

AKT2 Loss Impairs BRAF-Mutant Melanoma Metastasis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Strains
2.2. Xenografts
2.3. Luciferase Imaging
2.4. Metastasis Assays
2.5. Tumor Cell Isolation and Tissue Preparation
2.6. Cell Lines/Tissue Culture
2.7. Immunoblot and Immunoprecipitation Analysis
2.8. Wound Healing Assay
2.9. Migration/Invasion Assays
2.10. Anchorage-Independent Growth (Soft Agar) Assay
2.11. Seahorse Glycolytic Rate Assay
2.12. Quantitative RT-PCR
2.13. Cell Cycle Analysis
2.14. Statistical Analysis
3. Results
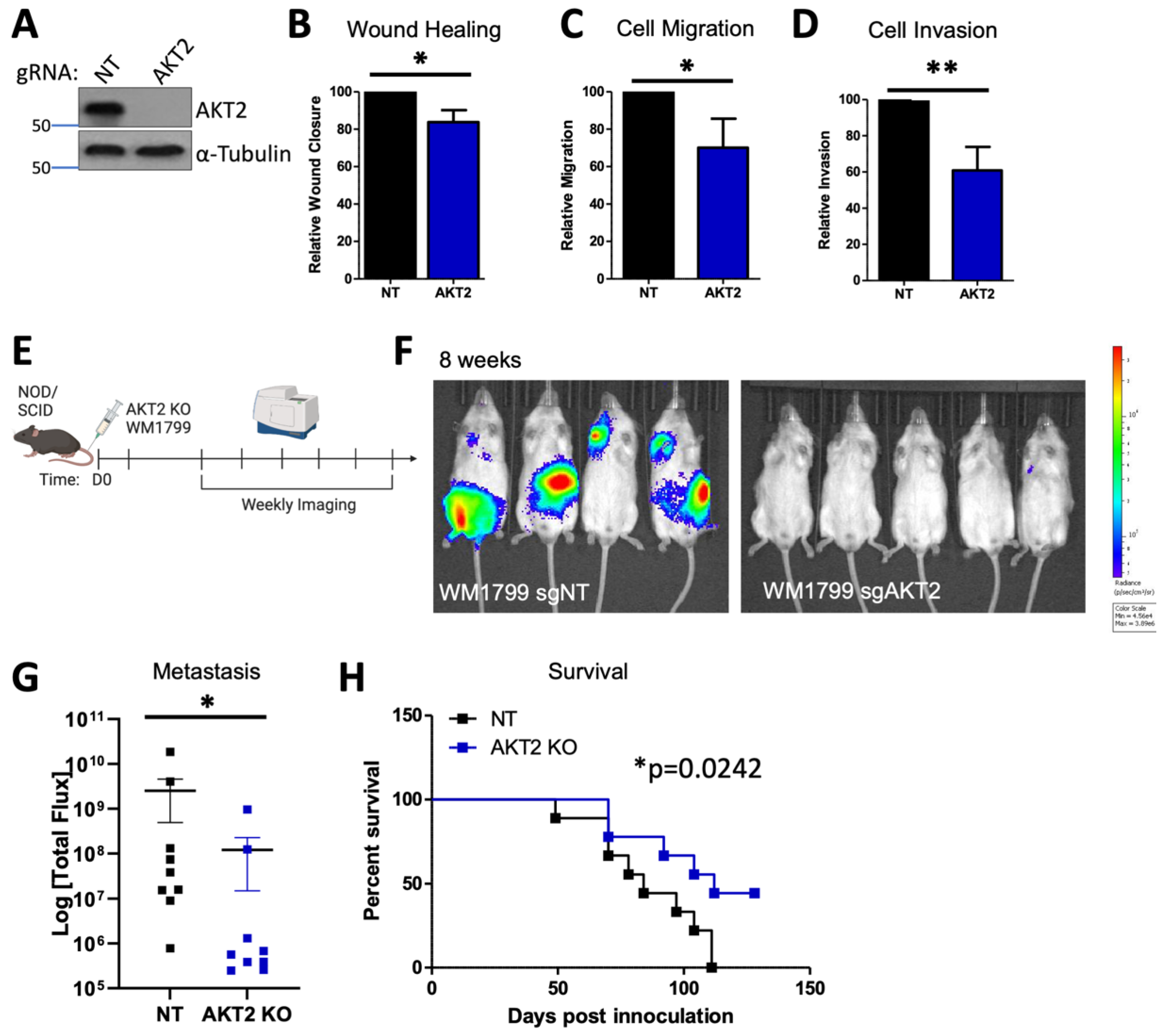
3.1. AKT2 Depletion Impairs Cell Migration and Invasion in Human Melanoma Cells
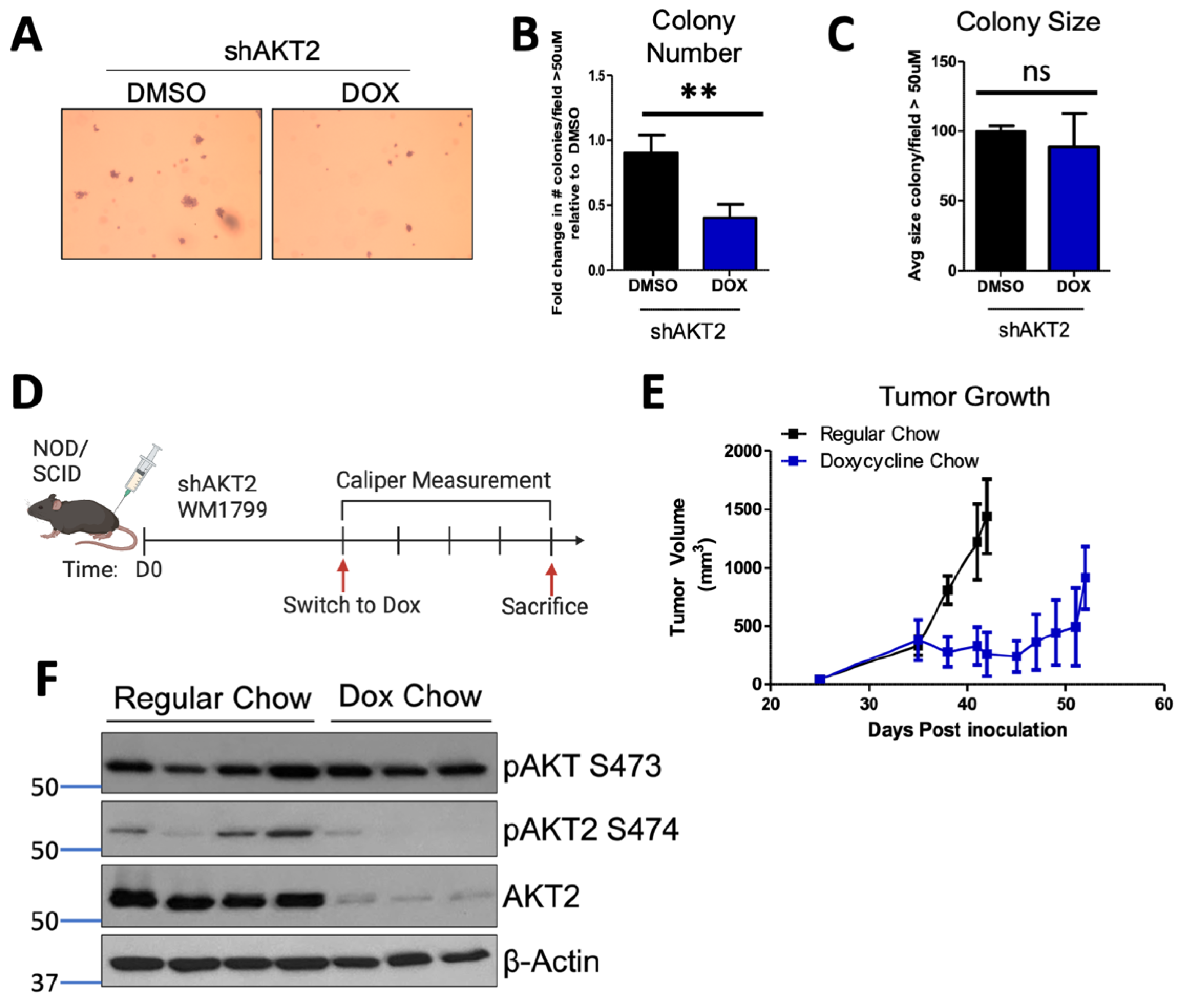
3.2. AKT2 Depletion Restricts Anchorage-Independent Growth In Vitro and In Vivo
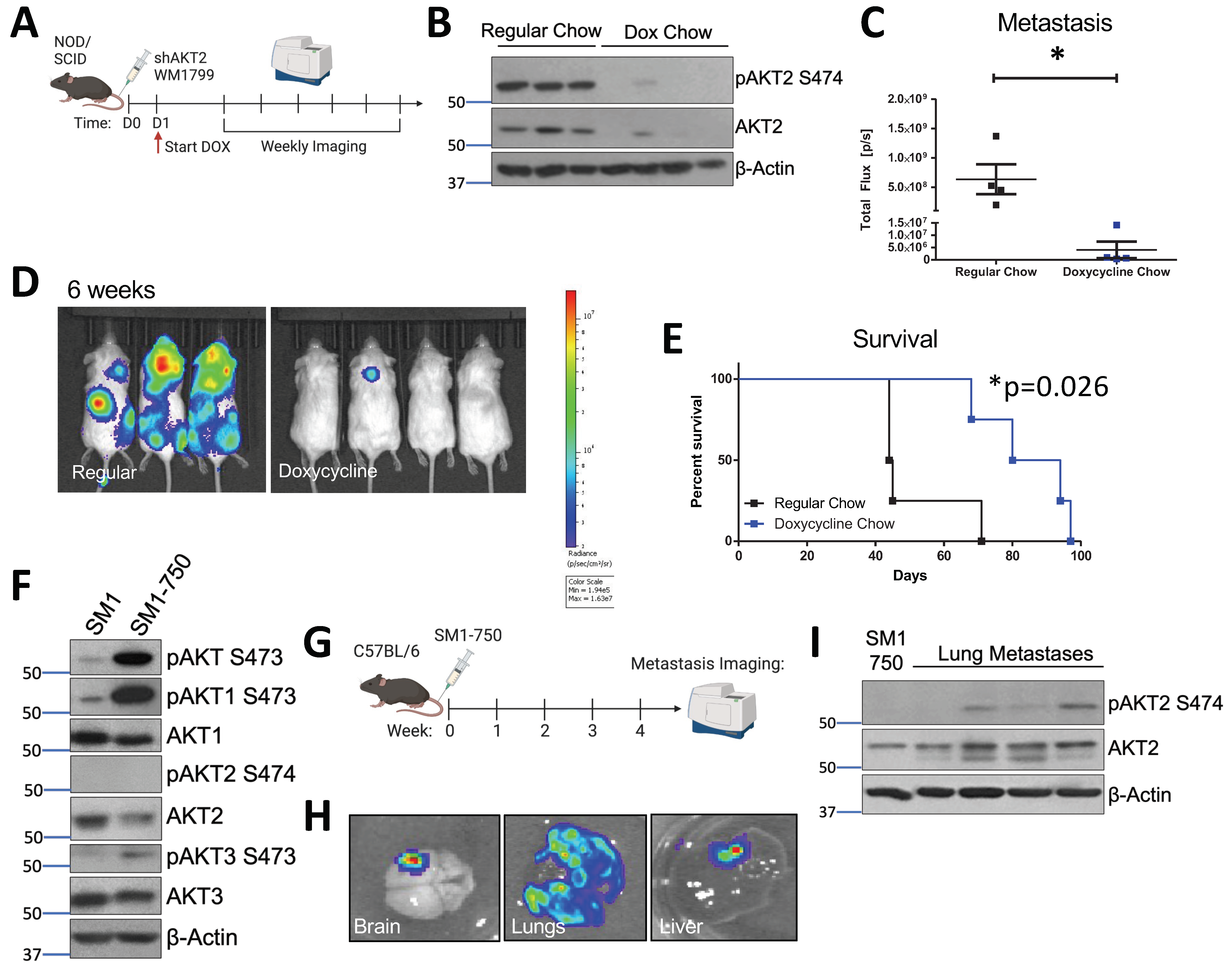
3.3. AKT2 Depletion Delays Metastatic Onset and Extends the Survival of Melanoma-Bearing Mice
3.4. AKT2 Phosphorylation Occurs in Metastatic Mouse Melanoma Lesions
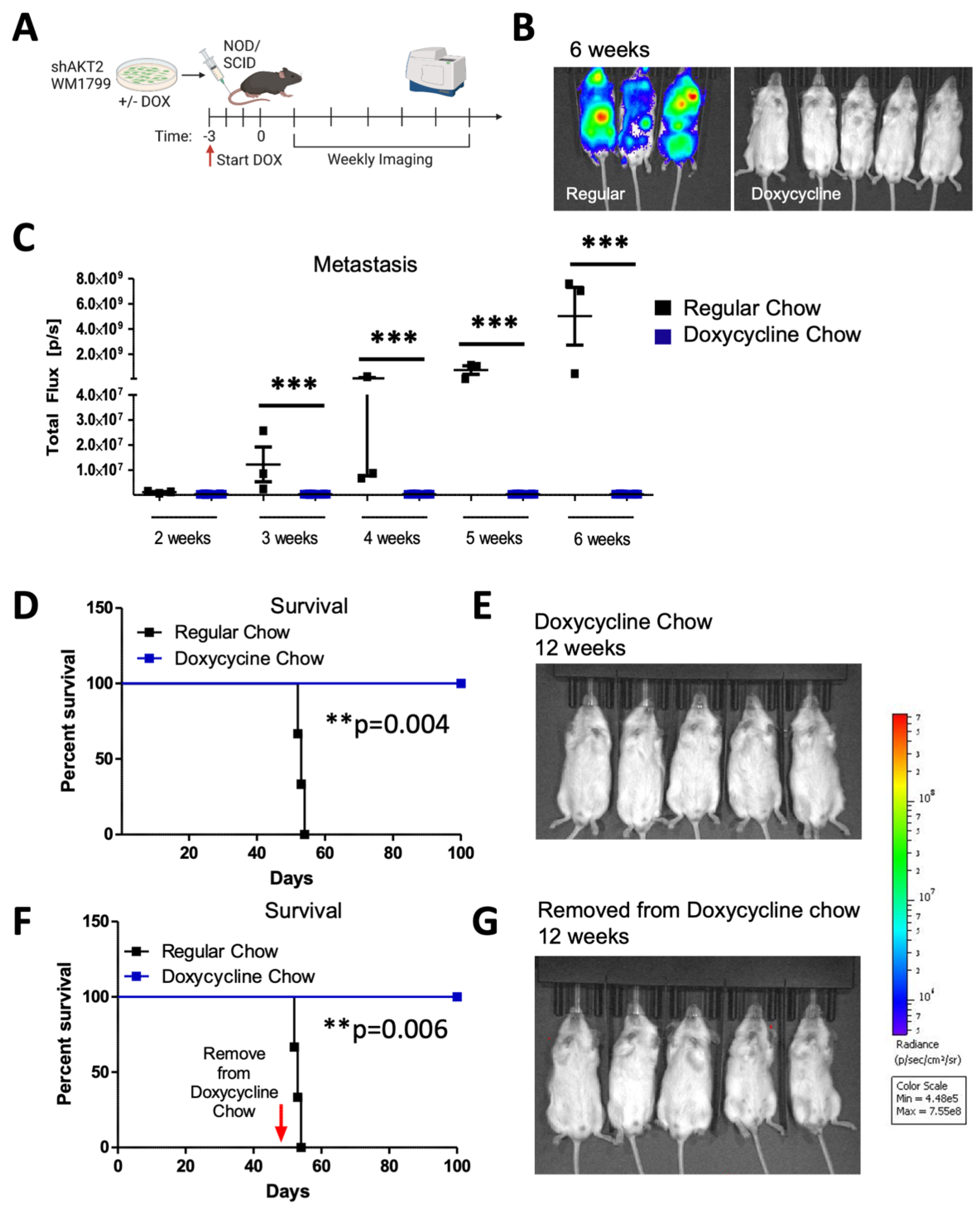
3.5. Prophylactic AKT2 Depletion Prevents Metastatic Cell Seeding
3.6. AKT2 Deletion Impairs Melanoma Migration, Invasion, and Metastasis
3.7. AKT1 Deletion Impairs Primary Tumor Formation
3.8. AKT1 Knockdown Impairs Cellular Proliferation and Anchorage-Independent Growth
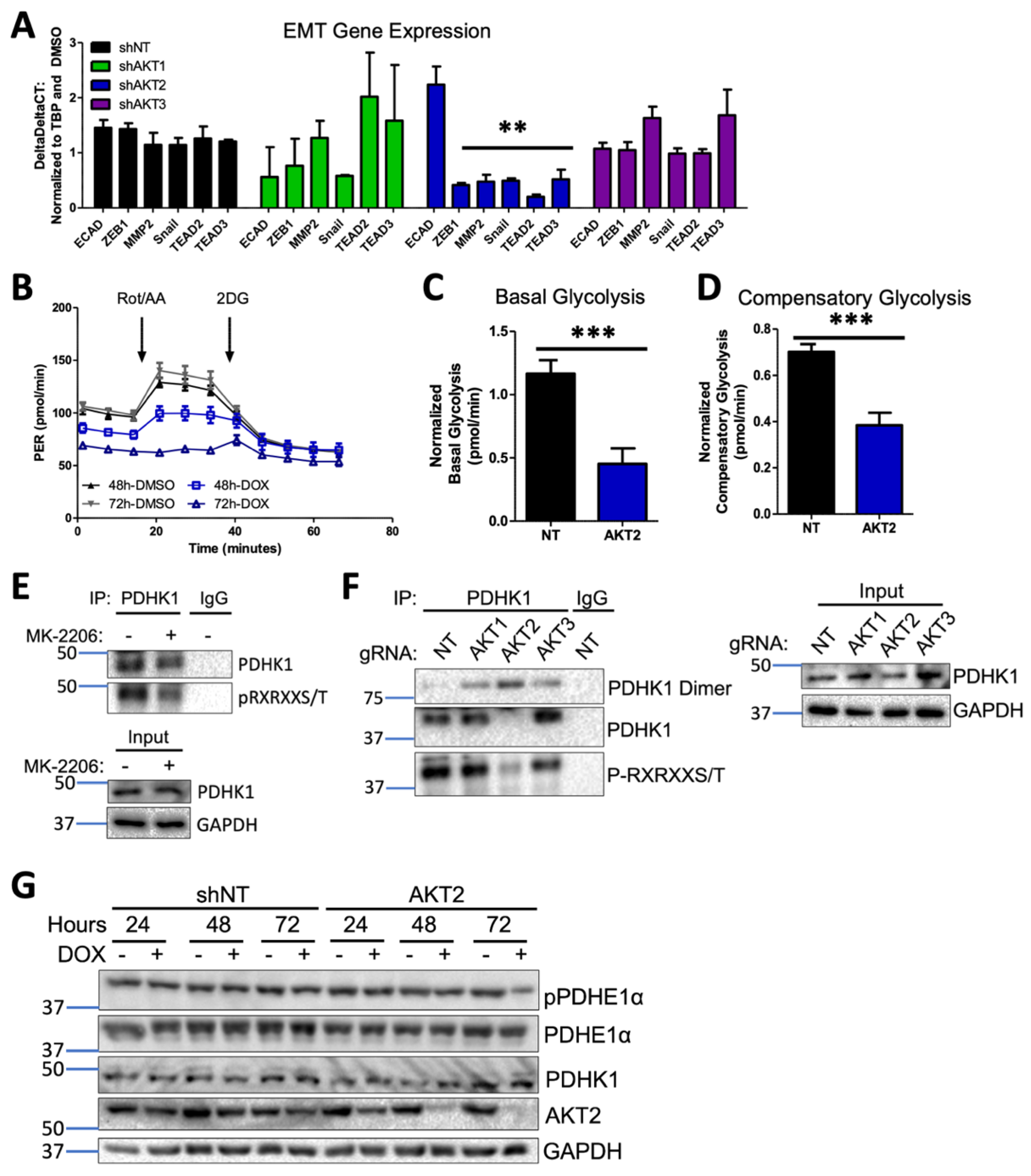
3.9. AKT2 Depletion Inhibits EMT and Impairs Glycolysis through PDHK1 Activity in Melanoma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Dai, D.L.; Martinka, M.; Li, G. Prognostic Significance of Activated Akt Expression in Melanoma: A Clinicopathologic Study of 292 Cases. J. Clin. Oncol. 2005, 23, 1473–1482. [Google Scholar] [CrossRef]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. BrafV600E Cooperates with Pten Loss to Induce Metastatic Melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Ibrahim, N.; Haluska, F.G. Molecular Pathogenesis of Cutaneous Melanocytic Neoplasms. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 551–579. [Google Scholar] [CrossRef]
- Bucheit, A.D.; Chen, G.; Siroy, A.; Tetzlaff, M.; Broaddus, R.; Milton, D.; Fox, P.; Bassett, R.; Hwu, P.; Gershenwald, J.E.; et al. Complete Loss of PTEN Protein Expression Correlates with Shorter Time to Brain Metastasis and Survival in Stage IIIB/C Melanoma Patients with BRAFV600 Mutations. Clin. Cancer Res. 2014, 20, 5527–5536. [Google Scholar] [CrossRef]
- Giles, K.M.; Rosenbaum, B.E.; Berger, M.; Izsak, A.; Li, Y.; Bochaca, I.I.; Vega-Saenz de Miera, E.; Wang, J.; Darvishian, F.; Zhong, H.; et al. Revisiting the Clinical and Biologic Relevance of Partial PTEN Loss in Melanoma. J. Investig. Dermatol. 2019, 139, 430–438. [Google Scholar] [CrossRef]
- Davies, M.A.; Katherine, S.-H.; Lin, E.; Tellez, C.; Deng, W.; Gopal, Y.N.; Woodman, S.E.; Calderone, T.C.; Ju, Z.; Lazar, A.J.; et al. Integrated Molecular and Clinical Analysis of AKT Activation in Metastatic Melanoma. Clin. Cancer Res. 2009, 15, 7538–7546. [Google Scholar] [CrossRef]
- Niessner, H.; Schmitz, J.; Tabatabai, G.; Schmid, A.M.; Calaminus, C.; Sinnberg, T.; Weide, B.; Eigentler, T.K.; Garbe, C.; Schittek, B.; et al. PI3K Pathway Inhibition Achieves Potent Antitumor Activity in Melanoma Brain Metastases In Vitro and In Vivo. Clin. Cancer Res. 2016, 22, 5818–5828. [Google Scholar] [CrossRef]
- Amaral, T.; Niessner, H.; Sinnberg, T.; Thomas, I.; Meiwes, A.; Garbe, C.; Garzarolli, M.; Rauschenberg, R.; Eigentler, T.; Meier, F. An Open-Label, Single-Arm, Phase II Trial of Buparlisib in Patients with Melanoma Brain Metastases Not Eligible for Surgery or Radiosurgery—The BUMPER Study. Neurooncol Adv. 2020, 2, vdaa140. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Gowda, R.; Sharma, A.; Noory, M.A.; Dinavahi, S.S.; Kardos, G.; Drabick, J.J.; Robertson, G.P. Improving Pharmacological Targeting of AKT in Melanoma. Cancer Lett. 2017, 404, 29–36. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K Inhibitors Are Finally Coming of Age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef]
- Halder, A.K.; Cordeiro, M.N.D.S. AKT Inhibitors: The Road Ahead to Computational Modeling-Guided Discovery. Int. J. Mol. Sci. 2021, 22, 3944. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef]
- Dummler, B.; Hemmings, B. Physiological Roles of PKB/Akt Isoforms in Development and Disease. Biochem. Soc. Trans. 2007, 35, 231–235. [Google Scholar] [CrossRef]
- Dummler, B.; Tschopp, O.; Hynx, D.; Yang, Z.-Z.; Dirnhofer, S. Hemmings Life with a Single Isoform of Akt: Mice Lacking Akt2 and Akt3 Are Viable but Display Impaired Glucose Homeostasis and Growth Deficiencies. Mol. Cell. Biol. 2006, 26, 8042–8051. [Google Scholar] [CrossRef]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/PKBα Is Required for Normal Growth but Dispensable for Maintenance of Glucose Homeostasis in Mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef]
- Cho, H.; Mu, J.; Kim, J.; Thorvaldsen, J.; Chu, Q.; Crenshaw, E.; Kaestner, K.; Bartolomei, M.; Shulman, G.; Birnbaum, M. Insulin Resistance and a Diabetes Mellitus-like Syndrome in Mice Lacking the Protein Kinase Akt2 (PKB Beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef]
- Gonzalez, E.; McGraw, T.E. The Akt Kinases: Isoform Specificity in Metabolism and Cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef]
- Testa, J.R.; Tsichlis, P.N. AKT Signaling in Normal and Malignant Cells. Oncogene 2005, 24, 7391–7393. [Google Scholar] [CrossRef]
- Dillon, R.L.; Muller, W.J. Distinct Biological Roles for the Akt Family in Mammary Tumor Progression. Cancer Res. 2010, 70, 4260–4264. [Google Scholar] [CrossRef]
- Irie, H.Y.; Pearline, R.V.; Grueneberg, D.; Hsia, M.; Ravichandran, P.; Kothari, N.; Natesan, S.; Brugge, J.S. Distinct Roles of Akt1 and Akt2 in Regulating Cell Migration and Epithelial-Mesenchymal Transition. J. Cell Biol. 2005, 171, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Maroulakou, I.G.; Oemler, W.; Naber, S.P.; Tsichlis, P.N. Akt1 Ablation Inhibits, Whereas Akt2 Ablation Accelerates, the Development of Mammary Adenocarcinomas in Mouse Mammary Tumor Virus (MMTV)-ErbB2/Neu and MMTV-Polyoma Middle T Transgenic Mice. Cancer Res. 2007, 67, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated Akt3 Activity Promotes Development of Malignant Melanoma. Cancer Res. 2004, 64, 7002–7010. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Dai, M.; Lu, A.; Yu, E.; Merlino, G. PHLPP1 Mediates Melanoma Metastasis Suppression through Repressing AKT2 Activation. Oncogene 2018, 37, 2225–2236. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Cho, J.H.; Robinson, J.P.; Arave, R.A.; Burnett, W.J.; Kircher, D.A.; Chen, G.; Davies, M.A.; Grossmann, A.H.; VanBrocklin, M.W.; McMahon, M.; et al. AKT1 Activation Promotes Development of Melanoma Metastases. Cell Rep. 2015, 13, 898–905. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832.e3. [Google Scholar] [CrossRef]
- Cheung, M.; Sharma, A.; Madhunapantula, S.V.; Robertson, G.P. Akt3 and Mutant V600EB-Raf Cooperate to Promote Early Melanoma Development. Cancer Res. 2008, 68, 3429–3439. [Google Scholar] [CrossRef]
- Nogueira, C.; Kim, K.-H.H.; Sung, H.; Paraiso, K.; Dannenberg, J.-H.H.; Bosenberg, M.; Chin, L.; Kim, M. Cooperative Interactions of PTEN Deficiency and RAS Activation in Melanoma Metastasis. Oncogene 2010, 29, 6222–6232. [Google Scholar] [CrossRef] [PubMed]
- Kircher, D.A.; Trombetti, K.A.; Silvis, M.R.; Parkman, G.L.; Fischer, G.M.; Angel, S.N.; Stehn, C.M.; Strain, S.C.; Grossmann, A.H.; Duffy, K.L.; et al. AKT1E17K Activates Focal Adhesion Kinase and Promotes Melanoma Brain Metastasis. Mol. Cancer Res. 2019, 17, 1787–1800. [Google Scholar] [CrossRef]
- Bayer, A.L.; Pietruska, J.; Farrell, J.; McRee, S.; Alcaide, P.; Hinds, P.W. AKT1 Is Required for a Complete Palbociclib-Induced Senescence Phenotype in BRAF-V600E-Driven Human Melanoma. Cancers 2022, 14, 572. [Google Scholar] [CrossRef]
- Chin, Y.; Yoshida, T.; Marusyk, A.; Beck, A.H.; Polyak, K.; Toker, A. Targeting Akt3 Signaling in Triple-Negative Breast Cancer. Cancer Res. 2014, 74, 964–973. [Google Scholar] [CrossRef]
- Davies, M.A. The Multi-Faceted Roles of the PI3K-AKT Pathway in Melanoma. J. Transl. Med. 2015, 13, 2039. [Google Scholar] [CrossRef] [PubMed]
- Koya, R.C.; Mok, S.; Otte, N.; Blacketor, K.J.; Begonya, C.-A.; Tumeh, P.C.; Minasyan, A.; Graham, N.A.; Graeber, T.G.; Chodon, T.; et al. BRAF Inhibitor Vemurafenib Improves the Antitumor Activity of Adoptive Cell Immunotherapy. Cancer Res. 2012, 72, 3928–3937. [Google Scholar] [CrossRef]
- Goel, V.; Ibrahim, N.; Jiang, G.; Singhal, M.; Fee, S.; Flotte, T.; Westmoreland, S.; Haluska, F.; Hinds, P.; Haluska, F. Melanocytic Nevus-like Hyperplasia and Melanoma in Transgenic BRAFV600E Mice. Cancer Res. 2009, 28, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Sheng, J.; Hu, M.G.; Haluska, F.G.; Cui, R.; Xu, Z.; Tsichlis, P.N.; Hu, G.-F.F.; Hinds, P.W. Loss of ARF Sensitizes Transgenic BRAFV600E Mice to UV-Induced Melanoma via Suppression of XPC. Cancer Res. 2013, 73, 4337–4348. [Google Scholar] [CrossRef]
- Pedri, D.; Karras, P.; Landeloos, E.; Marine, J.-C.; Rambow, F. Epithelial-to-Mesenchymal-like Transition Events in Melanoma. FEBS J. 2022, 289, 1352–1368. [Google Scholar] [CrossRef]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the Regulatory Landscape of Melanoma Reveals TEADS as Regulators of the Invasive Cell State. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef]
- Lu, J. The Warburg Metabolism Fuels Tumor Metastasis. Cancer Metastasis Rev. 2019, 38, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.-Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef]
- Masters, T.A.; Calleja, V.; Armoogum, D.A.; Marsh, R.J.; Applebee, C.J.; Laguerre, M.; Bain, A.J.; Larijani, B. Regulation of 3-Phosphoinositide–Dependent Protein Kinase 1 Activity by Homodimerization in Live Cells. Sci. Signal. 2010, 3, ra78. [Google Scholar] [CrossRef]
- Dhawan, P.; Singh, A.B.; Ellis, D.L.; Richmond, A. Constitutive Activation of Akt/Protein Kinase B in Melanoma Leads to up-Regulation of Nuclear Factor-kappaB and Tumor Progression. Cancer Res. 2002, 62, 7335–7342. [Google Scholar] [PubMed]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; Grant, M.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef]
- Luo, C.; Pietruska, J.R.; Sheng, J.; Bronson, R.T.; Hu, M.G.; Cui, R.; Hinds, P.W. Expression of Oncogenic BRAFV600E in Melanocytes Induces Schwannian Differentiation in Vivo. Pigment. Cell Melanoma Res. 2015, 28, 603–606. [Google Scholar] [CrossRef]
- Chen, G.; Chakravarti, N.; Aardalen, K.; Lazar, A.J.; Tetzlaff, M.; Wubberhorst, B.; Kim, S.-B.B.; Kopetz, S.; Ledoux, A.; Nanda, V.G.; et al. Molecular Profiling of Patient-Matched Brain and Extracranial Melanoma Metastases Implicates the PI3K Pathway as a Therapeutic Target. Clin. Cancer Res. 2014, 20, 5537–5546. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.-H.H. Twist Transcriptionally Up-Regulates AKT2 in Breast Cancer Cells Leading to Increased Migration, Invasion, and Resistance to Paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Polytarchou, C.; Hatziapostolou, M.; Kottakis, F.; Maroulakou, I.G.; Struhl, K.; Tsichlis, P.N. MicroRNAs Differentially Regulated by Akt Isoforms Control EMT and Stem Cell Renewal in Cancer Cells. Sci. Signal. 2009, 2, ra62. [Google Scholar] [CrossRef]
- Ganesh, K.; Massagué, J. Targeting Metastatic Cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef]
- Alonso, S.R.; Tracey, L.; Ortiz, P.; Pérez-Gómez, B.; Palacios, J.; Pollán, M.; Linares, J.; Serrano, S.; Sáez-Castillo, A.I.; Sánchez, L.; et al. A High-Throughput Study in Melanoma Identifies Epithelial-Mesenchymal Transition as a Major Determinant of Metastasis. Cancer Res. 2007, 67, 3450–3460. [Google Scholar] [CrossRef]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.; Yuan, X.; Balk, S.P.; Toker, A. PTEN-Deficient Tumors Depend on AKT2 for Maintenance and Survival. Cancer Discov. 2014, 4, 942–955. [Google Scholar] [CrossRef]
- Izraely, S.; Sagi-Assif, O.; Klein, A.; Meshel, T.; Tsarfaty, G.; Pasmanik-Chor, M.; Nahmias, C.; Couraud, P.-O.; Ateh, E.; Bryant, J.L.; et al. The Metastatic Microenvironment: Brain-Residing Melanoma Metastasis and Dormant Micrometastasis. Int. J. Cancer 2012, 131, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Rumio, C. Tumor Cell Glycolysis—At the Crossroad of Epithelial–Mesenchymal Transition and Autophagy. Cells 2022, 11, 1041. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Azambuja, A.P.; Simoes-Costa, M. Metabolic Reprogramming Promotes Neural Crest Migration via Yap/Tead Signaling. Dev. Cell 2020, 53, 199–211.e6. [Google Scholar] [CrossRef]
- Fischer, G.M.; Vashisht Gopal, Y.; McQuade, J.L.; Peng, W.; DeBerardinis, R.J.; Davies, M.A. Metabolic Strategies of Melanoma Cells: Mechanisms, Interactions with the Tumor Microenvironment, and Therapeutic Implications. Pigment. Cell Melanoma Res. 2017, 31, 11–30. [Google Scholar] [CrossRef]
- Polytarchou, C.; Iliopoulos, D.; Hatziapostolou, M.; Kottakis, F.; Maroulakou, I.; Struhl, K.; Tsichlis, P.N. Akt2 Regulates All Akt Isoforms and Promotes Resistance to Hypoxia through Induction of miR-21 upon Oxygen Deprivation. Cancer Res. 2011, 71, 4720–4731. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, M.; Fei, B.; Fang, X.; Ma, T.; Wang, D. miR-21-5p Targets PDHA1 to Regulate Glycolysis and Cancer Progression in Gastric Cancer. Oncol. Rep. 2018, 40, 2955–2963. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McRee, S.K.; Bayer, A.L.; Pietruska, J.; Tsichlis, P.N.; Hinds, P.W. AKT2 Loss Impairs BRAF-Mutant Melanoma Metastasis. Cancers 2023, 15, 4958. https://doi.org/10.3390/cancers15204958
McRee SK, Bayer AL, Pietruska J, Tsichlis PN, Hinds PW. AKT2 Loss Impairs BRAF-Mutant Melanoma Metastasis. Cancers. 2023; 15(20):4958. https://doi.org/10.3390/cancers15204958
Chicago/Turabian StyleMcRee, Siobhan K., Abraham L. Bayer, Jodie Pietruska, Philip N. Tsichlis, and Philip W. Hinds. 2023. "AKT2 Loss Impairs BRAF-Mutant Melanoma Metastasis" Cancers 15, no. 20: 4958. https://doi.org/10.3390/cancers15204958
APA StyleMcRee, S. K., Bayer, A. L., Pietruska, J., Tsichlis, P. N., & Hinds, P. W. (2023). AKT2 Loss Impairs BRAF-Mutant Melanoma Metastasis. Cancers, 15(20), 4958. https://doi.org/10.3390/cancers15204958