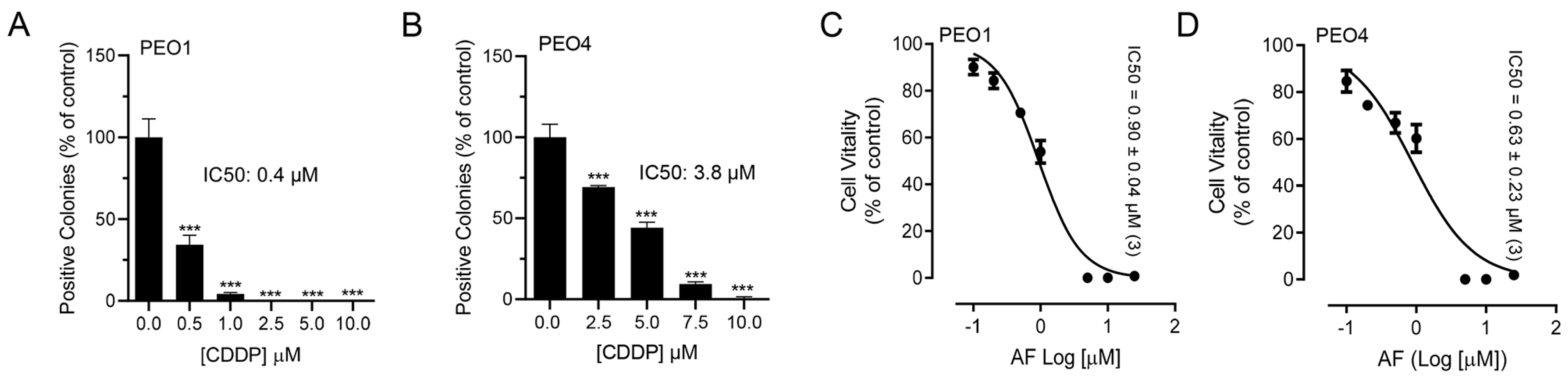
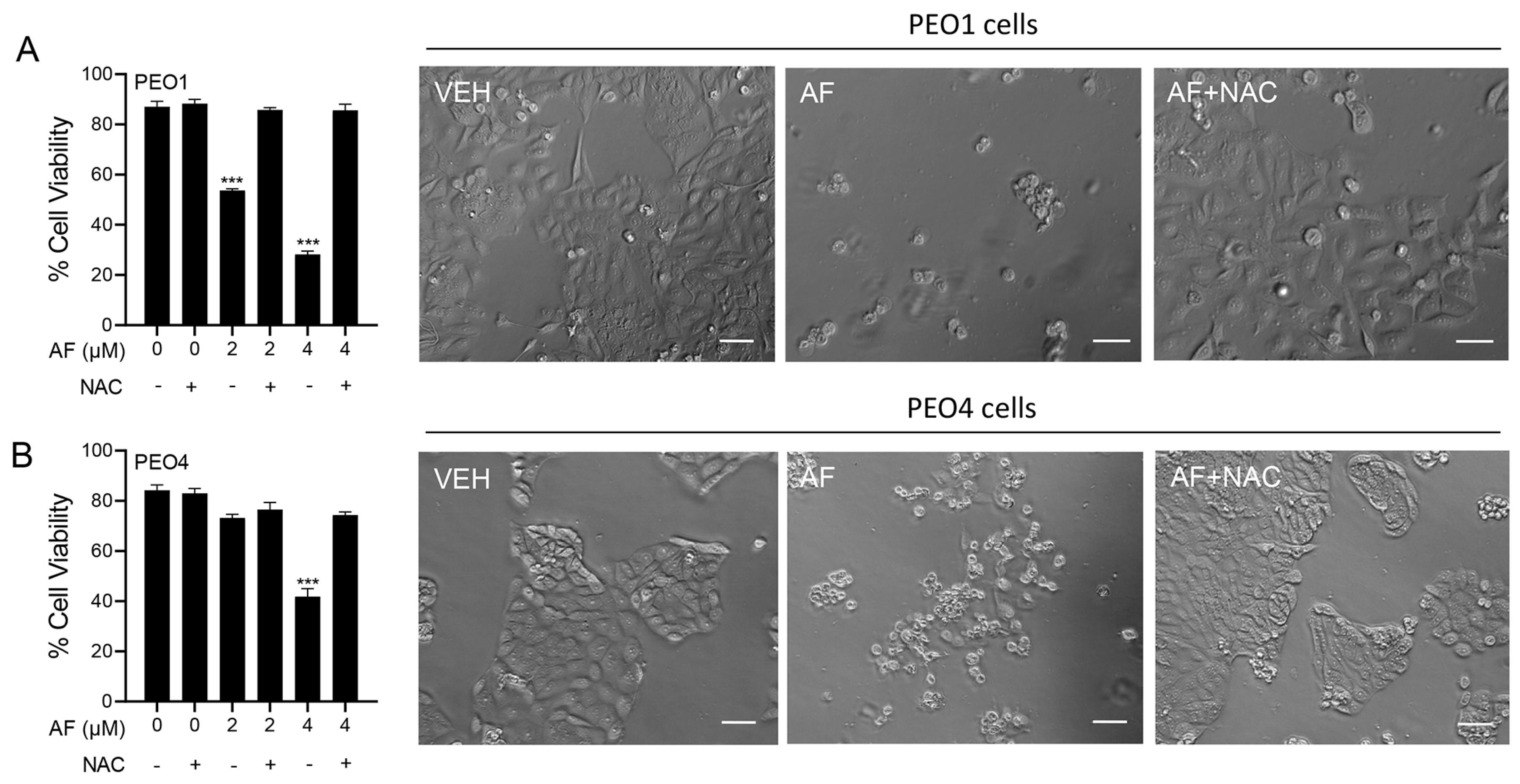
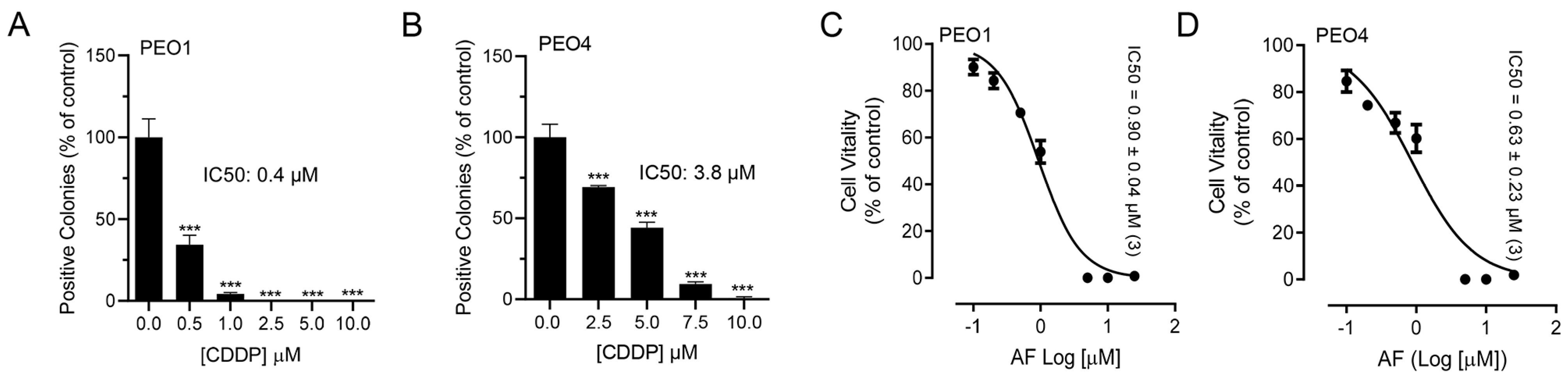
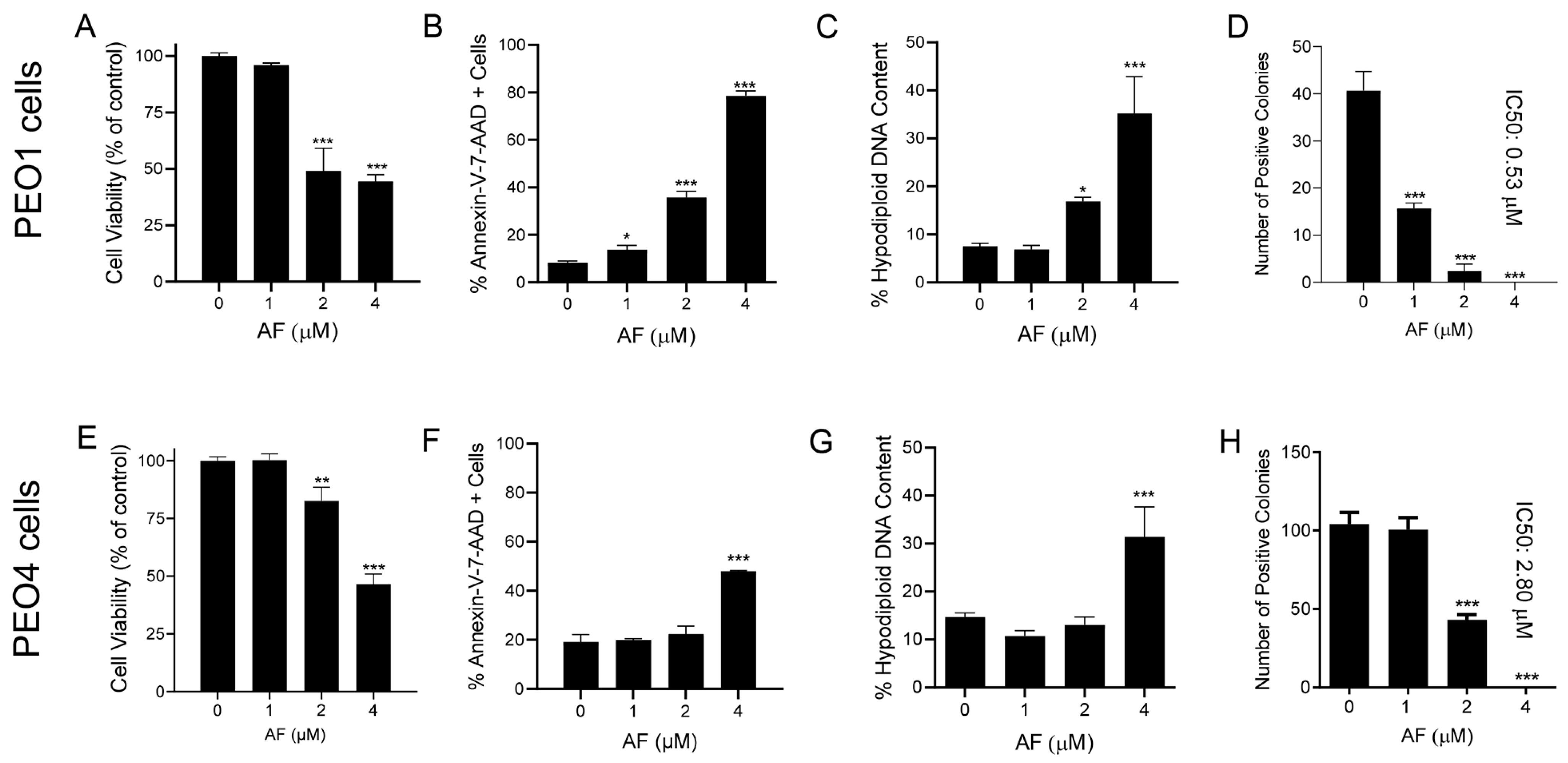
3.1. Auranofin Reduces the Vitality of HGSOC Cells Regardless of Their Sensitivities to Cisplatin
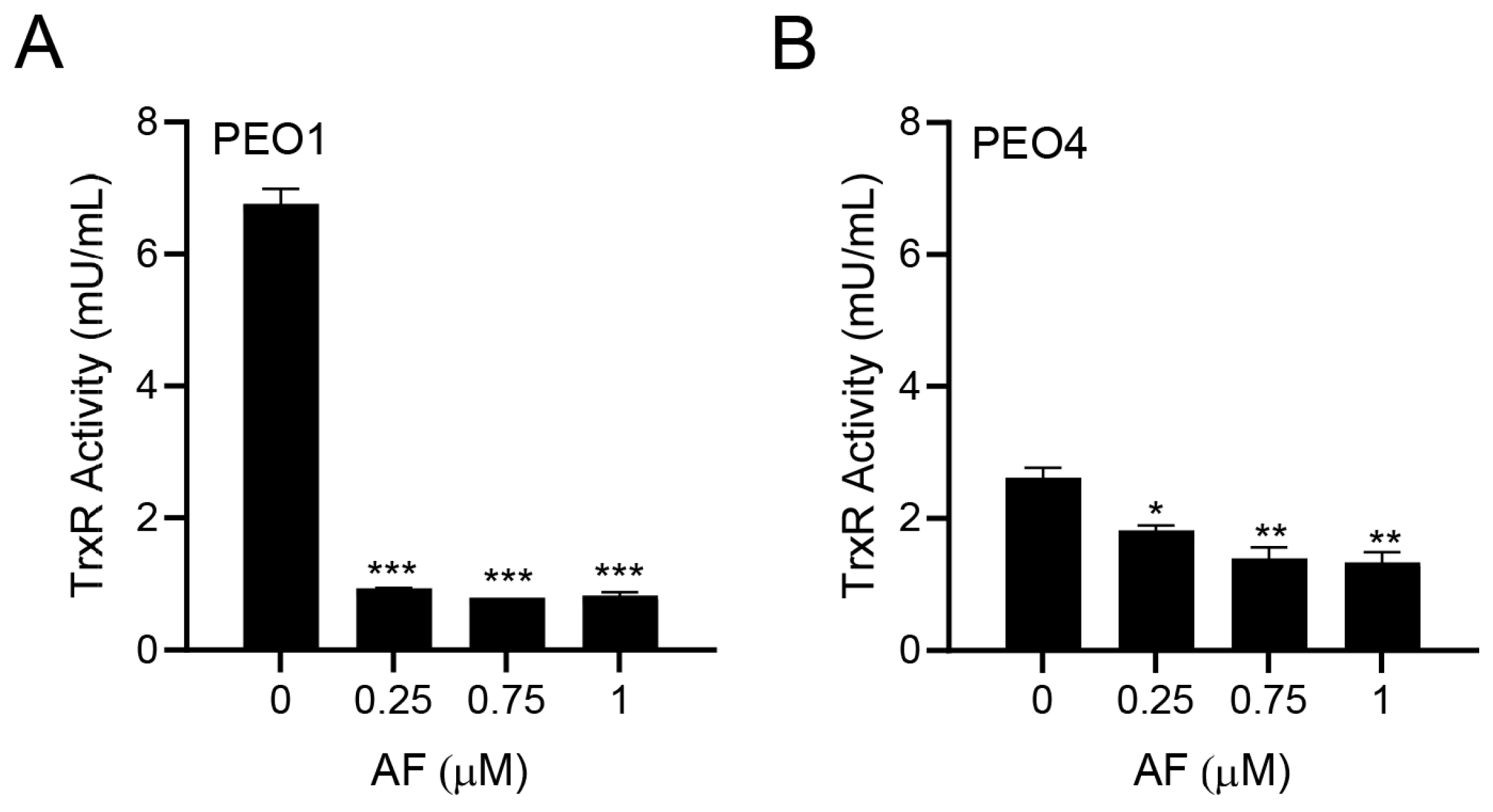
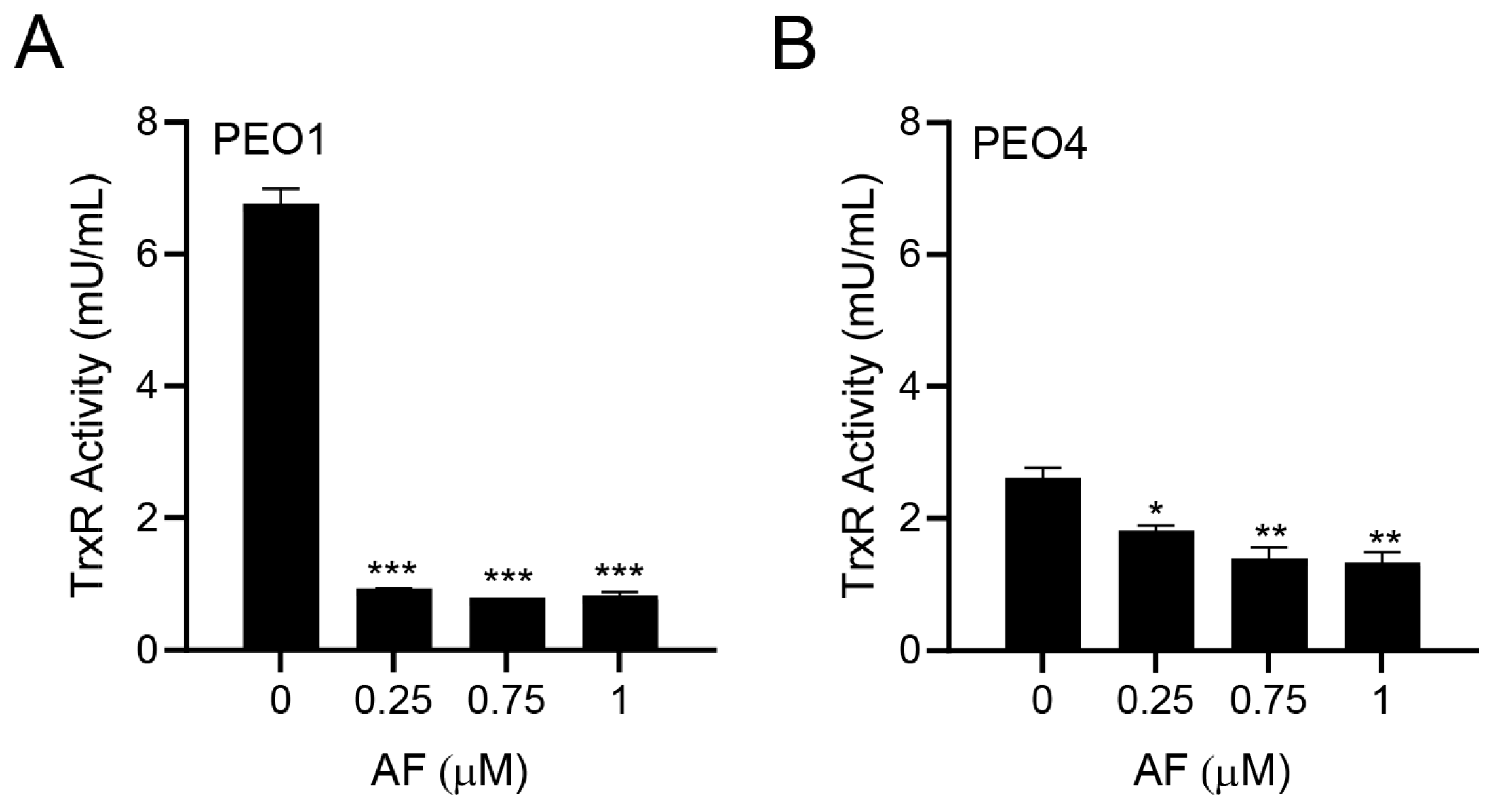
3.2. Auranofin Inhibits TrxR Activity
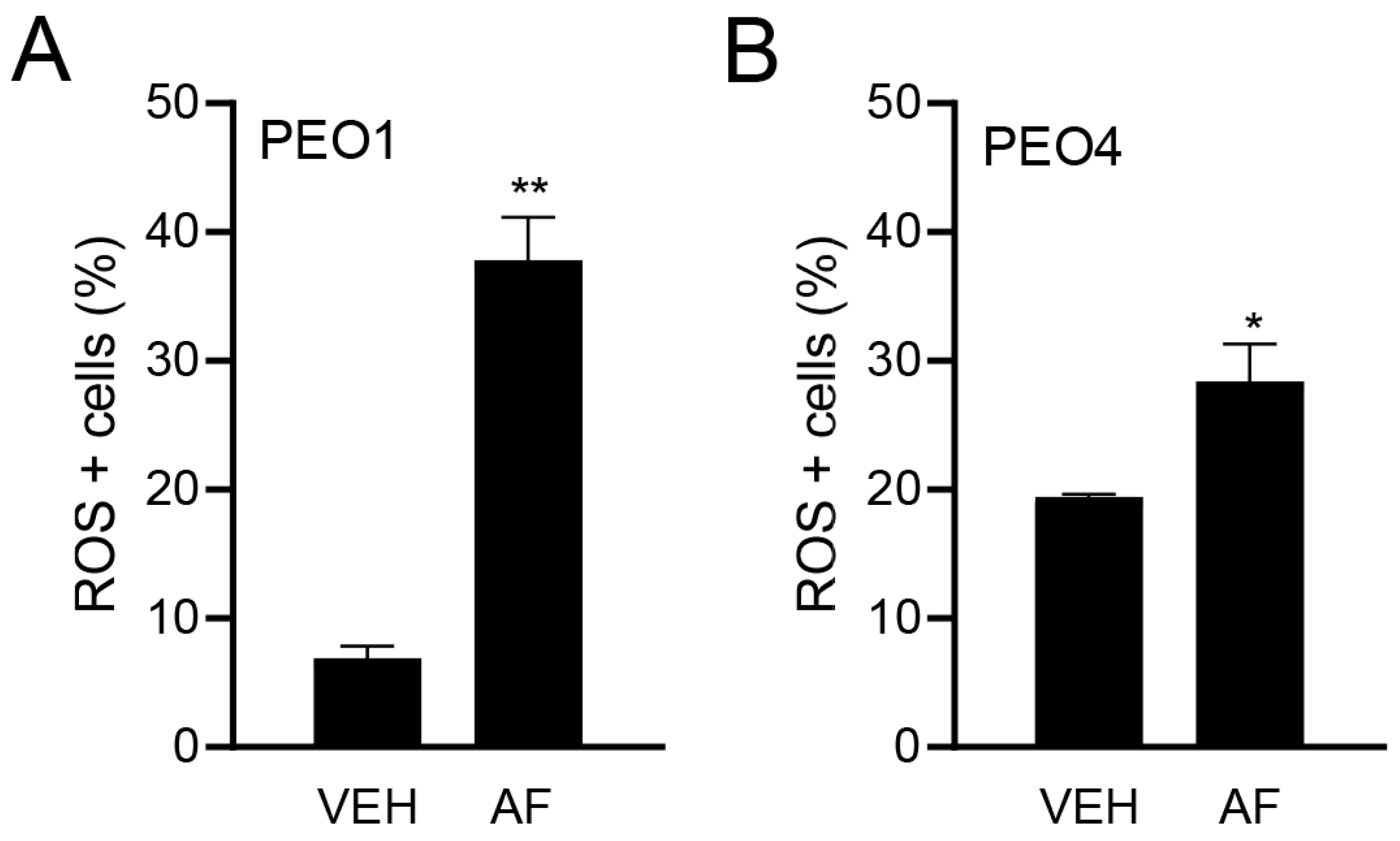
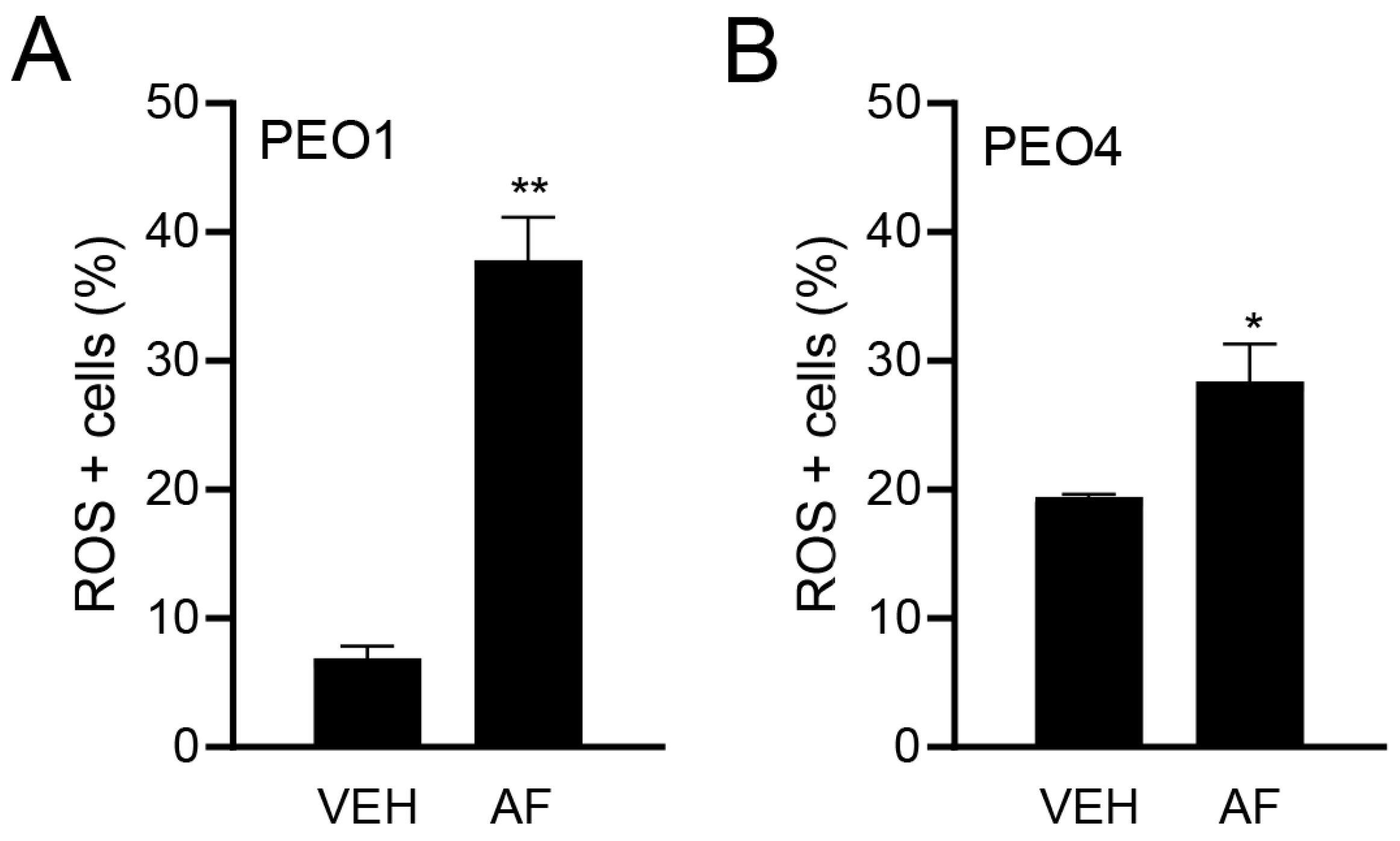
3.3. Auranofin Triggers the Accumulation of Reactive Oxygen Species
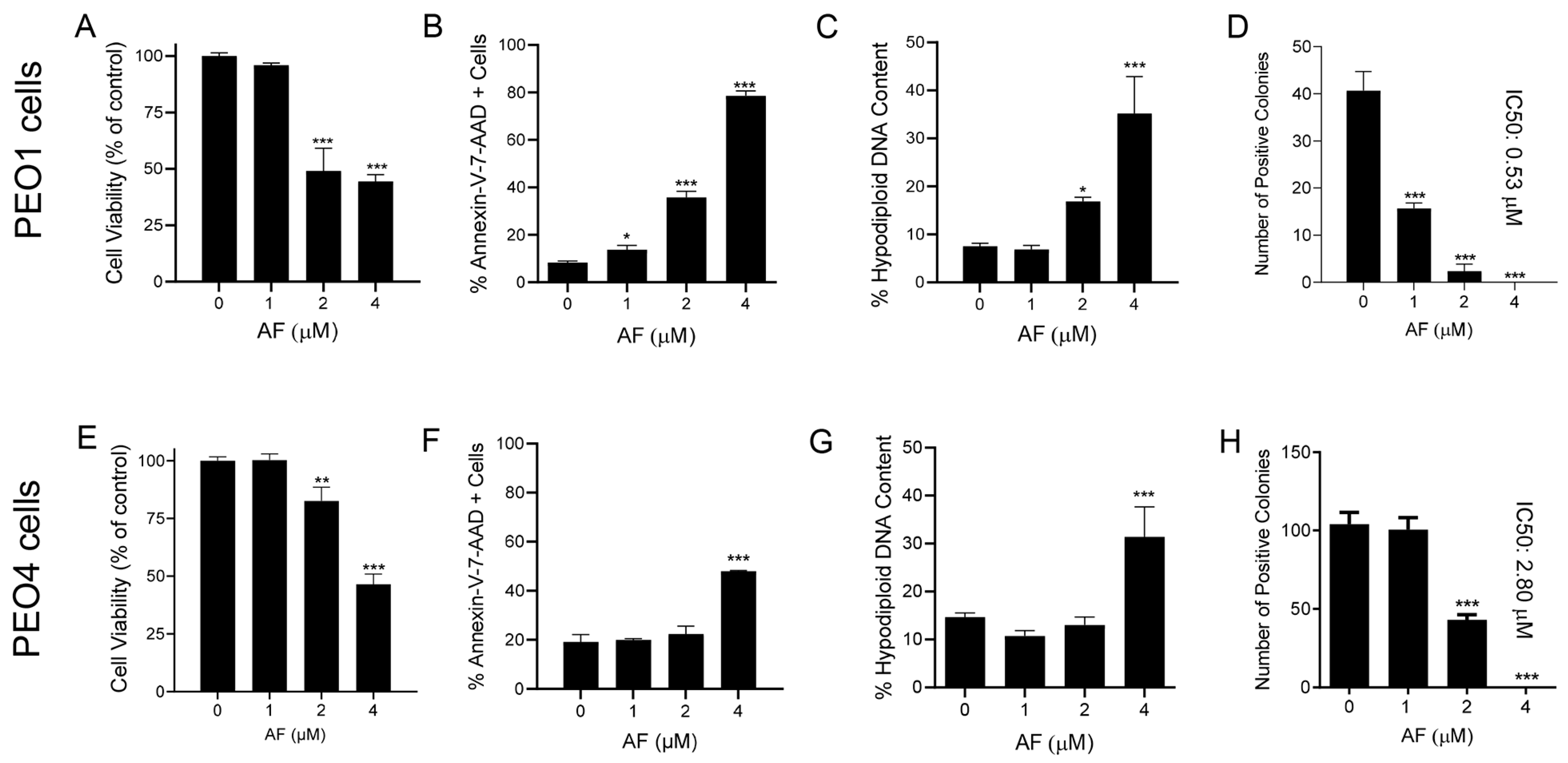
3.4. Auranofin Kills HGSOC Cells in Association with Induction of Apoptosis
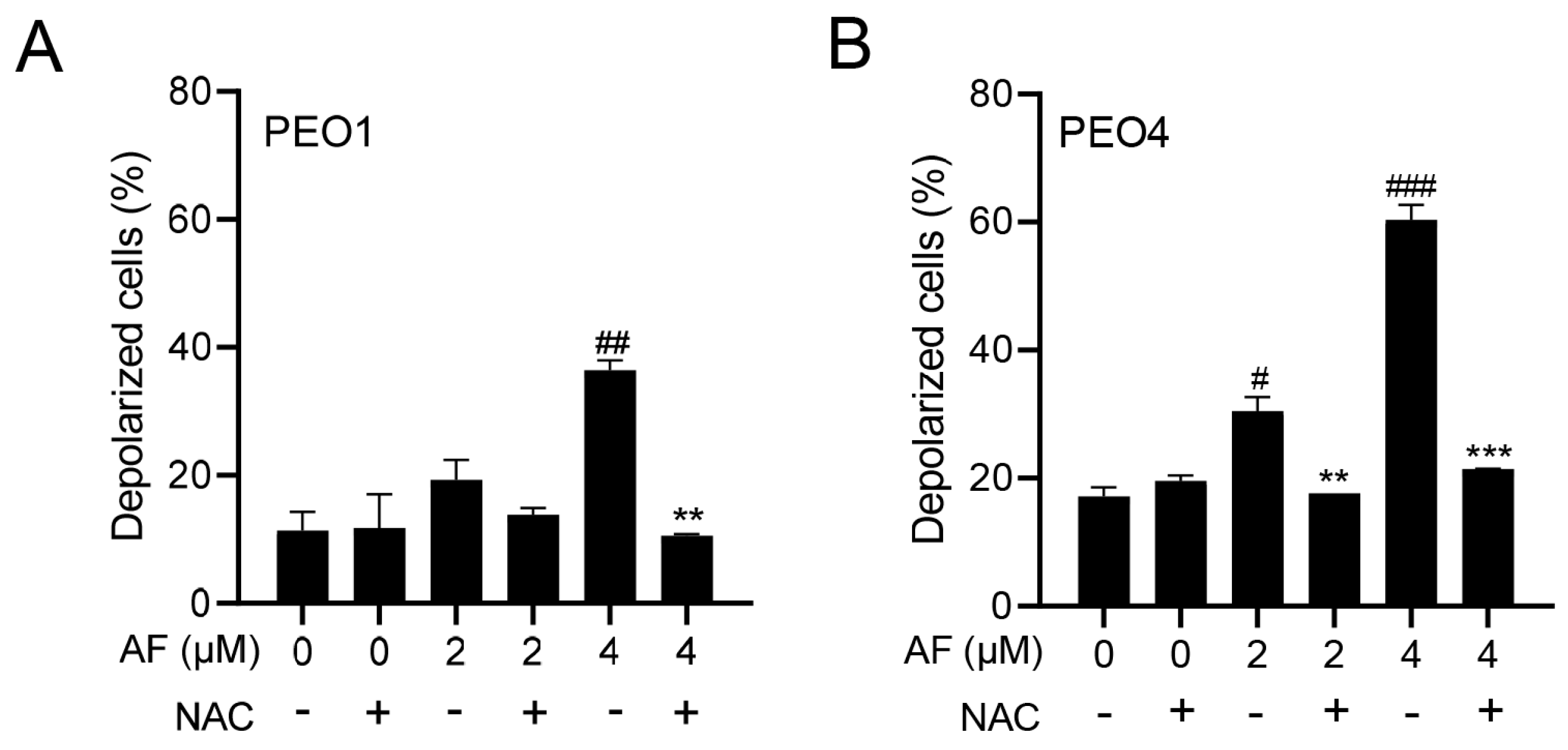
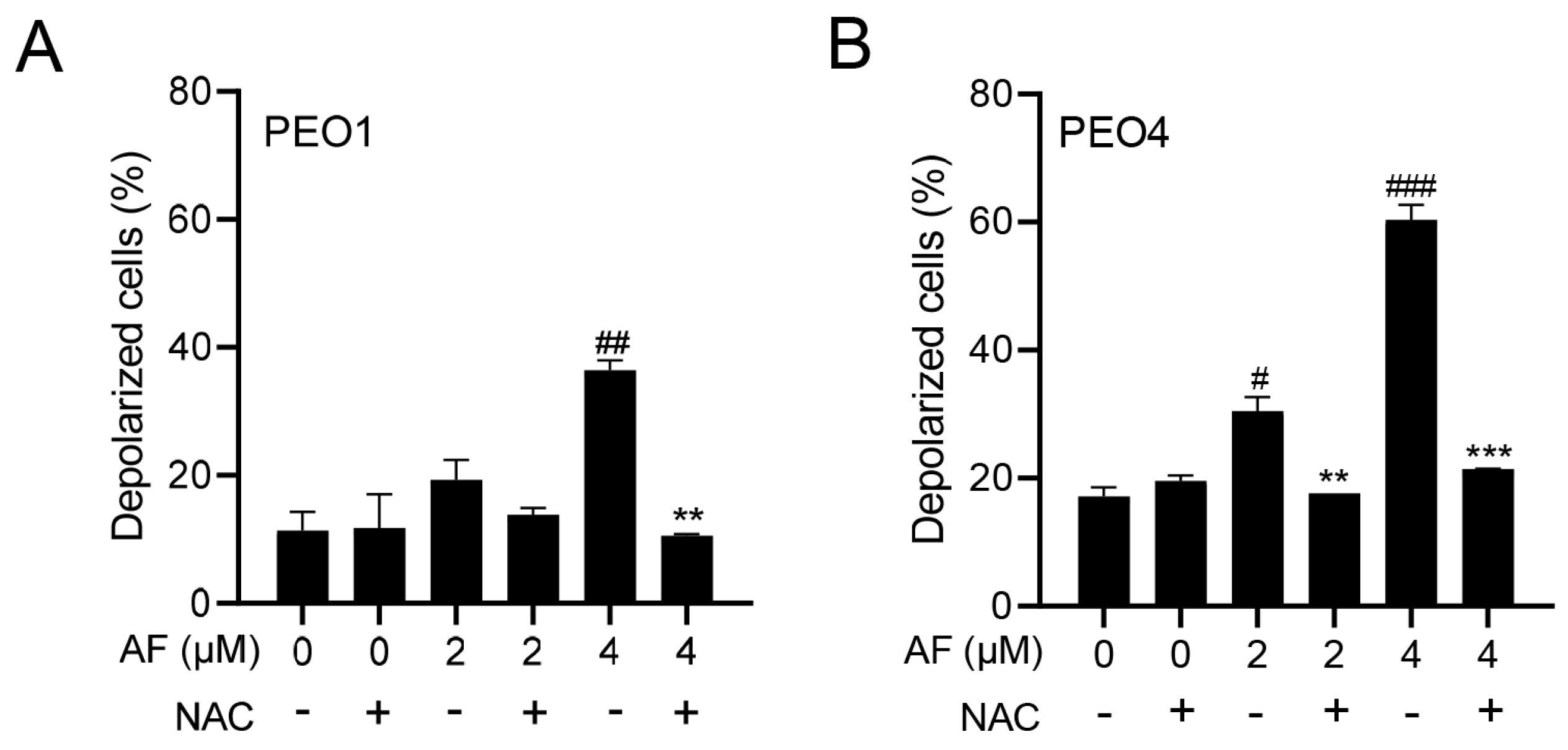
3.5. Auranofin Induces Dissipation of the Mitochondrial Potential, a Phenomenon That Is Prevented by the Presence of the ROS Scavenger N-acetyl Cysteine
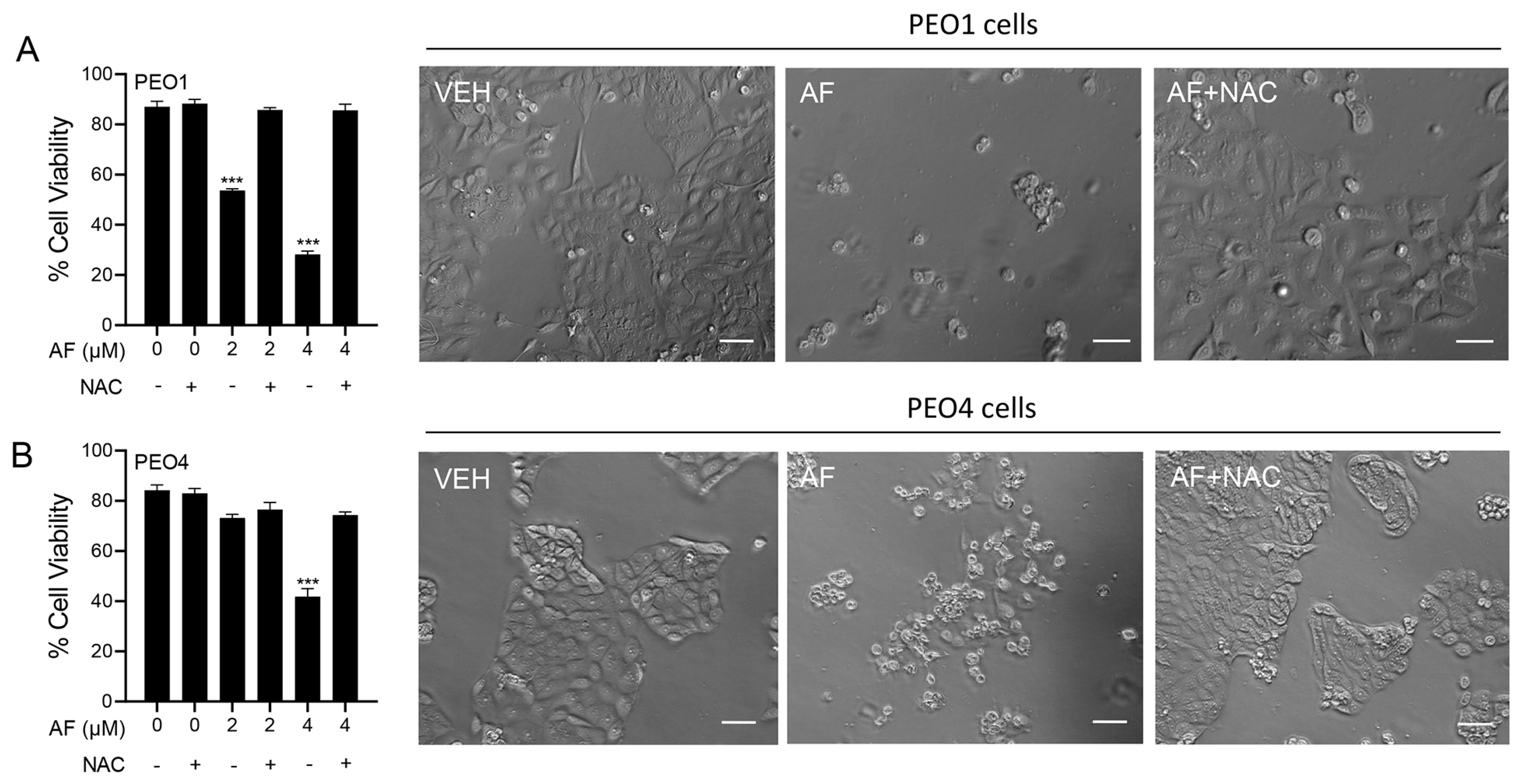
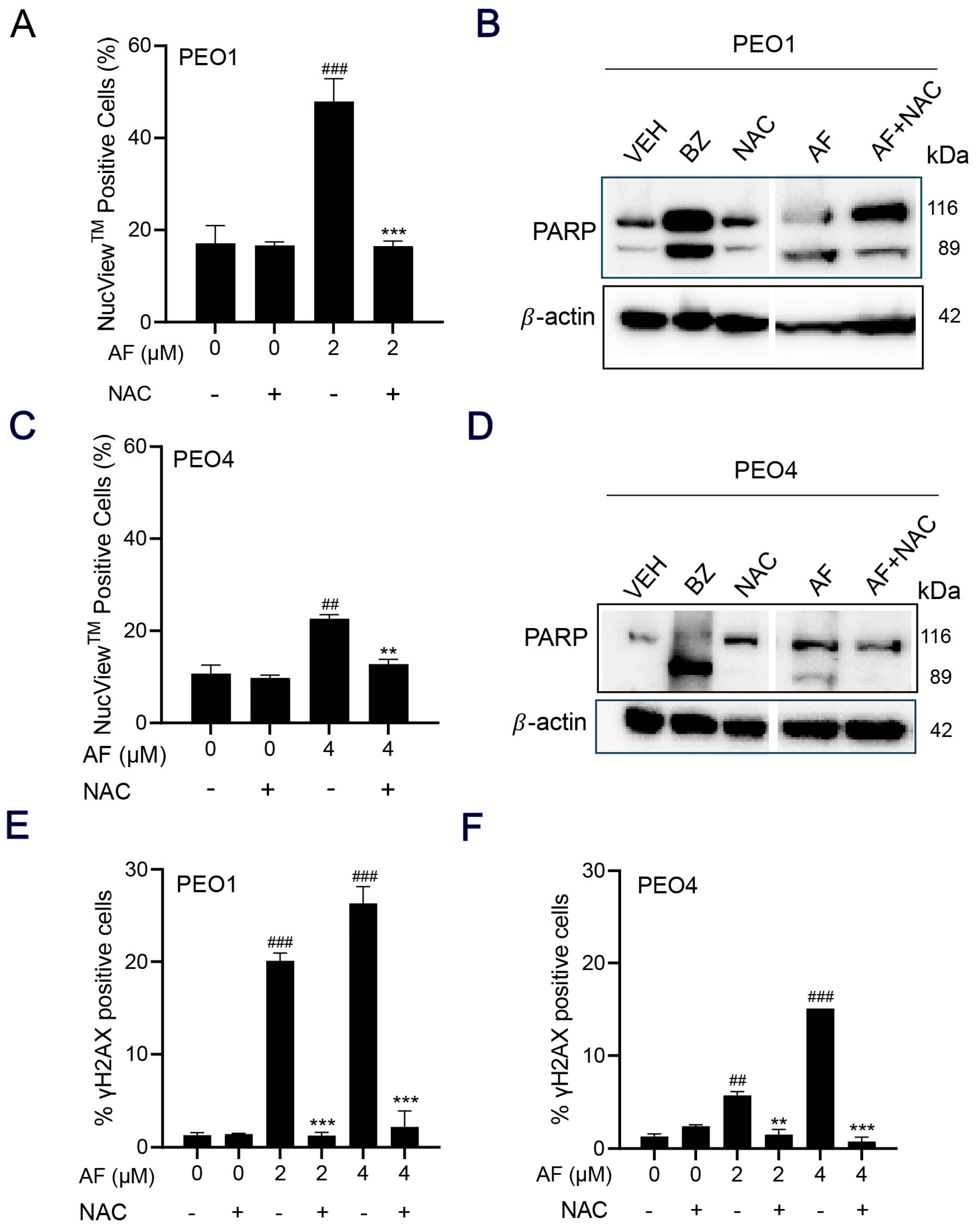
3.6. Auranofin-Induced Cell Death Is Prevented by N-acetyl Cysteine
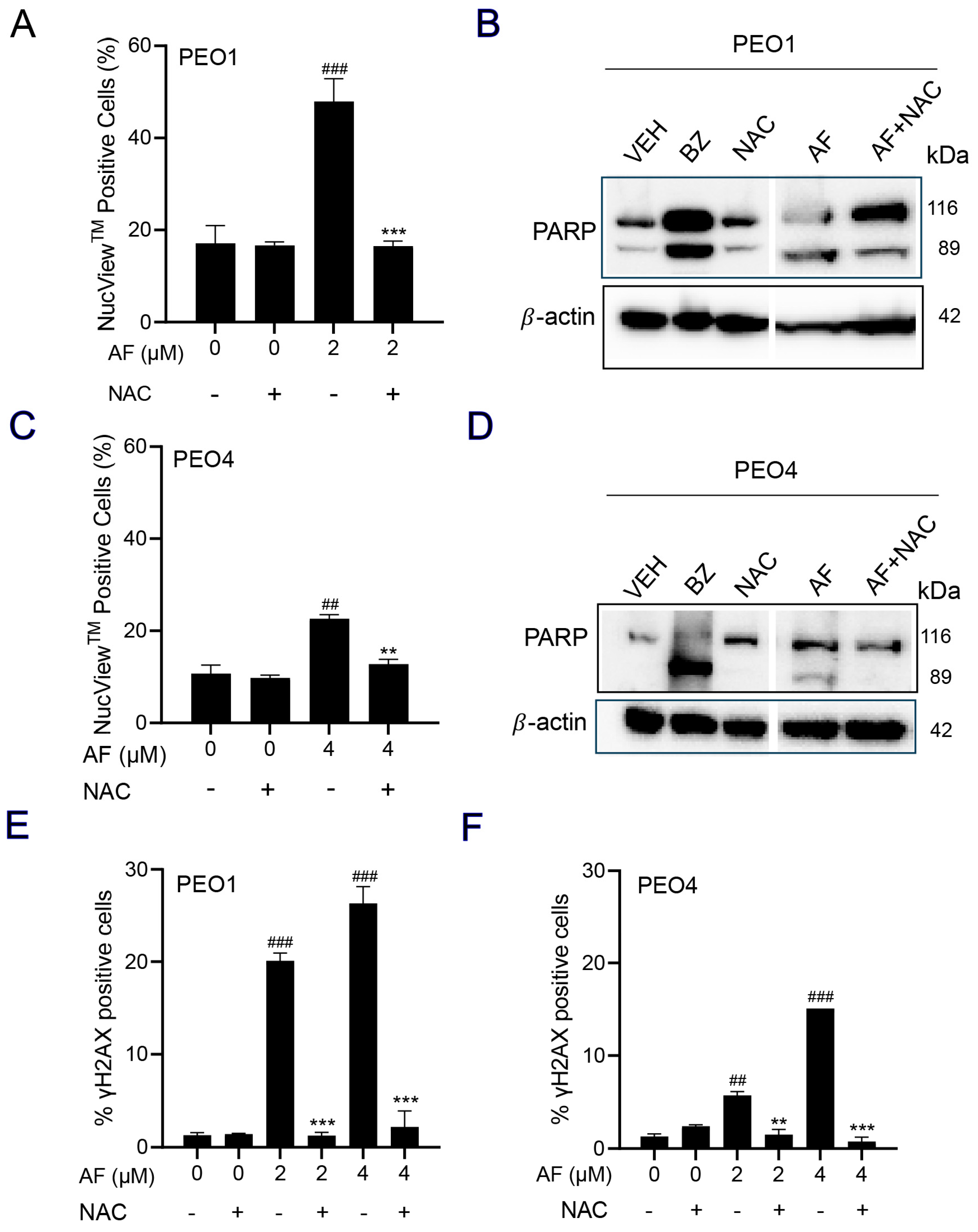
3.7. NAC Prevented Auranofin-Induced Caspase-3/7 Activation, Cleavage of PARP, and Induction of γH2AX
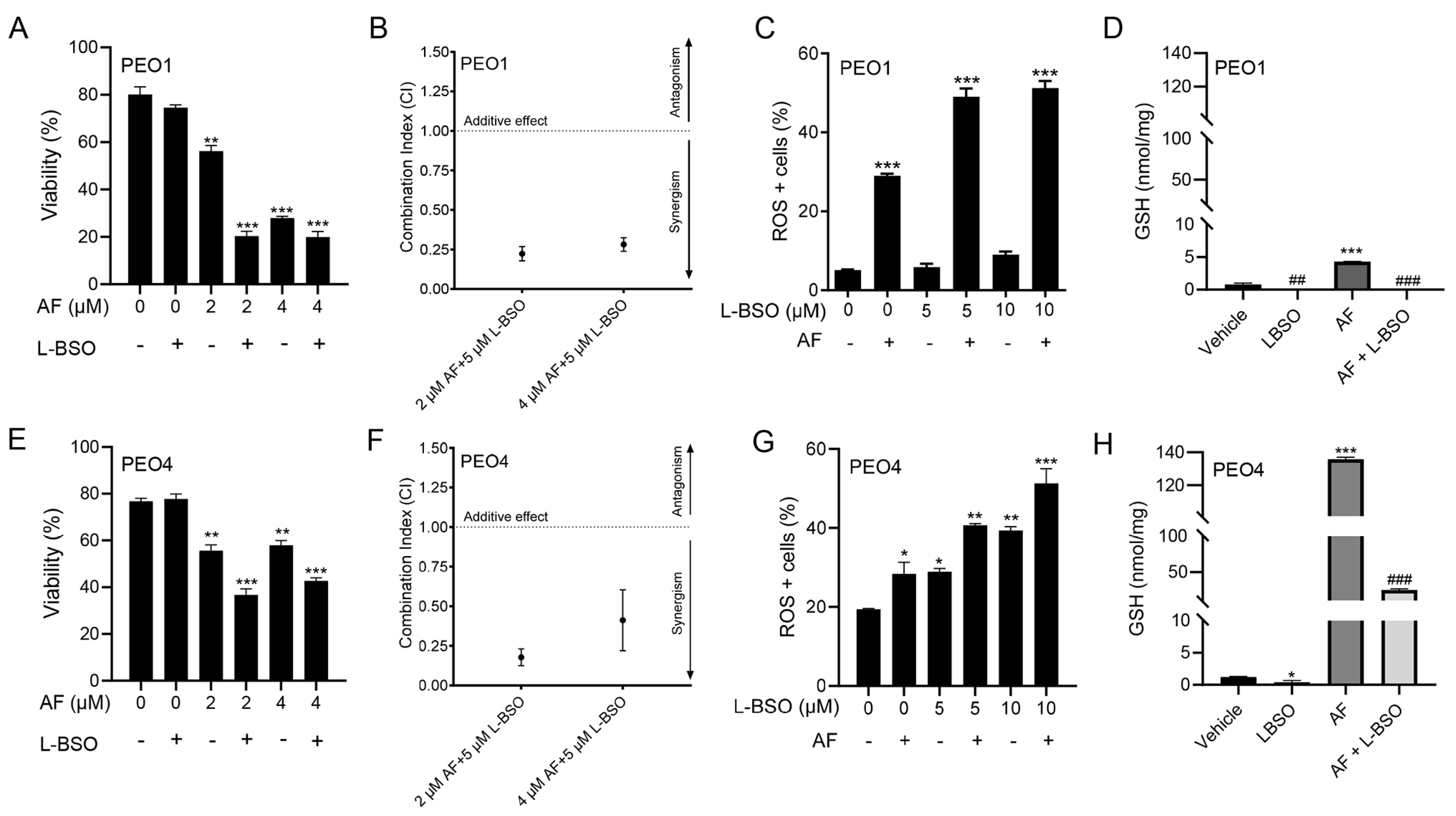
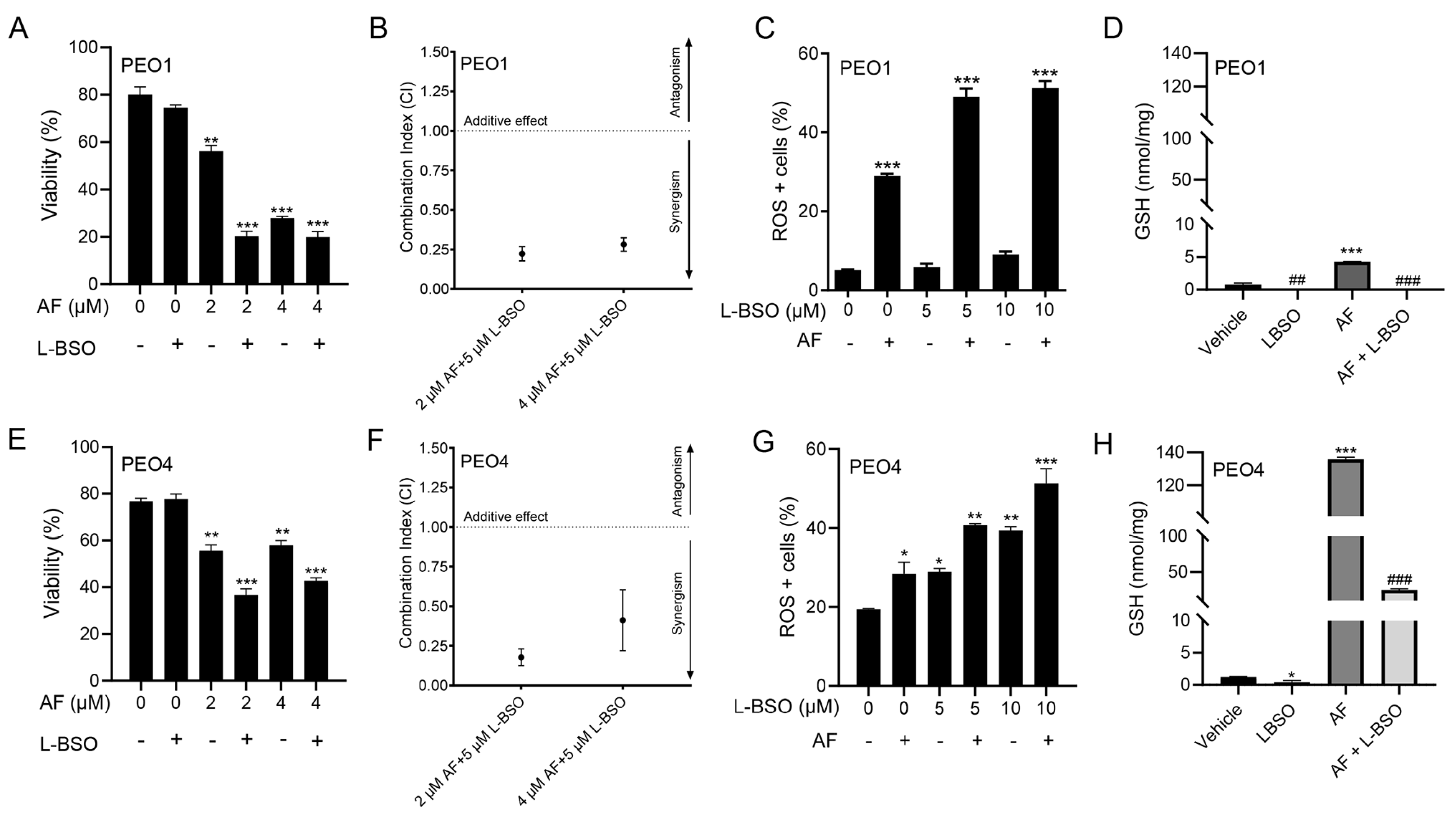
3.8. The Cytotoxic Effect of Auranofin and L-BSO against HGSOC Is Synergistic and Associates with Enhanced ROS Production and Reduced Levels of GSH
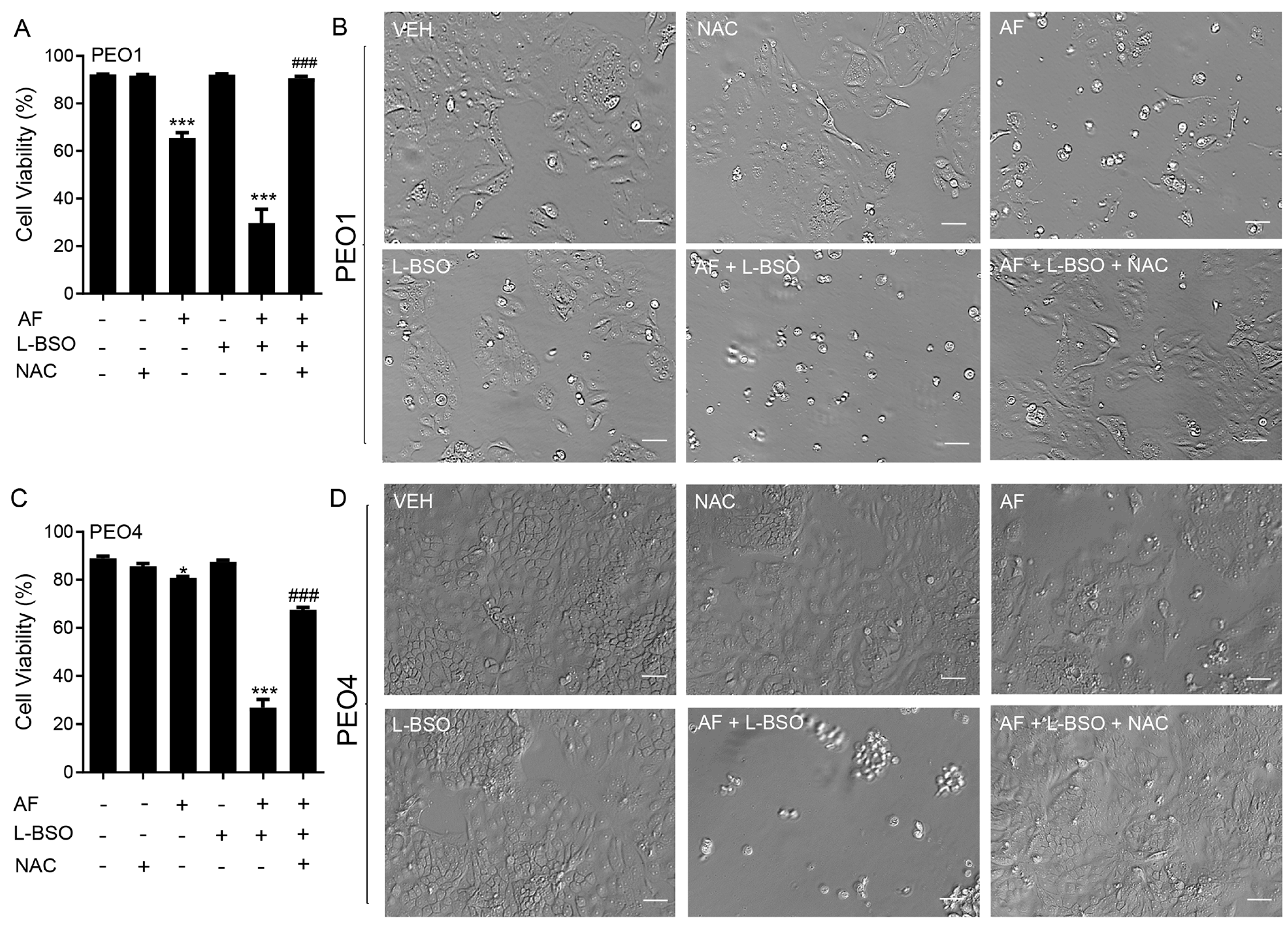
3.9. The Lethal Effect of Auranofin and L-BSO against HGSOC Is Prevented by the Presence of the ROS Scavenger NAC
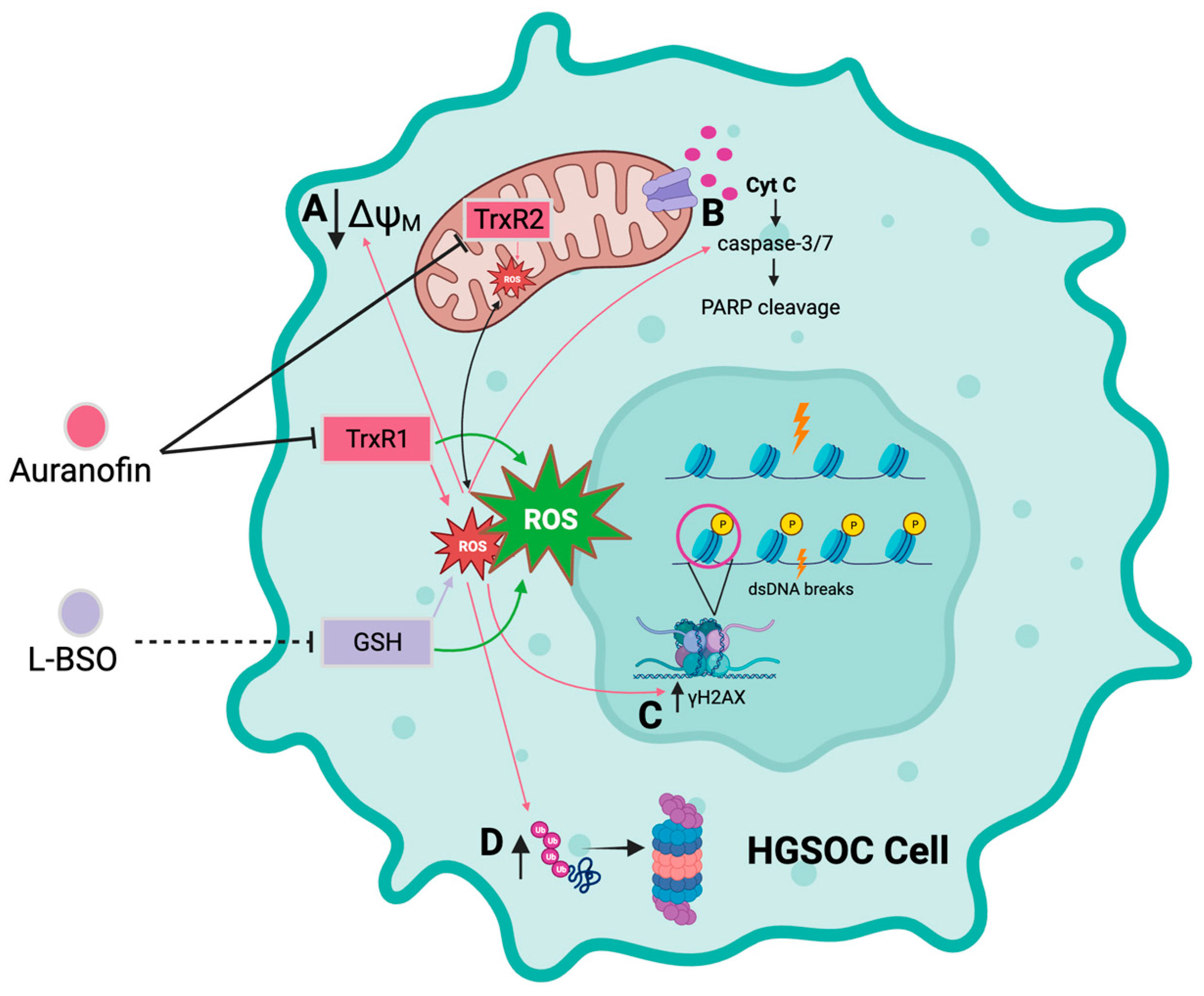
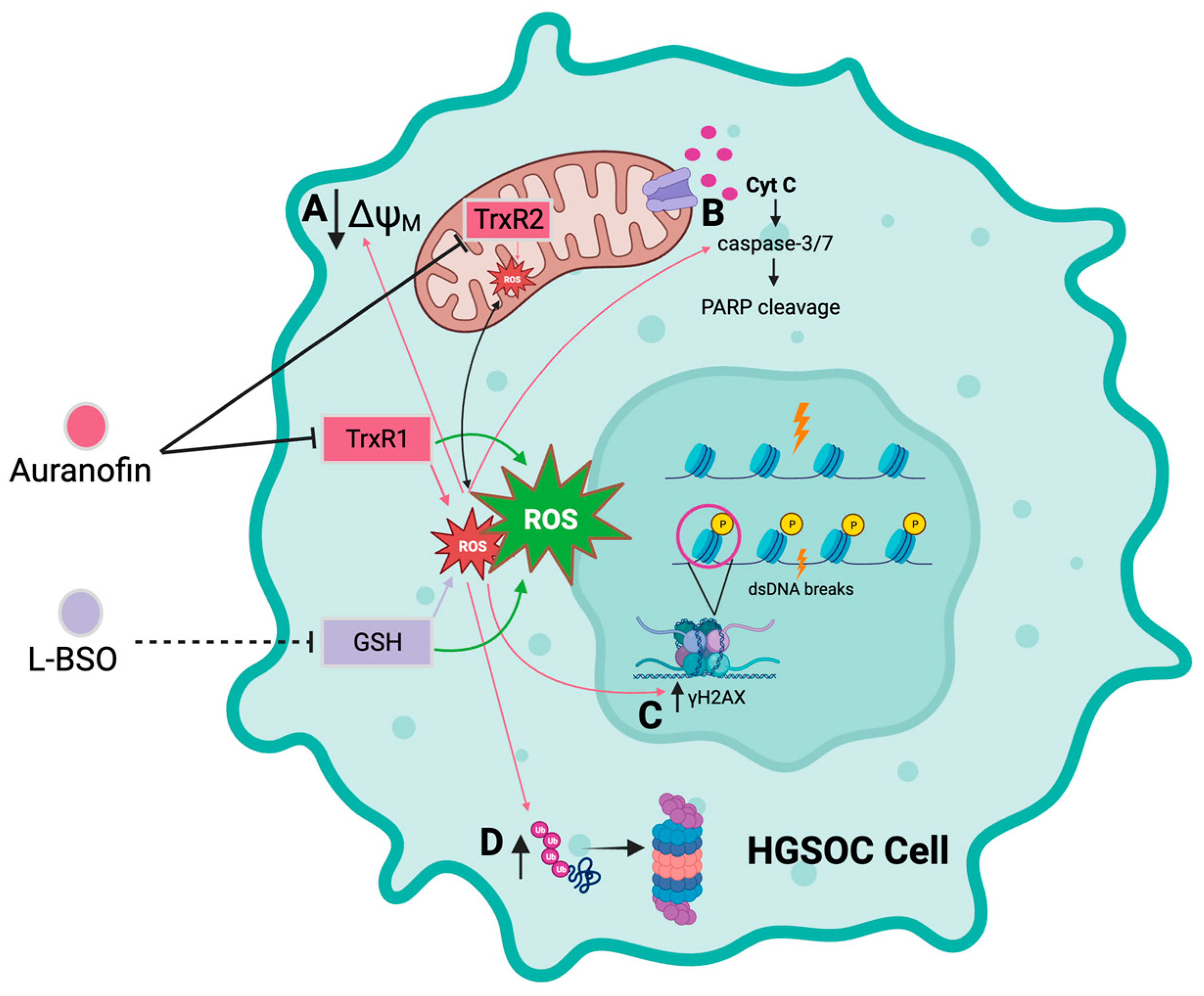
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ∆ψM | Transmembrane potential |
| 7-AAD | 7-Aminoactinomycin D |
| AF | Auranofin |
| ALL | Acute lymphoblastic leukemia |
| ATP | Adenosine triphosphate |
| BCA | Bicinchoninic acid |
| BRCA2 | Breast cancer type 2 susceptibility protein |
| BZ | Bortezomib |
| CI | Combination index |
| CLL | Chronic lymphocytic leukemia |
| CO2 | Carbon dioxide |
| DEVD | DNA-binding peptide; substrate for caspase-3 |
| DHE | Dihydroethidium |
| DNA | Deoxyribonucleic acid |
| DTNB | 5,5′-dithio-bis (2-nitrobenzoic acid) |
| DTT | Dithiothreitol |
| FBS | Fetal bovine serum |
| FDA | US Food and Drug Administration |
| GLOBOCAN | Global Cancer Observatory |
| GSH | Glutathione |
| H2AX | Histone variant H2AX |
| HCl | Hydrochloric acid |
| HEPES | 4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid |
| HGSOC | High-grade serous ovarian cancer |
| HIV | Human immunodeficiency virus |
| HRP | Horseradish peroxidase |
| IC50 | Half-maximal inhibitory concentration |
| IgG | Immunoglobulin G |
| L-BSO | L-buthionine sulfoximine |
| MTT | 3-(4,5-dimethylliazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NAC | N-acetyl-L-cysteine |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NSLC | Non-small cell lung cancer |
| OD | Optical density |
| PARP | Poly-adenosine diphosphate (ADP) ribose polymerase |
| PBS | Phosphate-buffered saline |
| PE | Phycoerythrin |
| P3Cy5 | Phycoerythrin-Cyanine®5 |
| PFA | Paraformaldehyde |
| PMSF | Phenylmethylsulfonyl fluoride |
| PS | Phosphatidylserine |
| ROS | Reactive oxygen species |
| SDS | Sodium dodecyl sulfate |
| TBS-T | Tris-buffered saline 0.1% Tween 20 |
| TNB | 5′-thio-2-nitrobenzoic acid |
| TrxR | Thioredoxin reductase |
| z-DEVD-fmk | Caspase-3 inhibitor |
References
- Lisio, M.-A.; Fu, L.; Goyeneche, A.; Gao, Z.-H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Group on Epidemiological Studies of Ovarian Cancer. Ovarian cancer and oral contraceptives: Collaborative reanalysis of data from 45 epidemiological studies including 23 257 women with ovarian cancer and 87 303 controls. Lancet 2008, 371, 303–314. [Google Scholar] [CrossRef]
- Yang, H.P.; Anderson, W.F.; Rosenberg, P.S.; Trabert, B.; Gierach, G.L.; Wentzensen, N.; Cronin, K.A.; Sherman, M.E. Ovarian cancer incidence trends in relation to changing patterns of menopausal hormone therapy use in the United States. J. Clin. Oncol. 2013, 31, 2146–2151. [Google Scholar] [CrossRef] [PubMed]
- Forgie, B.N.; Prakash, R.; Telleria, C.M. Revisiting the Anti-Cancer Toxicity of Clinically Approved Platinating Derivatives. Int. J. Mol. Sci. 2022, 23, 15410. [Google Scholar] [CrossRef]
- Campbell, R.; Shim, H.; Choi, J.; Park, M.; Byun, E.; Islam, S.; Song, S.H.; Kim, A. Implantable Cisplatin Synthesis Microdevice for Regional Chemotherapy. Adv. Healthc. Mater. 2021, 10, e2001582. [Google Scholar] [CrossRef]
- Yang, L.; Xie, H.J.; Li, Y.Y.; Wang, X.; Liu, X.X.; Mai, J. Molecular mechanisms of platinum-based chemotherapy resistance in ovarian cancer (Review). Oncol. Rep. 2022, 47, 82. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Khan, H.J.; Rohondia, S.O.; Othman Ahmed, Z.S.; Zalavadiya, N.; Dou, Q.P. Chapter 5—Increasing opportunities of drug repurposing for treating breast cancer by the integration of molecular, histological, and systemic approaches. In Drug Repurposing in Cancer Therapy; To, K.K.W., Cho, W.C.S., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 121–172. [Google Scholar]
- Telleria, C.M. Drug Repurposing for Cancer Therapy. J. Cancer Sci. Ther. 2012, 4, ix–xi. [Google Scholar] [CrossRef]
- Zhang, L.; Hapon, M.B.; Goyeneche, A.A.; Srinivasan, R.; Gamarra-Luques, C.D.; Callegari, E.A.; Drappeau, D.D.; Terpstra, E.J.; Pan, B.; Knapp, J.R.; et al. Mifepristone increases mRNA translation rate, triggers the unfolded protein response, increases autophagic flux, and kills ovarian cancer cells in combination with proteasome or lysosome inhibitors. Mol. Oncol. 2016, 10, 1099–1117. [Google Scholar] [CrossRef]
- Gamarra-Luques, C.D.; Goyeneche, A.A.; Hapon, M.B.; Telleria, C.M. Mifepristone prevents repopulation of ovarian cancer cells escaping cisplatin-paclitaxel therapy. BMC Cancer 2012, 12, 200. [Google Scholar] [CrossRef]
- Freeburg, E.M.; Goyeneche, A.A.; Telleria, C.M. Mifepristone abrogates repopulation of ovarian cancer cells in between courses of cisplatin treatment. Int. J. Oncol. 2009, 34, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Goyeneche, A.A.; Caron, R.W.; Telleria, C.M. Mifepristone inhibits ovarian cancer cell growth in vitro and in vivo. Clin. Cancer Res. 2007, 13, 3370–3379. [Google Scholar] [CrossRef]
- Subeha, M.R.; Goyeneche, A.A.; Bustamante, P.; Lisio, M.A.; Burnier, J.V.; Telleria, C.M. Nelfinavir Induces Cytotoxicity towards High-Grade Serous Ovarian Cancer Cells, Involving Induction of the Unfolded Protein Response, Modulation of Protein Synthesis, DNA Damage, Lysosomal Impairment, and Potentiation of Toxicity Caused by Proteasome Inhibition. Cancers 2021, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Sutton, B.M.; McGusty, E.; Walz, D.T.; DiMartino, M.J. Oral gold. Antiarthritic properties of alkylphosphinegold coordination complexes. J. Med. Chem. 1972, 15, 1095–1098. [Google Scholar] [CrossRef]
- Landini, I.; Lapucci, A.; Pratesi, A.; Massai, L.; Napoli, C.; Perrone, G.; Pinzani, P.; Messori, L.; Mini, E.; Nobili, S. Selection and characterization of a human ovarian cancer cell line resistant to auranofin. Oncotarget 2017, 8, 96062–96078. [Google Scholar] [CrossRef] [PubMed]
- Marzo, T.; Massai, L.; Pratesi, A.; Stefanini, M.; Cirri, D.; Magherini, F.; Becatti, M.; Landini, I.; Nobili, S.; Mini, E.; et al. Replacement of the Thiosugar of Auranofin with Iodide Enhances the Anticancer Potency in a Mouse Model of Ovarian Cancer. ACS Med. Chem. Lett. 2019, 10, 656–660. [Google Scholar] [CrossRef]
- Barnes, B.M.; Nelson, L.; Tighe, A.; Burghel, G.J.; Lin, I.H.; Desai, S.; McGrail, J.C.; Morgan, R.D.; Taylor, S.S. Distinct transcriptional programs stratify ovarian cancer cell lines into the five major histological subtypes. Genome Med. 2021, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Korch, C.; Spillman, M.A.; Jackson, T.A.; Jacobsen, B.M.; Murphy, S.K.; Lessey, B.A.; Jordan, V.C.; Bradford, A.P. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol. Oncol. 2012, 127, 241–248. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Wiegand, K.C.; Melnyk, N.; Chow, C.; Salamanca, C.; Prentice, L.M.; Senz, J.; Yang, W.; Spillman, M.A.; Cochrane, D.R.; et al. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 2013, 8, e72162. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, J.H.; Berek, J.S.; Hu, M.C. Auranofin displays anticancer activity against ovarian cancer cells through FOXO3 activation independent of p53. Int. J. Oncol. 2014, 45, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Oommen, D.; Yiannakis, D.; Jha, A.N. BRCA1 deficiency increases the sensitivity of ovarian cancer cells to auranofin. Mutat. Res. 2016, 784–785, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Blayney, J.K.; Davison, T.; McCabe, N.; Walker, S.; Keating, K.; Delaney, T.; Greenan, C.; Williams, A.R.; McCluggage, W.G.; Capes-Davis, A.; et al. Prior knowledge transfer across transcriptional data sets and technologies using compositional statistics yields new mislabelled ovarian cell line. Nucleic Acids Res. 2016, 44, e137. [Google Scholar] [CrossRef]
- Andrews, P.A.; Murphy, M.P.; Howell, S.B. Differential potentiation of alkylating and platinating agent cytotoxicity in human ovarian carcinoma cells by glutathione depletion. Cancer Res. 1985, 45, 6250–6253. [Google Scholar]
- Langdon, S.P.; Lawrie, S.S.; Hay, F.G.; Hawkes, M.M.; McDonald, A.; Hayward, I.P.; Schol, D.J.; Hilgers, J.; Leonard, R.C.; Smyth, J.F. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988, 48, 6166–6172. [Google Scholar]
- Cooke, S.L.; Ng, C.K.; Melnyk, N.; Garcia, M.J.; Hardcastle, T.; Temple, J.; Langdon, S.; Huntsman, D.; Brenton, J.D. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene 2010, 29, 4905–4913. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Selvaraju, K.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Arnér, E.S.J.; Linder, S. Repurposing of auranofin: Thioredoxin reductase remains a primary target of the drug. Biochimie 2019, 162, 46–54. [Google Scholar] [CrossRef]
- Freire Boullosa, L.; Van Loenhout, J.; Flieswasser, T.; De Waele, J.; Hermans, C.; Lambrechts, H.; Cuypers, B.; Laukens, K.; Bartholomeus, E.; Siozopoulou, V.; et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol. 2021, 42, 101949. [Google Scholar] [CrossRef]
- Bhatia, M.; McGrath, K.L.; Di Trapani, G.; Charoentong, P.; Shah, F.; King, M.M.; Clarke, F.M.; Tonissen, K.F. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2016, 8, 68–78. [Google Scholar] [CrossRef]
- Onodera, T.; Momose, I.; Kawada, M. Potential Anticancer Activity of Auranofin. Chem. Pharm. Bull. 2019, 67, 186–191. [Google Scholar] [CrossRef]
- Abdalbari, F.H.; Telleria, C.M. The gold complex auranofin: New perspectives for cancer therapy. Discov. Oncol. 2021, 12, 42. [Google Scholar] [CrossRef]
- You, B.R.; Park, W.H. Auranofin induces mesothelioma cell death through oxidative stress and GSH depletion. Oncol. Rep. 2016, 35, 546–551. [Google Scholar] [CrossRef]
- Oommen, D.; Dodd, N.J.F.; Yiannakis, D.; Moyeed, R.; Jha, A.N. Linking genotoxicity and cytotoxicity with membrane fluidity: A comparative study in ovarian cancer cell lines following exposure to auranofin. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2016, 809, 43–49. [Google Scholar] [CrossRef]
- Rios Perez, M.V.; Roife, D.; Dai, B.; Pratt, M.; Dobrowolski, R.; Kang, Y.; Li, X.; Augustine, J.J.; Zielinski, R.; Priebe, W.; et al. Antineoplastic effects of auranofin in human pancreatic adenocarcinoma preclinical models. Surg. Open Sci. 2019, 1, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Fhu, C.W.; Ali, A. Dysregulation of the Ubiquitin Proteasome System in Human Malignancies: A Window for Therapeutic Intervention. Cancers 2021, 13, 1513. [Google Scholar] [CrossRef] [PubMed]
- Guidi, F.; Landini, I.; Puglia, M.; Magherini, F.; Gabbiani, C.; Cinellu, M.A.; Nobili, S.; Fiaschi, T.; Bini, L.; Mini, E.; et al. Proteomic analysis of ovarian cancer cell responses to cytotoxic gold compounds. Metallomics 2012, 4, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Guo, Z.; Xia, X.; Liao, Y.; Zhang, F.; Huang, C.; Liu, Y.; Deng, X.; Jiang, L.; Wang, X.; et al. Auranofin lethality to prostate cancer includes inhibition of proteasomal deubiquitinases and disrupted androgen receptor signaling. Eur. J. Pharmacol. 2019, 846, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kwolek-Mirek, M.; Zadrag-Tecza, R. Comparison of methods used for assessing the viability and vitality of yeast cells. FEMS Yeast Res. 2014, 14, 1068–1079. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef]
- Galluzzi, L.; Blomgren, K.; Kroemer, G. Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 2009, 10, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003, 10, 709–717. [Google Scholar] [CrossRef]
- Kalyanaraman, B. NAC, NAC, Knockin’ on Heaven’s door: Interpreting the mechanism of action of N-acetylcysteine in tumor and immune cells. Redox Biol. 2022, 57, 102497. [Google Scholar] [CrossRef] [PubMed]
- Tagde, A.; Singh, H.; Kang, M.H.; Reynolds, C.P. The glutathione synthesis inhibitor buthionine sulfoximine synergistically enhanced melphalan activity against preclinical models of multiple myeloma. Blood Cancer J. 2014, 4, e229. [Google Scholar] [CrossRef]
- Cui, X.Y.; Park, S.H.; Park, W.H. Anti-Cancer Effects of Auranofin in Human Lung Cancer Cells by Increasing Intracellular ROS Levels and Depleting GSH Levels. Molecules 2022, 27, 5207. [Google Scholar] [CrossRef]
- Cui, X.Y.; Park, S.H.; Park, W.H. Auranofin inhibits the proliferation of lung cancer cells via necrosis and caspase-dependent apoptosis. Oncol. Rep. 2020, 44, 2715–2724. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, X.; Wang, L.; Zhang, R.; Pu, X.; Wu, S.; Li, L.; Tong, P.; Wang, J.; Meng, Q.H.; et al. Inhibition of Thioredoxin/Thioredoxin Reductase Induces Synthetic Lethality in Lung Cancers with Compromised Glutathione Homeostasis. Cancer Res. 2019, 79, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, V.; Heusel, J.R.; Fulda, S.; van Wijk, S.J.L. ATM inhibition enhances Auranofin-induced oxidative stress and cell death in lung cell lines. PLoS ONE 2020, 15, e0244060. [Google Scholar] [CrossRef]
- Seo, M.J.; Kim, I.Y.; Lee, D.M.; Park, Y.J.; Cho, M.-Y.; Jin, H.J.; Choi, K.S. Dual inhibition of thioredoxin reductase and proteasome is required for auranofin-induced paraptosis in breast cancer cells. Cell Death Dis. 2023, 14, 42. [Google Scholar] [CrossRef]
- Lee, J.E.; Kwon, Y.J.; Baek, H.S.; Ye, D.J.; Cho, E.; Choi, H.K.; Oh, K.S.; Chun, Y.J. Synergistic induction of apoptosis by combination treatment with mesupron and auranofin in human breast cancer cells. Arch. Pharm. Res. 2017, 40, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.K.; Shin, S.; Ye, D.J.; An, H.G.; Kwon, T.U.; Baek, H.S.; Kwon, Y.J.; Chun, Y.J. Combined treatment with auranofin and trametinib induces synergistic apoptosis in breast cancer cells. J. Toxicol. Environ. Health A 2021, 84, 84–94. [Google Scholar] [CrossRef]
- Marzo, T.; Cirri, D.; Gabbiani, C.; Gamberi, T.; Magherini, F.; Pratesi, A.; Guerri, A.; Biver, T.; Binacchi, F.; Stefanini, M.; et al. Auranofin, Et3PAuCl, and Et3PAuI Are Highly Cytotoxic on Colorectal Cancer Cells: A Chemical and Biological Study. ACS Med. Chem. Lett. 2017, 8, 997–1001. [Google Scholar] [CrossRef]
- Zou, P.; Chen, M.; Ji, J.; Chen, W.; Chen, X.; Ying, S.; Zhang, J.; Zhang, Z.; Liu, Z.; Yang, S.; et al. Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget 2015, 6, 36505–36521. [Google Scholar] [CrossRef] [PubMed]
- Sachweh, M.C.; Stafford, W.C.; Drummond, C.J.; McCarthy, A.R.; Higgins, M.; Campbell, J.; Brodin, B.; Arner, E.S.; Lain, S. Redox effects and cytotoxic profiles of MJ25 and auranofin towards malignant melanoma cells. Oncotarget 2015, 6, 16488–16506. [Google Scholar] [CrossRef]
- Karsa, M.; Kosciolek, A.; Bongers, A.; Mariana, A.; Failes, T.; Gifford, A.J.; Kees, U.R.; Cheung, L.C.; Kotecha, R.S.; Arndt, G.M.; et al. Exploiting the reactive oxygen species imbalance in high-risk paediatric acute lymphoblastic leukaemia through auranofin. Br. J. Cancer 2021, 125, 55–64. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Lopez, E.; Guo, H.; Zhang, H.; Liu, Y.; Chen, Z.; Huang, S.; Zhou, S.; Leeming, A.; et al. Repurposing auranofin to treat TP53-mutated or PTEN-deleted refractory B-cell lymphoma. Blood Cancer J. 2019, 9, 95. [Google Scholar] [CrossRef]
- Graczyk-Jarzynka, A.; Goral, A.; Muchowicz, A.; Zagozdzon, R.; Winiarska, M.; Bajor, M.; Trzeciecka, A.; Fidyt, K.; Krupka, J.A.; Cyran, J.; et al. Inhibition of thioredoxin-dependent H2O2 removal sensitizes malignant B-cells to pharmacological ascorbate. Redox Biol. 2019, 21, 101062. [Google Scholar] [CrossRef]
- Magherini, F.; Fiaschi, T.; Valocchia, E.; Becatti, M.; Pratesi, A.; Marzo, T.; Massai, L.; Gabbiani, C.; Landini, I.; Nobili, S.; et al. Antiproliferative effects of two gold(I)-N-heterocyclic carbene complexes in A2780 human ovarian cancer cells: A comparative proteomic study. Oncotarget 2018, 9, 28042–28068. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Comm. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van Ijcken, W.F.J.; Heine, A.A.J.; Smid, M.; et al. Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of In Vitro Morphological Subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Hyter, S.; Hirst, J.; Pathak, H.; Pessetto, Z.Y.; Koestler, D.C.; Raghavan, R.; Pei, D.; Godwin, A.K. Developing a genetic signature to predict drug response in ovarian cancer. Oncotarget 2018, 9, 14828–14848. [Google Scholar] [CrossRef]
- Sze, J.H.; Raninga, P.V.; Nakamura, K.; Casey, M.; Khanna, K.K.; Berners-Price, S.J.; Di Trapani, G.; Tonissen, K.F. Anticancer activity of a Gold(I) phosphine thioredoxin reductase inhibitor in multiple myeloma. Redox Biol. 2020, 28, 101310. [Google Scholar] [CrossRef]
- Fan, C.; Zheng, W.; Fu, X.; Li, X.; Wong, Y.S.; Chen, T. Enhancement of auranofin-induced lung cancer cell apoptosis by selenocystine, a natural inhibitor of TrxR1 in vitro and in vivo. Cell Death Dis. 2014, 5, e1191. [Google Scholar] [CrossRef]
- Hou, G.X.; Liu, P.P.; Zhang, S.; Yang, M.; Liao, J.; Yang, J.; Hu, Y.; Jiang, W.Q.; Wen, S.; Huang, P. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis. Cell Death Dis. 2018, 9, 89. [Google Scholar] [CrossRef]
- Chiappetta, G.; Gamberi, T.; Faienza, F.; Limaj, X.; Rizza, S.; Messori, L.; Filomeni, G.; Modesti, A.; Vinh, J. Redox proteome analysis of auranofin exposed ovarian cancer cells (A2780). Redox Biol. 2022, 52, 102294. [Google Scholar] [CrossRef] [PubMed]
- Habermann, K.J.; Grünewald, L.; van Wijk, S.; Fulda, S. Targeting redox homeostasis in rhabdomyosarcoma cells: GSH-depleting agents enhance auranofin-induced cell death. Cell Death Dis. 2017, 8, e3067. [Google Scholar] [CrossRef] [PubMed]
- Raninga, P.V.; Lee, A.C.; Sinha, D.; Shih, Y.Y.; Mittal, D.; Makhale, A.; Bain, A.L.; Nanayakarra, D.; Tonissen, K.F.; Kalimutho, M.; et al. Therapeutic cooperation between auranofin, a thioredoxin reductase inhibitor and anti-PD-L1 antibody for treatment of triple-negative breast cancer. Int. J. Cancer 2020, 146, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Jastrząb, A.; Skrzydlewska, E. Thioredoxin-dependent system. Application of inhibitors. J. Enzyme Inhib. Med. Chem. 2021, 36, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Guo, E.; Zhou, B.; Shan, W.; Huang, J.; Weng, D.; Wu, P.; Wang, C.; Wang, S.; Zhang, W.; et al. A reactive oxygen species scoring system predicts cisplatin sensitivity and prognosis in ovarian cancer patients. BMC Cancer 2019, 19, 1061. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [PubMed]
- Pedre, B.; Barayeu, U.; Ezeriņa, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H2S and sulfane sulfur species. Pharmacol. Therap 2021, 228, 107916. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Dorn, P.; Liu, S.; Deng, H.; Hall, S.R.R.; Peng, R.-W.; Schmid, R.A.; Marti, T.M. Cisplatin-resistant A549 non-small cell lung cancer cells can be identified by increased mitochondrial mass and are sensitive to pemetrexed treatment. Cancer Cell Int. 2019, 19, 317. [Google Scholar] [CrossRef]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef]
- Kleih, M.; Böpple, K.; Dong, M.; Gaißler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef] [PubMed]
- Montopoli, M.; Bellanda, M.; Lonardoni, F.; Ragazzi, E.; Dorigo, P.; Froldi, G.; Mammi, S.; Caparrotta, L. “Metabolic reprogramming” in ovarian cancer cells resistant to cisplatin. Curr. Cancer Drug Targets 2011, 11, 226–235. [Google Scholar] [CrossRef]
- Park, J.; Lee, J.; Choi, C. Mitochondrial Network Determines Intracellular ROS Dynamics and Sensitivity to Oxidative Stress through Switching Inter-Mitochondrial Messengers. PLoS ONE 2011, 6, e23211. [Google Scholar] [CrossRef]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S.D.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin Induces Lethal Oxidative and Endoplasmic Reticulum Stress and Exerts Potent Preclinical Activity against Chronic Lymphocytic Leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Jacquemont, C.l.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional Restoration of BRCA2 Protein by Secondary BRCA2 Mutations in BRCA2-Mutated Ovarian Carcinoma. Cancer Res. 2009, 69, 6381–6386. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Huang, H.; Dou, Q.P.; Liu, J. Inhibition of 19S proteasome-associated deubiquitinases by metal-containing compounds. Oncoscience 2015, 2, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.H. Regulation of Protein Degradation by Proteasomes in Cancer. J. Cancer Prev. 2018, 23, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Bailey, H.H. l-S,R-buthionine sulfoximine: Historical development and clinical issues. Chem.-Biol. Interact. 1998, 111–112, 239–254. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Kennedy, L.; Sandhu, J.K.; Harper, M.E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met. Based Drugs 2010, 2010, 1429. [Google Scholar] [CrossRef]
- Fazzari, F.; Chow, S.; Cheung, M.; Barghout, S.H.; Schimmer, A.D.; Chang, Q.; Hedley, D. Combined Targeting of the Glutathione and Thioredoxin Antioxidant Systems in Pancreatic Cancer. ACS Pharmacol. Transl. Sci. 2022, 5, 1070–1078. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdalbari, F.H.; Martinez-Jaramillo, E.; Forgie, B.N.; Tran, E.; Zorychta, E.; Goyeneche, A.A.; Sabri, S.; Telleria, C.M. Auranofin Induces Lethality Driven by Reactive Oxygen Species in High-Grade Serous Ovarian Cancer Cells. Cancers 2023, 15, 5136. https://doi.org/10.3390/cancers15215136
Abdalbari FH, Martinez-Jaramillo E, Forgie BN, Tran E, Zorychta E, Goyeneche AA, Sabri S, Telleria CM. Auranofin Induces Lethality Driven by Reactive Oxygen Species in High-Grade Serous Ovarian Cancer Cells. Cancers. 2023; 15(21):5136. https://doi.org/10.3390/cancers15215136
Chicago/Turabian StyleAbdalbari, Farah H., Elvis Martinez-Jaramillo, Benjamin N. Forgie, Estelle Tran, Edith Zorychta, Alicia A. Goyeneche, Siham Sabri, and Carlos M. Telleria. 2023. "Auranofin Induces Lethality Driven by Reactive Oxygen Species in High-Grade Serous Ovarian Cancer Cells" Cancers 15, no. 21: 5136. https://doi.org/10.3390/cancers15215136
APA StyleAbdalbari, F. H., Martinez-Jaramillo, E., Forgie, B. N., Tran, E., Zorychta, E., Goyeneche, A. A., Sabri, S., & Telleria, C. M. (2023). Auranofin Induces Lethality Driven by Reactive Oxygen Species in High-Grade Serous Ovarian Cancer Cells. Cancers, 15(21), 5136. https://doi.org/10.3390/cancers15215136