
Immunotherapy with Monoclonal Antibodies for Acute Myeloid Leukemia: A Work in Progress


Abstract
:Simple Summary
Abstract
1. Introduction
2. Immune Checkpoint Inhibition in Acute Myeloid Leukemia
2.1. PD-1/PDL-1 Blockade
2.2. PD-1 Inhibitors
2.2.1. Nivolumab
2.2.2. Pembrolizumab
2.2.3. Tislelizumab
2.3. PD-L1 Inhibitors

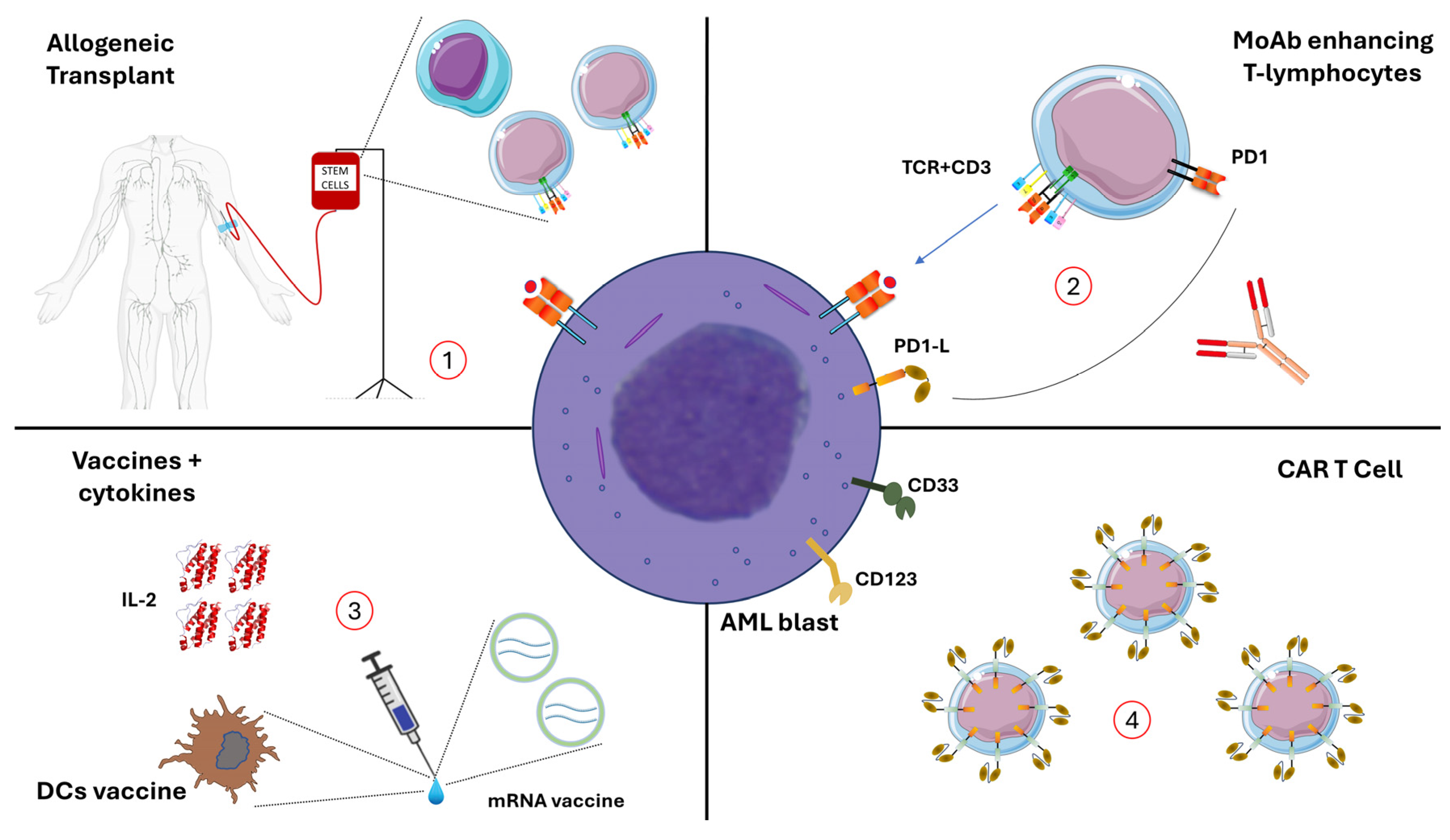
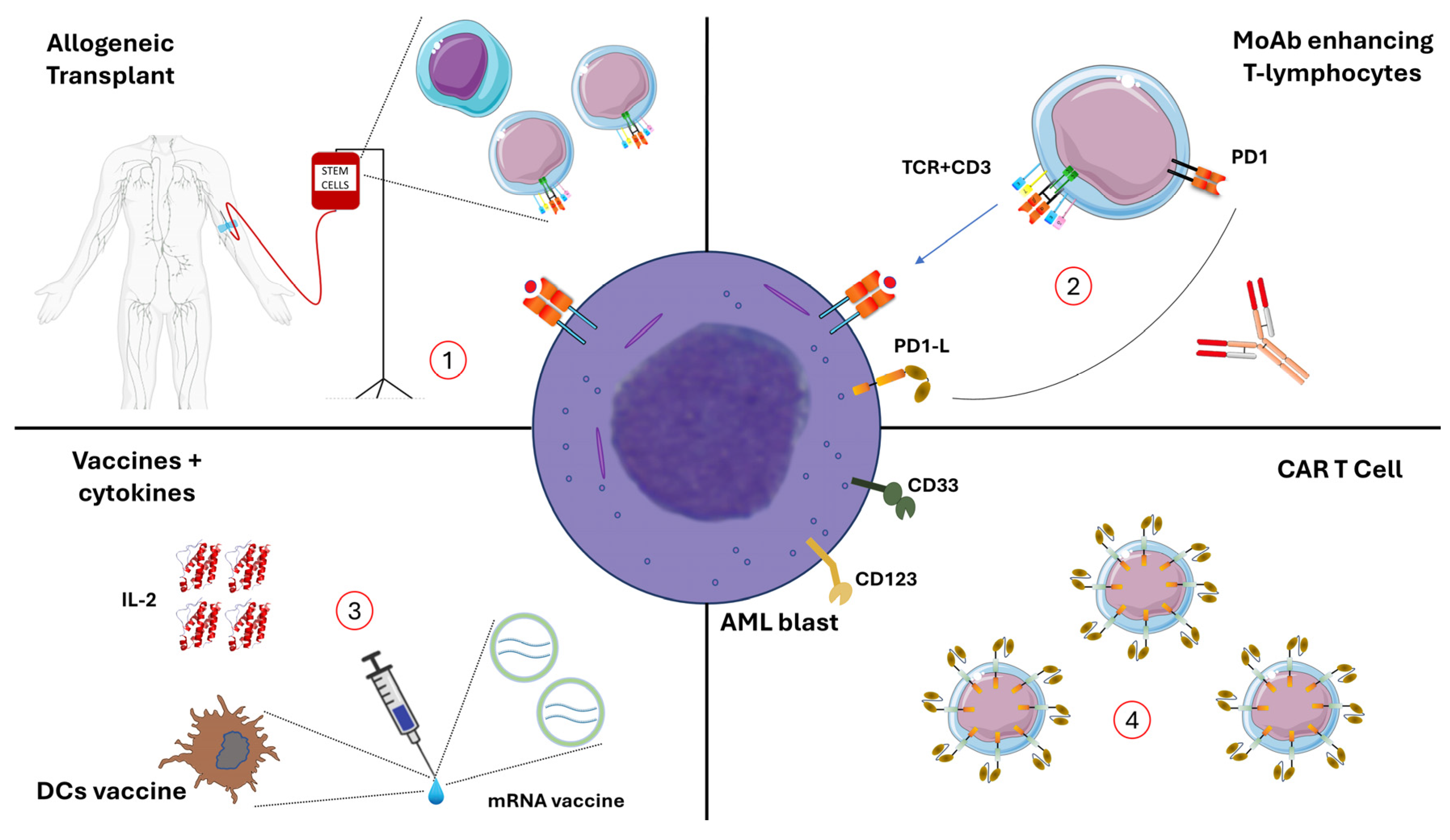{kind=link}
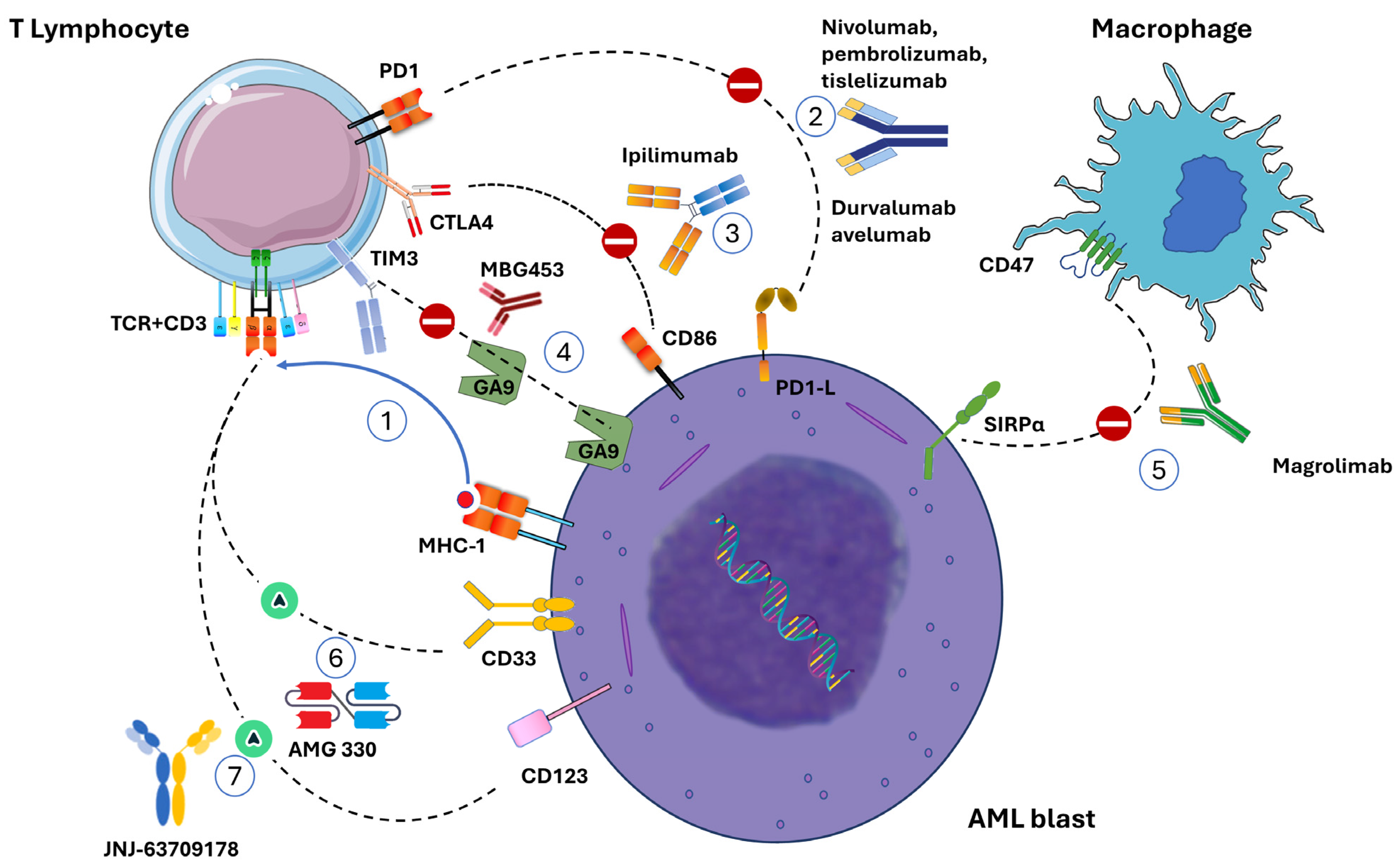
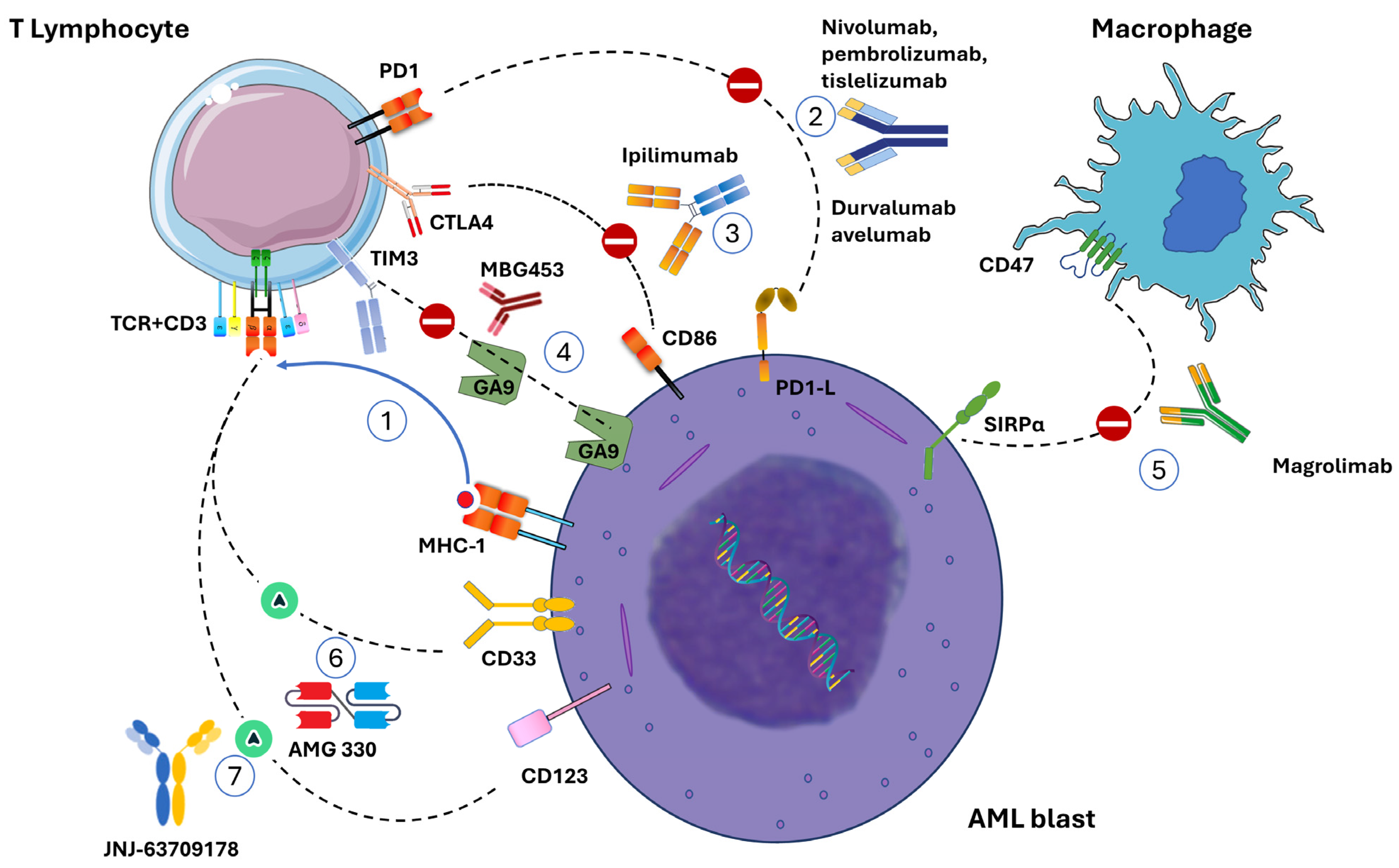{kind=link}
| Reference | Therapeutic Approach | Type of AML | Number of Patients | Response | Survival | AEs |
|---|---|---|---|---|---|---|
| Daver et al. [28] | Azacitidine + nivolumab | R/R AML | 70 | ORR: 33% CR/CRi: 22% | Median OS: 9.2 months | irAEs grade 3–4: 11% (n = 8) |
| Daver et al. [29] | Azacitidine + nivolumab + ipilimumab | R/R AML | 24 | ORR: 46% CR/CRi: 36% | / | NA |
| Davids et al. [30] | Nivolumab | AML and myeloid malignancies after transplant | 10 AML 19 myeloid malignancies | / | Median OS: 21.4 months 1-year OS: 56% | DLTs 1 mg/kg (ir-AE): 33% (n = 2/6) DLTs 0.5 mg/kg (ir-AE): 18% (n = 4/22) ir-AEs: 9% (n = 2/22) fatal GVHD: 9% (n = 2/22) |
| Ravandi et al. [31] | Nivolumab + idarubicin + cytarabine | ND-AML and HR-MDS | 42 AML 2 HR-MDS | composite CR: 78% | Median OS: 18.5 months | Early mortality: 5% irAEs grade 3/4: 13% 19 patients underwent allo-SCT GVHD: 68% (n = 13) |
| Liu et al. [32] | Nivolumab | Maintenance on AML in first CR, CR or CRi | 26 | / | Median OS: 53.9 months; 2-year OS: 60.0% | ORR: 32% Os rate 1 year: 56% |
| Gojo et al. [34] | Pembrolizumab + azacitidine | Newly diagnosed AML and R/R AML | 37 R/R AML 22 de novo AML | ORR: 55%, with 14% CR/CRi in R/R AML ORR: 94% with 47% CR/CRi in de novo AML | Median OS for de novo AML: 13.1 months | irAE n: 9 (24%) and 5 (11%) pts in Cohort 1, and 3 (14%) and 4 (18%) pts in Cohort 2 |
| Goswami et al. [35] | Pembrolizumab + decitabine | R/R AML | 10 | / | Median OS: 10 months | irAE n: 9. Trhombocytopenia: 80%. Neutropenia: 30%. |
| Zeidner et al. [36] | Pembrolizumab + high-dose cytarabine | R/R AML | 37 | ORR: 48% with a composite CR 38% | median OS: 13.2 months | Rare grade ≥ 3 ir-AEs after pembrolizumab: maculopapular rash (n = 2; 5%) |
| Gao et al. [43] | Tislelizumab + azacitidine or decitabine + CAG regimen (cytarabine, aclarubicin, G-CSF) | R/R AML | 27 | ORR: 63% | Median OS: 9.7 months | Grade 2–3 ir-AEs: 14.8% (n = 4) |
| Zeidan et al. [48] | Arm A: Azacitidine + durvalumab Arm B: Azacitidine | Untreated MDS or Elderly AML 65 year) | 129 | ORR Arm A: 31.3% Arm B: 35.4% DoR Arm A: 24.6 weeks Arm B: 51.7 weeks | mOS: Arm A: 13.0 month Arm B: 14.4 month | Arm A: constipation (57.8%) and thrombocytopenia (42.2%) |
| Zeng [45] | Avelumab + decitabine | De novo AML r/r AML | 7 | CR: 14% (n = 1) SD: 42% (n = 3) | febrile neutropenia (86%), hypoxia (57%), heart failure (29%), and pneumonitis (29%) | |
| Saxena [46] | Avelumab + azacitidine | r/r AML | 19 | CR/Cri: 10.5% | median OS: 4.8 months | trAEs grade ≥ 3: neutropenia, 10% (n = 2) Anemia: 10% (n = 2) irAEs grade 2/3: 10% (n = 2) |
3. CTLA-4 Inhibition
Ipilimumab
4. CD47-SIRPα Blockade
Magrolimab and Other CD47/SIRPα Inhibitors
| Reference | Therapeutic Approach | NCT Number/Reference | Stage | Indication Type of AML | Number of Patients | AEs | Available Results | Survival | Next Phase Planned |
|---|---|---|---|---|---|---|---|---|---|
| Chao et al. [62] | Magrolimab SA | NCT02678338 CAMELLIA trial | Phase 1 | r/r AML HS MDS | 15 (NCT 20) | Anemia 47% [70] | SD: 73% Lower bone marrow blast counts: 40% | ||
| Daver et al. [65] | Magrolimab + azacitidine | NCT03248479 | Phase 1b | AML unfit for intensive chemotherapy, naïve | 52 | Nausea, constipation or diarrhea, anemia (34.5%) | n = 34 CR/Cri: 56% Of which MRD- (IF): 37% ORR: TP53 Mutant AML: 71% (15/21) | TP53 mutant OS: 12.9 months TP53 WT: 18.9 months | Yes phase 3 ENHANCE-2 |
| Magrolimab + azacitidine or venetoclax + azacitidine or 7 + 3 (DA) | NCT04778397 ENHANCE-2 | Phase 3 | AML untreated TP53-mutant | 87 | N/A | N/A | N/A | Ongoing | |
| Daver et al. [67]. | Magrolimab + venetoclax + azacitidine | NCT04435691 | Phase 1/2 | Older/unfit or high-risk r/r AML | 74 | Grade 3 anemia in 23% (18/79) | ORR de novo AML: 81% (35/43) TP53 mutations: 74% (20/27) Responses in R/R AML were scarce (ORR 11%), this study arm closed | Median OS was not reached in newly diagnosed non-secondary AML patients; the median OS was 7.6 months among untreated secondary AML with TP53 mutation | |
| Ongoing [71] | Evorpacept + azacitidine | NCT04417517 ASPEN02 trial | Phase 1/2 | High-risk MDS | 65 (planned) | No severe treatment-emergent AEs | Blast reduction | NA | |
| Garcia-Manero [68] | Evorpacept + venetoclax + azacitidine | NCT04755244 ASPEN05 trial | Phase 1/2 | De novo AML r/r AML | 97 (planned) Data on 12 | neutropenia, anemia (6 each; 43%) | Objective responses 6 pts | NA | Yes |
| Qi J. [69] | Lemzoparlimab monotherapy or lemzoparlimab + azacitidine | NCT04202003 | Phase 1/2 | R/R AML and MDS | 105 (planned) 8 with available data | No SAE | 1 morphologic leukemia-free state |
5. The TIM-3/Galectin9 Signaling
6. The LAG-3/MHC Pathway
7. The CD27/CD70 Axis
8. Bispecific Antibodies (T Cell Engager): BiTE
| NCT Number/Reference | Target | Therapeutic Approach | Developmental Stage | Indication | No. of Patients | AEs | Available Results | Survival |
|---|---|---|---|---|---|---|---|---|
| NCT02152956 VOYAGE Uy, G.L. et al. [96] | CD123xCD3 | Flotetuzumab monotherapy | Phase 1 Phase 1/2 | PIF/ER AML | 88 (246 planned) | Cytokine release syndromes (CRS) and infusion-related reactions, mostly G 1–2 | CR/CRi 18.5% (5 of 27) | mOS 10.2 months |
| NCT02715011 Boyiadzis, M. [97] | CD123xCD3 | JNJ-63709178 monotherapy | Phase 1 | r/r AML | 62 | TEAEs ≥ 3 were observed in 11 (65%) patients. ISR: 100%. CRS: 27%. | 1 (1.6%) SD. Discontinued. | NA |
| Ravandi, F. et al. [98] | CD123xCD3 | Vibecotamab monotherapy | Phase 1 | LAM, (ALL-B, BPDCN) | 104 | CRS in 62 of 106 patients (85% grade 1–2, 15% grade ≥ 3) | ORR del 14.8% 7/51. CR (2), CRi (3). | |
| NCT05285813 Short, N.J. [99] | CD123xCD3 | Vibecotamab monotherapy | Phase 2 | AML MRD + MDS/CMML post-HMA | 13 (planned 40) | N/A Ongoing | N/A | N/A |
| NCT02520427 Ravandi et al. [100] | CD33xCD3 | AMG330 monotherapy | Phase 1 | r/r AML MRD + AML MDS | 96 | CRS: 13% (67% ≥ grade 3) Nausea: 20% | CR/CRiCRS: 9/42 | |
| NCT03224819 Subklewe et al. [101] | CD33xCD3 | AMG 673 monotherapy | Phase 1 | r/r AML | 30 | CRS: 50% AEs: 37% (50% grade ≥ 3) abnormal hepatic enzymes (n = 5, 17%), CRS (n = 4, 13%), leukopenia (n = 4, 13%), thrombocytopenia (n = 2, 7%), and febrile neutropenia (n = 2, 7%) | Bone marrow blast reductions in 17 (44%), 1 pz CRi | |
| NCT3144245 [94,102], | CD33xCD3 | AMV564 monotherapy | Phase 1 | r/r AML | 36 | Grade ≥ 3 treatment-emergent AE: anemia, in 4 (11%) | Bone marrow blast reduction in 17 patients (49%) 1 CR, 1 CRi | N/A |
| NCT03038230 [96], Mascarenhas J. et al. | CD3xCLL-1 | MCLA-117 monotherapy | Phase 1 | r/r AML newly diagnosed elderly untreated AMLHR-MDS | 50 | TEAEs were pyrexia (32%), CRS (32%), chills (22%), infusion site phlebitis (14%), vomiting (12%), and nausea (10%) | Out of 26 evaluable patients, 4 showed ≥50% blast reduction including 1 with morphological leukemia-free state | N/A |
9. Cellular Therapies (CAR-T)
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sasidharan Nair, V.; Elkord, E. Immune Checkpoint Inhibitors in Cancer Therapy: A Focus on T-Regulatory Cells. Immunol. Cell Biol. 2018, 96, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zinzani, P.L.; Lee, H.J.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Pembrolizumab in Relapsed or Refractory Hodgkin Lymphoma: 2-Year Follow-up of KEYNOTE-087. Blood 2019, 134, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, J.; Ramchandren, R.; Santoro, A.; Paszkiewicz-Kozik, E.; Gasiorowski, R.; Johnson, N.A.; Fogliatto, L.M.; Goncalves, I.; De Oliveira, J.S.R.; Buccheri, V.; et al. Pembrolizumab versus Brentuximab Vedotin in Relapsed or Refractory Classical Hodgkin Lymphoma (KEYNOTE-204): An Interim Analysis of a Multicentre, Randomised, Open-Label, Phase 3 Study. Lancet Oncol. 2021, 22, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Bigenwald, C.; Ribrag, V.; Lazarovici, J.; Jardin, F.; Sarkozy, C. Pembrolizumab in the Treatment of Refractory Primary Mediastinal Large B-Cell Lymphoma: Safety and Efficacy. Expert Rev. Anticancer Ther. 2021, 21, 941–956. [Google Scholar] [CrossRef]
- El Chaer, F.; Hourigan, C.S.; Zeidan, A.M. How I Treat AML in 2023 Incorporating the Updated Classifications and Guidelines. Blood 2023, 141, 2813–2823. [Google Scholar] [CrossRef]
- Molica, M.; Perrone, S. Molecular Targets for the Treatment of AML in the Forthcoming 5th World Health Organization Classification of Haematolymphoid Tumours. Expert Rev. Hematol. 2022, 15, 973–986. [Google Scholar] [CrossRef]
- Maschmeyer, G.; De Greef, J.; Mellinghoff, S.C.; Nosari, A.; Thiebaut-Bertrand, A.; Bergeron, A.; Franquet, T.; Blijlevens, N.M.A.; Maertens, J.A. European Conference on Infections in Leukemia (ECIL) Infections Associated with Immunotherapeutic and Molecular Targeted Agents in Hematology and Oncology. A Position Paper by the European Conference on Infections in Leukemia (ECIL). Leukemia 2019, 33, 844–862. [Google Scholar] [CrossRef]
- Maschmeyer, G.; Bullinger, L.; Garcia-Vidal, C.; Herbrecht, R.; Maertens, J.; Menna, P.; Pagano, L.; Thiebaut-Bertrand, A.; Calandra, T. Infectious Complications of Targeted Drugs and Biotherapies in Acute Leukemia. Clinical Practice Guidelines by the European Conference on Infections in Leukemia (ECIL), a Joint Venture of the European Group for Blood and Marrow Transplantation (EBMT), the European Organization for Research and Treatment of Cancer (EORTC), the International Immunocompromised Host Society (ICHS) and the European Leukemia Net (ELN). Leukemia 2022, 36, 1215–1226. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Christopher, M.J.; Petti, A.A.; Rettig, M.P.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O’Laughlin, M.; Fronick, C.C.; et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med. 2018, 379, 2330–2341. [Google Scholar] [CrossRef]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The Distribution of T-Cell Subsets and the Expression of Immune Checkpoint Receptors and Ligands in Patients with Newly Diagnosed and Relapsed Acute Myeloid Leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Munger, M.E.; Veenstra, R.G.; Weigel, B.J.; Hirashima, M.; Munn, D.H.; Murphy, W.J.; Azuma, M.; Anderson, A.C.; Kuchroo, V.K.; et al. Coexpression of Tim-3 and PD-1 Identifies a CD8+ T-Cell Exhaustion Phenotype in Mice with Disseminated Acute Myelogenous Leukemia. Blood 2011, 117, 4501–4510. [Google Scholar] [CrossRef] [PubMed]
- Vago, L.; Gojo, I. Immune Escape and Immunotherapy of Acute Myeloid Leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as Tools and Targets in Cancer Therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed]
- Molica, M.; Perrone, S.; Mazzone, C.; Niscola, P.; Cesini, L.; Abruzzese, E.; De Fabritiis, P. CD33 Expression and Gentuzumab Ozogamicin in Acute Myeloid Leukemia: Two Sides of the Same Coin. Cancers 2021, 13, 3214. [Google Scholar] [CrossRef]
- Raeber, M.E.; Sahin, D.; Karakus, U.; Boyman, O. A Systematic Review of Interleukin-2-Based Immunotherapies in Clinical Trials for Cancer and Autoimmune Diseases. eBioMedicine 2023, 90, 104539. [Google Scholar] [CrossRef]
- Barbullushi, K.; Rampi, N.; Serpenti, F.; Sciumè, M.; Fabris, S.; De Roberto, P.; Fracchiolla, N.S. Vaccination Therapy for Acute Myeloid Leukemia: Where Do We Stand? Cancers 2022, 14, 2994. [Google Scholar] [CrossRef]
- Vanhooren, J.; Dobbelaere, R.; Derpoorter, C.; Deneweth, L.; Van Camp, L.; Uyttebroeck, A.; De Moerloose, B.; Lammens, T. CAR-T in the Treatment of Acute Myeloid Leukemia: Barriers and How to Overcome Them. Hemasphere 2023, 7, e937. [Google Scholar] [CrossRef]
- Mardiana, S.; Gill, S. CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 697. [Google Scholar] [CrossRef]
- Tamura, H.; Dan, K.; Tamada, K.; Nakamura, K.; Shioi, Y.; Hyodo, H.; Wang, S.-D.; Dong, H.; Chen, L.; Ogata, K. Expression of Functional B7-H2 and B7.2 Costimulatory Molecules and Their Prognostic Implications in de Novo Acute Myeloid Leukemia. Clin. Cancer Res. 2005, 11, 5708–5717. [Google Scholar] [CrossRef]
- Zhang, Z.-F.; Zhang, Q.-T.; Xin, H.-Z.; Gan, S.-L.; Ma, J.; Liu, Y.-F.; Xie, X.-S.; Sun, H. Expression of Programmed Death Ligand-1 (PD-L1) in Human Acute Leukemia and Its Clinical Significance. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2015, 23, 930–934. [Google Scholar] [PubMed]
- Krönig, H.; Kremmler, L.; Haller, B.; Englert, C.; Peschel, C.; Andreesen, R.; Blank, C.U. Interferon-Induced Programmed Death-Ligand 1 (PD-L1/B7-H1) Expression Increases on Human Acute Myeloid Leukemia Blast Cells during Treatment. Eur. J. Haematol. 2014, 92, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 Inhibits the Effector Phase of Tumor Rejection by T Cell Receptor (TCR) Transgenic CD8+ T Cells. Cancer Res. 2004, 64, 1140–1145. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 Associate with Immunoreceptor Tyrosine-Based Switch Motif of Programmed Death 1 upon Primary Human T Cell Stimulation, but Only Receptor Ligation Prevents T Cell Activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gajewski, T.F.; Kline, J. PD-1/PD-L1 Interactions Inhibit Antitumor Immune Responses in a Murine Acute Myeloid Leukemia Model. Blood 2009, 114, 1545–1552. [Google Scholar] [CrossRef]
- Zhou, Q.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Weigel, B.J.; Riddle, M.; Sharpe, A.H.; Vallera, D.A.; Azuma, M.; Levine, B.L.; et al. Program Death-1 Signaling and Regulatory T Cells Collaborate to Resist the Function of Adoptively Transferred Cytotoxic T Lymphocytes in Advanced Acute Myeloid Leukemia. Blood 2010, 116, 2484–2493. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef]
- Daver, N.G.; Garcia-Manero, G.; Konopleva, M.Y.; Alfayez, M.; Pemmaraju, N.; Kadia, T.M.; DiNardo, C.D.; Cortes, J.E.; Ravandi, F.; Abbas, H.; et al. Azacitidine (AZA) with Nivolumab (Nivo), and AZA with Nivo + Ipilimumab (Ipi) in Relapsed/Refractory Acute Myeloid Leukemia: A Non-Randomized, Prospective, Phase 2 Study. Blood 2019, 134, 830. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Costello, C.; Herrera, A.F.; Locke, F.L.; Maegawa, R.O.; Savell, A.; Mazzeo, M.; Anderson, A.; Boardman, A.P.; et al. A Multicenter Phase 1 Study of Nivolumab for Relapsed Hematologic Malignancies after Allogeneic Transplantation. Blood 2020, 135, 2182–2191. [Google Scholar] [CrossRef]
- Ravandi, F.; Assi, R.; Daver, N.; Benton, C.B.; Kadia, T.; Thompson, P.A.; Borthakur, G.; Alvarado, Y.; Jabbour, E.J.; Konopleva, M.; et al. Idarubicin, Cytarabine, and Nivolumab in Patients with Newly Diagnosed Acute Myeloid Leukaemia or High-Risk Myelodysplastic Syndrome: A Single-Arm, Phase 2 Study. Lancet Haematol. 2019, 6, e480–e488. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sharon, E.; Karrison, T.G.; Zha, Y.; Fulton, N.; Streicher, H.; Sweet, K.; Yaghmour, G.; Liu, J.J.; Jonas, B.A.; et al. Randomized Phase II Study to Assess the Role of Nivolumab As Single Agent to Eliminate Minimal Residual Disease and Maintain Remission in Acute Myelogenous Leukemia (AML) Patients after Chemotherapy (NCI9706 Protocol; REMAIN Trial). Blood 2022, 140, 1716–1719. [Google Scholar] [CrossRef]
- Oran, B.; Garcia-Manero, G.; Saliba, R.M.; Alfayez, M.; Al-Atrash, G.; Ciurea, S.O.; Jabbour, E.J.; Mehta, R.S.; Popat, U.R.; Ravandi, F.; et al. Posttransplantation Cyclophosphamide Improves Transplantation Outcomes in Patients with AML/MDS Who Are Treated with Checkpoint Inhibitors. Cancer 2020, 126, 2193–2205. [Google Scholar] [CrossRef]
- Gojo, I.; Stuart, R.K.; Webster, J.; Blackford, A.; Varela, J.C.; Morrow, J.; DeZern, A.E.; Foster, M.C.; Levis, M.J.; Coombs, C.C.; et al. Multi-Center Phase 2 Study of Pembroluzimab (Pembro) and Azacitidine (AZA) in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML) and in Newly Diagnosed (≥65 Years) AML Patients. Blood 2019, 134, 832. [Google Scholar] [CrossRef]
- Goswami, M.; Gui, G.; Dillon, L.W.; Lindblad, K.E.; Thompson, J.; Valdez, J.; Kim, D.-Y.; Ghannam, J.Y.; Oetjen, K.A.; Destefano, C.B.; et al. Pembrolizumab and Decitabine for Refractory or Relapsed Acute Myeloid Leukemia. J. Immunother. Cancer 2022, 10, e003392. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F.; Vincent, B.G.; Ivanova, A.; Moore, D.; McKinnon, K.P.; Wilkinson, A.D.; Mukhopadhyay, R.; Mazziotta, F.; Knaus, H.A.; Foster, M.C.; et al. Phase II Trial of Pembrolizumab after High-Dose Cytarabine in Relapsed/Refractory Acute Myeloid Leukemia. Blood Cancer Discov. 2021, 2, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.R.; Solh, M.M.; Morris, L.E.; Holland, H.K.; Bachier-Rodriguez, L.; Zhang, X.; Guzowski, C.; Jackson, K.C.; Brown, S.; Bashey, A. Phase 2 Study of PD-1 Blockade Following Autologous Transplantation for Patients with AML Ineligible for Allogeneic Transplant. Blood Adv. 2023, 7, 5215–5224. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Ribrag, V.; Zhang, Y.; Farooqui, M.; Marinello, P.; Smith, B.D. Pembrolizumab for Myelodysplastic Syndromes after Failure of Hypomethylating Agents in the Phase 1b KEYNOTE-013 Study. Leuk. Lymphoma 2022, 63, 1660–1668. [Google Scholar] [CrossRef]
- Chien, K.S.; Kim, K.; Nogueras-Gonzalez, G.M.; Borthakur, G.; Naqvi, K.; Daver, N.G.; Montalban-Bravo, G.; Cortes, J.E.; DiNardo, C.D.; Jabbour, E.; et al. Phase II Study of Azacitidine with Pembrolizumab in Patients with Intermediate-1 or Higher-risk Myelodysplastic Syndrome. Br. J. Haematol. 2021, 195, 378–387. [Google Scholar] [CrossRef]
- Zhang, T.; Song, X.; Xu, L.; Ma, J.; Zhang, Y.; Gong, W.; Zhang, Y.; Zhou, X.; Wang, Z.; Wang, Y.; et al. The Binding of an Anti-PD-1 Antibody to FcγRΙ Has a Profound Impact on Its Biological Functions. Cancer Immunol. Immunother. 2018, 67, 1079–1090. [Google Scholar] [CrossRef]
- Desai, J.; Deva, S.; Lee, J.S.; Lin, C.-C.; Yen, C.-J.; Chao, Y.; Keam, B.; Jameson, M.; Hou, M.-M.; Kang, Y.-K.; et al. Phase IA/IB Study of Single-Agent Tislelizumab, an Investigational Anti-PD-1 Antibody, in Solid Tumors. J. Immunother. Cancer 2020, 8, e000453. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Guo, J.; Zhang, Q.; Pan, H.; Yuan, Y.; Bai, Y.; Liu, T.; Zhou, Q.; Zhao, J.; Shu, Y.; et al. Tislelizumab in Chinese Patients with Advanced Solid Tumors: An Open-Label, Non-Comparative, Phase 1/2 Study. J. Immunother. Cancer 2020, 8, e000437. [Google Scholar] [CrossRef]
- Gao, X.-N.; Su, Y.-F.; Li, M.; Jing, Y.; Wang, J.; Xu, L.; Zhang, L.-L.; Wang, A.; Wang, Y.-Z.; Zheng, X.; et al. Single-Center Phase 2 Study of PD-1 Inhibitor Combined with DNA Hypomethylation Agent + CAG Regimen in Patients with Relapsed/Refractory Acute Myeloid Leukemia. Cancer Immunol. Immunother. 2023, 72, 2769–2782. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Boss, I.; Beach, C.L.; Copeland, W.B.; Thompson, E.; Fox, B.A.; Hasle, V.E.; Hellmann, A.; Taussig, D.C.; Tormo, M.; et al. A Randomized Phase 2 Trial of Azacitidine with or without Durvalumab as First-Line Therapy for Older Patients with AML. Blood Adv. 2022, 6, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Mineishi, S.; Claxton, D.; Zhu, J.; Zhao, C.; Jia, B.; Ehmann, W.C.; Rybka, W.B.; Naik, S.; Songdej, N.; et al. A Phase I Clinical Trial of Avelumab in Combination with Decitabine as First Line Treatment of Unfit Patients with Acute Myeloid Leukemia. Am. J. Hematol. 2021, 96, E46. [Google Scholar] [CrossRef]
- Saxena, K.; Herbrich, S.M.; Pemmaraju, N.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Pierce, S.A.; Jabbour, E.; Wang, S.A.; Bueso-Ramos, C.; et al. A Phase 1b/2 Study of Azacitidine with PD-L1 Antibody Avelumab in Relapsed/Refractory Acute Myeloid Leukemia. Cancer 2021, 127, 3761–3771. [Google Scholar] [CrossRef]
- Stahl, M.; DeVeaux, M.; Montesinos, P.; Itzykson, R.; Ritchie, E.K.; Sekeres, M.A.; Barnard, J.D.; Podoltsev, N.A.; Brunner, A.M.; Komrokji, R.S.; et al. Hypomethylating Agents in Relapsed and Refractory AML: Outcomes and Their Predictors in a Large International Patient Cohort. Blood Adv. 2018, 2, 923–932. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Salimi, T.; Epstein, R.S. Real-World Use and Outcomes of Hypomethylating Agent Therapy in Higher-Risk Myelodysplastic Syndromes: Why Are We Not Achieving the Promise of Clinical Trials? Future Oncol. 2021, 17, 5163–5175. [Google Scholar] [CrossRef]
- Giannopoulos, K. Targeting Immune Signaling Checkpoints in Acute Myeloid Leukemia. J. Clin. Med. 2019, 8, 236. [Google Scholar] [CrossRef]
- Alatrash, G.; Daver, N.; Mittendorf, E.A. Targeting Immune Checkpoints in Hematologic Malignancies. Pharmacol. Rev. 2016, 68, 1014–1025. [Google Scholar] [CrossRef]
- Costello, R.T.; Mallet, F.; Sainty, D.; Maraninchi, D.; Gastaut, J.A.; Olive, D. Regulation of CD80/B7-1 and CD86/B7-2 Molecule Expression in Human Primary Acute Myeloid Leukemia and Their Role in Allogenic Immune Recognition. Eur. J. Immunol. 1998, 28, 90–103. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.-B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.S.; Flamand, Y.; Tomlinson, B.K.; Keng, M.; Mendez, L.M.; Khaled, S.; Bashey, A.; Brunner, A.M.; Savell, A.; Neuberg, D.; et al. Safety and Efficacy of Decitabine Plus Ipilimumab in Relapsed or Refractory MDS/AML in the Post-BMT or Transplant Naïve Settings. Blood 2020, 136, 15–17. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Knaus, H.A.; Robinson, T.M.; Towlerton, A.M.H.; Warren, E.H.; Zeidner, J.F.; Blackford, A.L.; Duffield, A.S.; Rizzieri, D.; Frattini, M.G.; et al. A Multi-Center Phase I Trial of Ipilimumab in Patients with Myelodysplastic Syndromes Following Hypomethylating Agent Failure. Clin. Cancer Res. 2018, 24, 3519–3527. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, M.; Brossart, P.; Cant, C.; Cella, M.; Colonna, M.; Brugger, W.; Kanz, L.; Ullrich, A.; Bühring, H.J. Signal-Regulatory Protein Alpha (SIRPalpha) but Not SIRPbeta Is Involved in T-Cell Activation, Binds to CD47 with High Affinity, and Is Expressed on Immature CD34+CD38− Hematopoietic Cells. Blood 2001, 97, 2741–2749. [Google Scholar] [CrossRef]
- Tsai, R.K.; Discher, D.E. Inhibition of “Self” Engulfment through Deactivation of Myosin-II at the Phagocytic Synapse between Human Cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin Is the Dominant Pro-Phagocytic Signal on Multiple Human Cancers and Is Counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63ra94. [Google Scholar] [CrossRef]
- Jiang, H.; Fu, R.; Wang, H.; Li, L.; Liu, H.; Shao, Z. CD47 Is Expressed Abnormally on Hematopoietic Cells in Myelodysplastic Syndrome. Leuk. Res. 2013, 37, 907–910. [Google Scholar] [CrossRef]
- Ostendorf, B.N.; Flenner, E.; Flörcken, A.; Westermann, J. Phenotypic Characterization of Aberrant Stem and Progenitor Cell Populations in Myelodysplastic Syndromes. PLoS ONE 2018, 13, e0197823. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef]
- Jiang, Z.; Sun, H.; Yu, J.; Tian, W.; Song, Y. Targeting CD47 for Cancer Immunotherapy. J. Hematol. Oncol. 2021, 14, 180. [Google Scholar] [CrossRef]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.-P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2019, 9, 1380. [Google Scholar] [CrossRef]
- Feng, D.; Gip, P.; McKenna, K.M.; Zhao, F.; Mata, O.; Choi, T.S.; Duan, J.; Sompalli, K.; Majeti, R.; Weissman, I.L.; et al. Combination Treatment with 5F9 and Azacitidine Enhances Phagocytic Elimination of Acute Myeloid Leukemia. Blood 2018, 132, 2729. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, Q.; Weng, C.; Ramage, C.L.; Nishida, Y.; Chao, M.; Maute, R.L.; Herbrich, S.; Zhang, W.; Andreeff, M.; et al. Combined Blockade of CD47-Sirpa Interaction By 5F9 (Magrolimab) and Azacitidine/Venetoclax Therapy Facilitates Macrophage-Mediated Anti-Leukemia Efficacy in AML Pre-Clinical Models. Blood 2021, 138, 510. [Google Scholar] [CrossRef]
- Daver, N.G.; Vyas, P.; Kambhampati, S.; Al Malki, M.M.; Larson, R.A.; Asch, A.S.; Mannis, G.; Chai-Ho, W.; Tanaka, T.N.; Bradley, T.J.; et al. Tolerability and Efficacy of the Anticluster of Differentiation 47 Antibody Magrolimab Combined with Azacitidine in Patients with Previously Untreated AML: Phase Ib Results. J. Clin. Oncol. 2023, 1–12. [Google Scholar] [CrossRef]
- Daver, N.; Vyas, P.; Chao, M.; Xing, G.; Renard, C.; Ramsingh, G.; Sallman, D.A.; Wei, A.H. A Phase 3, Randomized, Open-Label Study Evaluating the Safety and Efficacy of Magrolimab in Combination with Azacitidine in Previously Untreated Patients with TP53-Mutant Acute Myeloid Leukemia. Blood 2021, 138, 3426. [Google Scholar] [CrossRef]
- Daver, N.; Senapati, J.; Maiti, A.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Pemmaraju, N.; Jabbour, E.; Montalban-Bravo, G.; Tang, G.; et al. Phase I/II Study of Azacitidine (AZA) with Venetoclax (VEN) and Magrolimab (Magro) in Patients (Pts) with Newly Diagnosed (ND) Older/Unfit or High-Risk Acute Myeloid Leukemia (AML) and Relapsed/Refractory (R/R) AML. Blood 2022, 140, 141–144. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Przespolewski, A.; Abaza, Y.; Byrne, M.; Fong, A.P.; Jin, F.; Forgie, A.J.; Tsiatis, A.C.; Guan, S.; Erba, H.P. Evorpacept (ALX148), a CD47-Blocking Myeloid Checkpoint Inhibitor, in Combination with Azacitidine and Venetoclax in Patients with Acute Myeloid Leukemia (ASPEN-05): Results from Phase 1a Dose Escalation Part. Blood 2022, 140, 9046–9047. [Google Scholar] [CrossRef]
- Qi, J.; Li, J.; Jiang, B.; Jiang, B.; Liu, H.; Cao, X.; Zhang, M.; Meng, Y.; Ma, X.; Jia, Y.; et al. A Phase I/IIa Study of Lemzoparlimab, a Monoclonal Antibody Targeting CD47, in Patients with Relapsed and/or Refractory Acute Myeloid Leukemia (AML) and Myelodysplastic Syndrome (MDS): Initial Phase I Results. Blood 2020, 136, 30–31. [Google Scholar] [CrossRef]
- Brierley, C.K.; Staves, J.; Roberts, C.; Johnson, H.; Vyas, P.; Goodnough, L.T.; Murphy, M.F. The Effects of Monoclonal anti-CD47 on RBCs, Compatibility Testing, and Transfusion Requirements in Refractory Acute Myeloid Leukemia. Transfusion 2019, 59, 2248–2254. [Google Scholar] [CrossRef]
- ALX Oncology Inc. A Phase 1/2 Study of Evorpacept (ALX148) in Combination with Azacitidine in Patients with Higher Risk Myelodysplastic Syndrome (MDS) (ASPEN-02). 2023. Available online: Clinicaltrials.gov (accessed on 1 January 2020).
- Ferris, R.L.; Lu, B.; Kane, L.P. Too Much of a Good Thing? Tim-3 and TCR Signaling in T Cell Exhaustion. J. Immunol. 2014, 193, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-Infiltrating and Peripheral Blood T-Cell Immunophenotypes Predict Early Relapse in Localized Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of Tissue Inflammation by the Immune Receptor Tim-3 Expressed on Innate Immune Cells. Science 2007, 318, 1141–1143. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-Cell Exhaustion in the Tumor Microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 Pathways to Reverse T Cell Exhaustion and Restore Anti-Tumor Immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Hobo, W.; Hutten, T.J.A.; Schaap, N.P.M.; Dolstra, H. Immune Checkpoint Molecules in Acute Myeloid Leukaemia: Managing the Double-Edged Sword. Br. J. Haematol. 2018, 181, 38–53. [Google Scholar] [CrossRef]
- Darwish, N.H.E.; Sudha, T.; Godugu, K.; Elbaz, O.; Abdelghaffar, H.A.; Hassan, E.E.A.; Mousa, S.A. Acute Myeloid Leukemia Stem Cell Markers in Prognosis and Targeted Therapy: Potential Impact of BMI-1, TIM-3 and CLL-1. Oncotarget 2016, 7, 57811–57820. [Google Scholar] [CrossRef]
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 Is a Promising Target to Selectively Kill Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2010, 7, 708–717. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Komrokji, R.S.; Brunner, A.M. TIM-3 Pathway Dysregulation and Targeting in Cancer. Expert Rev. Anticancer Ther. 2021, 21, 523–534. [Google Scholar] [CrossRef]
- Zeidan, A.; Esteve, J.; Kim, H.-J.; Miyazaki, Y.; Platzbecker, U.; Schuh, A.; Westermann, J.; Malek, K.; Scott, J.; Niolat, J.; et al. AML-187: The STIMULUS Clinical Trial Program: Evaluating Combination Therapy with MBG453 in Patients with Higher-Risk Myelodysplastic Syndrome (HR-MDS) or Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2020, 20, S188. [Google Scholar] [CrossRef]
- Brunner, A.; Borate, U.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Wermke, M.; Janssen, J.; Traer, E.; et al. AML-190: Anti-TIM-3 Antibody MBG453 in Combination with Hypomethylating Agents (HMAs) in Patients with High-Risk Myelodysplastic Syndrome (HR-MDS) and Acute Myeloid Leukemia: A Phase 1 Study. Clin. Lymphoma Myeloma Leuk. 2020, 20, S188–S189. [Google Scholar] [CrossRef]
- Li, N.; Workman, C.J.; Martin, S.M.; Vignali, D.A.A. Biochemical Analysis of the Regulatory T Cell Protein Lymphocyte Activation Gene-3 (LAG-3; CD223). J. Immunol. 2004, 173, 6806–6812. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; Black, R.A.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases Regulate T-Cell Proliferation and Effector Function via LAG-3. EMBO J. 2007, 26, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Noviello, M.; Manfredi, F.; Ruggiero, E.; Perini, T.; Oliveira, G.; Cortesi, F.; De Simone, P.; Toffalori, C.; Gambacorta, V.; Greco, R.; et al. Bone Marrow Central Memory and Memory Stem T-Cell Exhaustion in AML Patients Relapsing after HSCT. Nat. Commun. 2019, 10, 1065. [Google Scholar] [CrossRef]
- Chen, Y.; Tan, J.; Huang, S.; Huang, X.; Huang, J.; Chen, J.; Yu, Z.; Lu, Y.; Weng, J.; Du, X.; et al. Higher Frequency of the CTLA-4+ LAG-3+ T-Cell Subset in Patients with Newly Diagnosed Acute Myeloid Leukemia. Asia-Pac. J. Clin. Oncol. 2020, 16, e12–e18. [Google Scholar] [CrossRef]
- Hintzen, R.Q.; Lens, S.M.; Lammers, K.; Kuiper, H.; Beckmann, M.P.; van Lier, R.A. Engagement of CD27 with Its Ligand CD70 Provides a Second Signal for T Cell Activation. J. Immunol. 1995, 154, 2612–2623. [Google Scholar] [CrossRef]
- Bowman, M.R.; Crimmins, M.A.; Yetz-Aldape, J.; Kriz, R.; Kelleher, K.; Herrmann, S. The Cloning of CD70 and Its Identification as the Ligand for CD27. J. Immunol. 1994, 152, 1756–1761. [Google Scholar] [CrossRef]
- Jacobs, J.; Deschoolmeester, V.; Zwaenepoel, K.; Rolfo, C.; Silence, K.; Rottey, S.; Lardon, F.; Smits, E.; Pauwels, P. CD70: An Emerging Target in Cancer Immunotherapy. Pharmacol. Ther. 2015, 155, 1–10. [Google Scholar] [CrossRef]
- Riether, C.; Schürch, C.M.; Bührer, E.D.; Hinterbrandner, M.; Huguenin, A.-L.; Hoepner, S.; Zlobec, I.; Pabst, T.; Radpour, R.; Ochsenbein, A.F. CD70/CD27 Signaling Promotes Blast Stemness and Is a Viable Therapeutic Target in Acute Myeloid Leukemia. J. Exp. Med. 2017, 214, 359–380. [Google Scholar] [CrossRef]
- Riether, C.; Pabst, T.; Höpner, S.; Bacher, U.; Hinterbrandner, M.; Banz, Y.; Müller, R.; Manz, M.G.; Gharib, W.H.; Francisco, D.; et al. Targeting CD70 with Cusatuzumab Eliminates Acute Myeloid Leukemia Stem Cells in Patients Treated with Hypomethylating Agents. Nat. Med. 2020, 26, 1459–1467. [Google Scholar] [CrossRef]
- Pabst, T.; Vey, N.; Adès, L.; Bacher, U.; Bargetzi, M.; Fung, S.; Gaidano, G.; Gandini, D.; Hultberg, A.; Johnson, A.; et al. Results from a Phase I/II Trial of Cusatuzumab Combined with Azacitidine in Patients with Newly Diagnosed Acute Myeloid Leukemia Who Are Ineligible for Intensive Chemotherapy. Haematologica 2023, 108, 1793–1802. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Bargou, R.C. T Cell-Engaging Therapies—BiTEs and Beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef] [PubMed]
- Bras, A.E.; Haas, V.; Stigt, A.; Jongen-Lavrencic, M.; Beverloo, H.B.; Marvelde, J.G.; Zwaan, C.M.; Dongen, J.J.M.; Leusen, J.H.W.; Velden, V.H.J. CD123 Expression Levels in 846 Acute Leukemia Patients Based on Standardized Immunophenotyping. Cytometry 2019, 96, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; Van Dongen, G.A.M.S.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Walsum, M.S.; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The Novel AML Stem Cell–Associated Antigen CLL-1 Aids in Discrimination between Normal and Leukemic Stem Cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as Salvage Immunotherapy for Refractory Acute Myeloid Leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Desai, P.; Daskalakis, N.; Donnellan, W.; Ferrante, L.; Goldberg, J.D.; Grunwald, M.R.; Guttke, C.; Li, X.; Perez-Simon, J.A.; et al. First-in-Human Study of JNJ-63709178, a CD123/CD3 Targeting Antibody, in Relapsed/Refractory Acute Myeloid Leukemia. Clin. Transl. Sci. 2023, 16, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Bashey, A.; Stock, W.; Foran, J.M.; Mawad, R.; Egan, D.; Blum, W.; Yang, A.; Pastore, A.; Johnson, C.; et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of Vibecotamab (XmAb14045), a CD123 x CD3 T Cell-Engaging Bispecific Antibody; Initial Results of a Phase 1 Study. Blood 2020, 136, 4–5. [Google Scholar] [CrossRef]
- Short, N.J.; Bachireddy, P.; Huang, X.; Hwang, H.; Leng, X.; Lee, J.; Nguyen, D.; Garcia-Manero, G.; Dinardo, C.D.; Borthakur, G.; et al. A Phase II Study of Vibecotamab, a CD3-CD123 Bispecific T-Cell Engaging Antibody, for MRD-Positive AML and MDS after Hypomethylating Agent Failure. J. Clin. Oncol. 2023, 41, TPS7076. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated Results from Phase I Dose-Escalation Study of AMG 330, a Bispecific T-Cell Engager Molecule, in Patients with Relapsed/Refractory Acute Myeloid Leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Subklewe, M.; Stein, A.; Walter, R.B.; Bhatia, R.; Wei, A.H.; Ritchie, D.; Bücklein, V.; Vachhani, P.; Dai, T.; Hindoyan, A.; et al. Preliminary Results from a Phase 1 First-in-Human Study of AMG 673, a Novel Half-Life Extended (HLE) Anti-CD33/CD3 BiTE® (Bispecific T-Cell Engager) in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 2019, 134, 833. [Google Scholar] [CrossRef]
- Westervelt, P.; Cortes, J.E.; Altman, J.K.; Long, M.; Oehler, V.G.; Gojo, I.; Guenot, J.; Chun, P.; Roboz, G.J. Phase 1 First-in-Human Trial of AMV564, a Bivalent Bispecific (2:2) CD33/CD3 T-Cell Engager, in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML). Blood 2019, 134, 834. [Google Scholar] [CrossRef]
- Cheng, P.; Chen, X.; Dalton, R.; Calescibetta, A.; So, T.; Gilvary, D.; Ward, G.; Smith, V.; Eckard, S.; Fox, J.A.; et al. Immunodepletion of MDSC by AMV564, a Novel Bivalent, Bispecific CD33/CD3 T Cell Engager, Ex Vivo in MDS and Melanoma. Mol. Ther. 2022, 30, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Cortes, J.; Huls, G.; Venditti, A.; Breems, D.; De Botton, S. Update from the Ongoing Phase I Multinational Study of MCLA-117, a Bispecific CLEC12A x CD3 T-Cell Engager, in Patients (Pts) with Acute Myelogenous Leukemia (AML); European Hematology Association: The Hague, The Netherlands, 2020. [Google Scholar]
- Ngai, L.L.; Ma, C.Y.; Maguire, O.; Do, A.D.; Robert, A.; Logan, A.C.; Griffiths, E.A.; Nemeth, M.J.; Green, C.; Pourmohamad, T.; et al. Bimodal Expression of Potential Drug Target CLL-1 (CLEC12A) on CD34+ Blasts of AML Patients. Eur. J. Haematol. 2021, 107, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Schorr, C.; Perna, F. Targets for Chimeric Antigen Receptor T-Cell Therapy of Acute Myeloid Leukemia. Front. Immunol. 2022, 13, 1085978. [Google Scholar] [CrossRef] [PubMed]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, T.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T Cells for Treatment of Relapsed/Refractory Acute Myelogenous Leukemia. Leukemia 2021, 35, 3282–3286. [Google Scholar] [CrossRef] [PubMed]
- Cummins, K.D.; Frey, N.; Nelson, A.M.; Schmidt, A.; Luger, S.; Isaacs, R.E.; Lacey, S.F.; Hexner, E.; Melenhorst, J.J.; June, C.H.; et al. Treating Relapsed/Refractory (RR) AML with Biodegradable Anti-CD123 CAR Modified T Cells. Blood 2017, 130, 1359. [Google Scholar]
- Naik, S.; Madden, R.M.; Lipsitt, A.; Lockey, T.; Bran, J.; Rubnitz, J.E.; Klco, J.; Shulkin, B.; Patil, S.L.; Schell, S.; et al. Safety and Anti-Leukemic Activity of CD123-CAR T Cells in Pediatric Patients with AML: Preliminary Results from a Phase 1 Trial. Blood 2022, 140, 4584–4585. [Google Scholar] [CrossRef]
- Gurney, M.; O’Dwyer, M. Realizing Innate Potential: CAR-NK Cell Therapies for Acute Myeloid Leukemia. Cancers 2021, 13, 1568. [Google Scholar] [CrossRef]
- Kang, S.; Li, Y.; Qiao, J.; Meng, X.; He, Z.; Gao, X.; Yu, L. Antigen-Specific TCR-T Cells for Acute Myeloid Leukemia: State of the Art and Challenges. Front. Oncol. 2022, 12, 787108. [Google Scholar] [CrossRef]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch Me If You Can: How AML and Its Niche Escape Immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef]
- Pimenta, D.B.; Varela, V.A.; Datoguia, T.S.; Caraciolo, V.B.; Lopes, G.H.; Pereira, W.O. The Bone Marrow Microenvironment Mechanisms in Acute Myeloid Leukemia. Front. Cell Dev. Biol. 2021, 9, 764698. [Google Scholar] [CrossRef] [PubMed]
- Philipp, N.; Kazerani, M.; Nicholls, A.; Vick, B.; Wulf, J.; Straub, T.; Scheurer, M.; Muth, A.; Hänel, G.; Nixdorf, D.; et al. T-Cell Exhaustion Induced by Continuous Bispecific Molecule Exposure Is Ameliorated by Treatment-Free Intervals. Blood 2022, 140, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molica, M.; Perrone, S.; Andriola, C.; Rossi, M. Immunotherapy with Monoclonal Antibodies for Acute Myeloid Leukemia: A Work in Progress. Cancers 2023, 15, 5060. https://doi.org/10.3390/cancers15205060
Molica M, Perrone S, Andriola C, Rossi M. Immunotherapy with Monoclonal Antibodies for Acute Myeloid Leukemia: A Work in Progress. Cancers. 2023; 15(20):5060. https://doi.org/10.3390/cancers15205060
Chicago/Turabian StyleMolica, Matteo, Salvatore Perrone, Costanza Andriola, and Marco Rossi. 2023. "Immunotherapy with Monoclonal Antibodies for Acute Myeloid Leukemia: A Work in Progress" Cancers 15, no. 20: 5060. https://doi.org/10.3390/cancers15205060
APA StyleMolica, M., Perrone, S., Andriola, C., & Rossi, M. (2023). Immunotherapy with Monoclonal Antibodies for Acute Myeloid Leukemia: A Work in Progress. Cancers, 15(20), 5060. https://doi.org/10.3390/cancers15205060