
Concurrent Inhibition of Akt and ERK Using TIC-10 Can Overcome Venetoclax Resistance in Mantle Cell Lymphoma

 , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Development of Venetoclax-Resistant Cell Lines
2.2. Cell Proliferation and Apoptosis Assays
2.3. BH3 Profiling
2.4. Nucleic Acid Extraction
2.5. Reverse Transcriptase-Quantitative PCR
2.6. AcceSssIble Assay
2.7. Genome-Wide Methylation and Chromatin Accessibility Analysis
2.8. Magnetic Cell Sorting of MCL Cells from Co-Cultures for RNA Extraction
2.9. Whole Exome Sequencing
2.10. Transcriptome Analysis
2.11. Gene Set Enrichment Analysis
2.12. Statistical Analysis
3. Results
3.1. Generation of In Vitro Models of Acquired and Microenvironmentally Induced Venetoclax Resistance
3.2. Venetoclax Resistance Is Associated with Changes in BH3-Mediated Apoptotic Priming
3.3. Acquired Venetoclax Resistance Is Associated with Very Few Genetic Events but an Extensive Epigenetic Reprogramming
3.4. Epigenetic and Transcriptomic Changes Highlight the Role of the PI3K/Akt Pathway in Venetoclax Resistance
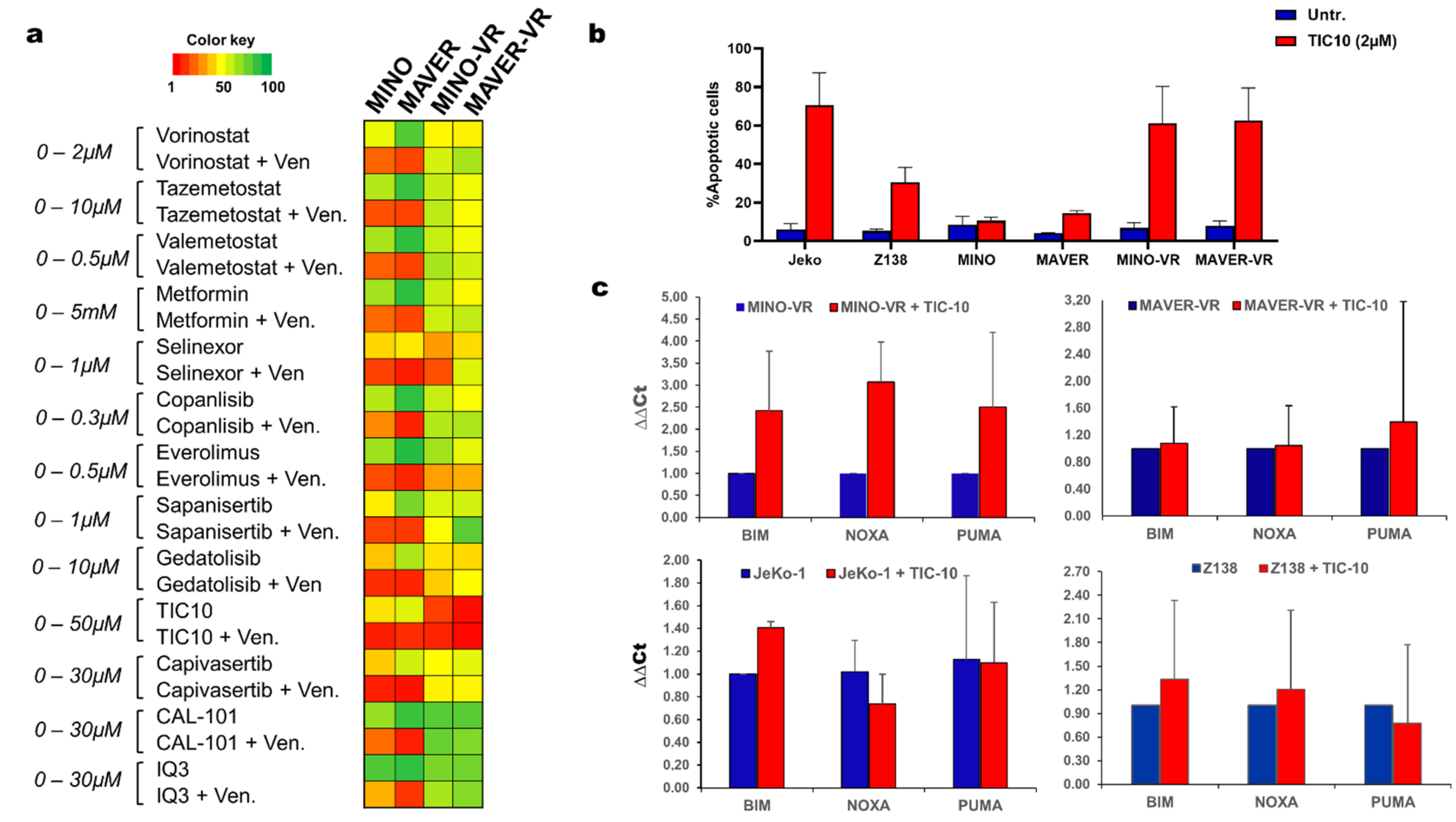
3.5. Pharmacological Inhibition of Akt and ERK Is Toxic to Venetoclax-Resistant Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sethi, S.; Epstein-Peterson, Z.; Kumar, A.; Ho, C. Current Knowledge in Genetics, Molecular Diagnostic Tools, and Treatments for Mantle Cell Lymphomas. Front. Oncol. 2021, 11, 739441. [Google Scholar] [CrossRef] [PubMed]
- Eskelund, C.W.; Kolstad, A.; Jerkeman, M.; Räty, R.; Laurell, A.; Eloranta, S.; Smedby, K.E.; Husby, S.; Pedersen, L.B.; Andersen, N.S.; et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): Prolonged remissions without survival plateau. Br. J. Haematol. 2016, 175, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Eskelund, C.W.; Dimopoulos, K.; Kolstad, A.; Glimelius, I.; Räty, R.; Gjerdrum, L.M.R.; Sonnevi, K.; Josefsson, P.; Nilsson-Ehle, H.; Bentzen, H.H.N.; et al. Detailed Long-Term Follow-Up of Patients Who Relapsed After the Nordic Mantle Cell Lymphoma Trials: MCL2 and MCL3. Hemasphere 2020, 5, e510. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Kahl, B. Targeting BCL-2 in Hematologic Malignancies. Target. Oncol. 2018, 13, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Lin, V.S.; Anderson, M.A.; Huang, D.C.S.; Roberts, A.W.; Seymour, J.F.; Tam, C.S.L. Venetoclax for the treatment of mantle cell lymphoma. Ann. Lymphoma 2019, 3, 4. [Google Scholar] [CrossRef]
- Tam, C.S.; Anderson, M.A.; Pott, C.; Agarwal, R.; Handunnetti, S.; Hicks, R.J.; Burbury, K.; Turner, G.; Di Iulio, J.; Bressel, M.; et al. Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. New Engl. J. Med. 2018, 378, 1211–1223. [Google Scholar] [CrossRef]
- Agarwal, R.; Dawson, M.A.; Dreyling, M.; Tam, C.S. Understanding resistance mechanisms to BTK and BCL2 inhibitors in mantle cell lymphoma: Implications for design of clinical trials. Leuk. Lymphoma 2018, 59, 2769–2781. [Google Scholar] [CrossRef]
- Jayappa, K.D.; Portell, C.A.; Gordon, V.L.; Capaldo, B.J.; Bekiranov, S.; Axelrod, M.J.; Brett, L.K.; Wulfkuhle, J.D.; Gallagher, R.I.; Petricoin, E.F.; et al. Erratum: Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017, 1, 933–946. [Google Scholar] [CrossRef]
- Jayappa, K.D.; Gordon, V.L.; Morris, C.G.; Wilson, B.; Shetty, B.D.; Cios, K.J.; Arora, P.C.; Isaac, K.M.; Saha, S.; Bender, T.P.; et al. Extrinsic interactions in the microenvironment in vivo activate an antiapoptotic multidrug-resistant phenotype in CLL. Blood Adv. 2021, 5, 3497–3510. [Google Scholar] [CrossRef]
- Dousset, C.; Le Coq, J.; Touzeau, C.; Le Gouill, S.; Maïga, S.; Gomez-Bougie, P.; Moreau, P.; Chiron, D.; Pellat-Deceunynck, C.; Moreau-Aubry, A.; et al. BH3 profiling as a tool to identify acquired resistance to venetoclax in multiple myeloma. Br. J. Haematol. 2017, 179, 684–688. [Google Scholar] [CrossRef]
- Bhatt, S.; Pioso, M.S.; Olesinski, E.A.; Yilma, B.; Ryan, J.A.; Mashaka, T.; Leutz, B.; Adamia, S.; Zhu, H.; Kuang, Y.; et al. Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 872–890.e6. [Google Scholar] [CrossRef]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef]
- Zhao, S.; Kanagal-Shamanna, R.; Navsaria, L.; Ok, C.Y.; Zhang, S.; Nomie, K.; Han, G.; Hao, D.; Hill, H.A.; Jiang, C.; et al. Efficacy of venetoclax in high risk relapsed mantle cell lymphoma (MCL)—Outcomes and mutation profile from venetoclax resistant MCL patients. Am. J. Hematol. 2020, 95, 623–629. [Google Scholar] [CrossRef]
- Zhao, X.; Ren, Y.; Lawlor, M.; Shah, B.D.; Park, P.M.; Lwin, T.; Wang, X.; Liu, K.; Wang, M.; Gao, J.; et al. BCL2 Amplicon Loss and Transcriptional Remodeling Drives ABT-199 Resistance in B Cell Lymphoma Models. Cancer Cell 2019, 35, 752–766.e9. [Google Scholar] [CrossRef]
- Agarwal, R.; Chan, Y.-C.; Tam, C.S.; Hunter, T.; Vassiliadis, D.; Teh, C.E.; Thijssen, R.; Yeh, P.; Wong, S.Q.; Ftouni, S.; et al. Dynamic molecular monitoring reveals that SWI–SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med. 2019, 25, 119–129. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Helbo, A.S.; Munch-Petersen, H.F.; Sjö, L.; Christensen, J.; Kristensen, L.S.; Asmar, F.; Hermansen, N.E.U.; O’Connel, C.; Gimsing, P.; et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol. Oncol. 2017, 12, 180–195. [Google Scholar] [CrossRef]
- Ryan, J.; Letai, A. BH3 profiling in whole cells by fluorimeter or FACS. Methods 2013, 61, 156–164. [Google Scholar] [CrossRef]
- Becket, E.; Chopra, S.; Duymich, C.E.; Lin, J.J.; You, J.S.; Pandiyan, K.; Nichols, P.W.; Siegmund, K.D.; Charlet, J.; Weisenberger, D.J.; et al. Identification of DNA Methylation–Independent Epigenetic Events Underlying Clear Cell Renal Cell Carcinoma. Cancer Res. 2016, 76, 1954–1964. [Google Scholar] [CrossRef]
- Fortin, J.-P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017, 33, 558–560. [Google Scholar] [CrossRef]
- McCartney, D.L.; Walker, R.M.; Morris, S.W.; McIntosh, A.M.; Porteous, D.J.; Evans, K.L. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom. Data 2016, 9, 22–24. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.V.; Huang, S.; Zhang, H.; Zhang, J.; Bell, T.; Zhou, S.; Pogue, E.; Ding, Z.; Lam, L.; Westin, J.; et al. Strategic Therapeutic Targeting to Overcome Venetoclax Resistance in Aggressive B-cell Lymphomas. Clin. Cancer Res. 2018, 24, 3967–3980. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Anderson, M.A.; Gong, J.-N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Guièze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernández-Sánchez, M.; Zurita, A.M.; Sun, J.; Hacken, E.T.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384.e13. [Google Scholar] [CrossRef] [PubMed]
- Bojarczuk, K.; Sasi, B.K.; Gobessi, S.; Innocenti, I.; Pozzato, G.; Laurenti, L.; Efremov, D. BCR signaling inhibitors differ in their ability to overcome Mcl-1–mediated resistance of CLL B cells to ABT-199. Blood 2016, 127, 3192–3201. [Google Scholar] [CrossRef]
- Zhang, Q.; Riley-Gillis, B.; Han, L.; Jia, Y.; Lodi, A.; Zhang, H.; Ganesan, S.; Pan, R.; Konoplev, S.N.; Sweeney, S.R.; et al. Activation of RAS/MAPK pathway confers MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct. Target. Ther. 2022, 7, 51. [Google Scholar] [CrossRef]
- Tromp, J.M.; Geest, C.R.; Breij, E.C.; Elias, J.A.; van Laar, J.; Luijks, D.M.; Kater, A.P.; Beaumont, T.; van Oers, M.H.; Eldering, E. Tipping the Noxa/Mcl-1 Balance Overcomes ABT-737 Resistance in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2012, 18, 487–498. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593-12. [Google Scholar] [CrossRef]
- Touzeau, C.; Dousset, C.; Le Gouill, S.; Sampath, D.; Leverson, J.D.; Souers, A.J.; Maïga, S.; Béné, M.C.; Moreau, P.; Deceunynck, C.; et al. The Bcl-2 specific BH3 mimetic ABT-199: A promising targeted therapy for t(11;14) multiple myeloma. Leukemia 2014, 28, 210–212. [Google Scholar] [CrossRef]
- Herling, C.D.; Abedpour, N.; Weiss, J.; Schmitt, A.; Jachimowicz, R.D.; Merkel, O.; Cartolano, M.; Oberbeck, S.; Mayer, P.; Berg, V.; et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat. Commun. 2018, 9, 727. [Google Scholar] [CrossRef]
- Wang, P.; Yu, J.; Zhang, L. The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 4054–4059. [Google Scholar] [CrossRef]
- Huang, S.; Liu, Y.; Chen, Z.; Wang, M.; Jiang, V.C. PIK-75 overcomes venetoclax resistance via blocking PI3K-AKT signaling and MCL-1 expression in mantle cell lymphoma. Am. J. Cancer Res. 2022, 12, 1102–1115. [Google Scholar]
- Mireuta, M.; Darnel, A.; Pollak, M. IGFBP-2 expression in MCF-7 cells is regulated by the PI3K/AKT/mTOR pathway through Sp1-induced increase in transcription. Growth Factors 2010, 28, 243–255. [Google Scholar] [CrossRef]
- Wilhelm, F.; Kässner, F.; Schmid, G.; Kratzsch, J.; Laner, A.; Wabitsch, M.; Körner, A.; Kiess, W.; Garten, A. Phosphatidylinositol 3-kinase (PI3K) signalling regulates insulin-like-growth factor binding protein-2 (IGFBP-2) production in human adipocytes. Growth Horm. IGF Res. 2015, 25, 115–120. [Google Scholar] [CrossRef]
- Kühnl, A.; Kaiser, M.; Neumann, M.; Fransecky, L.; Heesch, S.; Radmacher, M.; Marcucci, G.; Bloomfield, C.D.; Hofmann, W.-K.; Thiel, E.; et al. High expression of IGFBP2 is associated with chemoresistance in adult acute myeloid leukemia. Leuk. Res. 2011, 35, 1585–1590. [Google Scholar] [CrossRef]
- Allen, J.E.; Krigsfeld, G.; Mayes, P.A.; Patel, L.; Dicker, D.T.; Patel, A.S.; Dolloff, N.G.; Messaris, E.; Scata, K.A.; Wang, W.; et al. Dual Inactivation of Akt and ERK by TIC10 Signals Foxo3a Nuclear Translocation, TRAIL Gene Induction, and Potent Antitumor Effects. Sci. Transl. Med. 2013, 5, 171ra17. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Venetoclax IC50 (µM) |
|---|---|
| Parental cell lines | |
| JeKo-1 | 5.003 |
| Z-138 | 6.071 |
| MINO | 0.154 |
| MAVER-1 | 0.055 |
| Long-term (LT) cultured cell lines | |
| MINO-LT | 0.050 |
| MAVER1-LT | 1.320 |
| Venetoclax-resistant (VR) cell lines | |
| MINO-VR | 17.55 |
| MAVER1-VR | 22.63 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granau, A.M.; Andersen, P.A.; Jakobsen, T.; Taouxi, K.; Dalila, N.; Mogensen, J.B.; Kristensen, L.S.; Grønbæk, K.; Dimopoulos, K. Concurrent Inhibition of Akt and ERK Using TIC-10 Can Overcome Venetoclax Resistance in Mantle Cell Lymphoma. Cancers 2023, 15, 510. https://doi.org/10.3390/cancers15020510
Granau AM, Andersen PA, Jakobsen T, Taouxi K, Dalila N, Mogensen JB, Kristensen LS, Grønbæk K, Dimopoulos K. Concurrent Inhibition of Akt and ERK Using TIC-10 Can Overcome Venetoclax Resistance in Mantle Cell Lymphoma. Cancers. 2023; 15(2):510. https://doi.org/10.3390/cancers15020510
Chicago/Turabian StyleGranau, Agnete Marie, Pilar Aarøe Andersen, Theresa Jakobsen, Konstantina Taouxi, Nawar Dalila, Johanne Bay Mogensen, Lasse Sommer Kristensen, Kirsten Grønbæk, and Konstantinos Dimopoulos. 2023. "Concurrent Inhibition of Akt and ERK Using TIC-10 Can Overcome Venetoclax Resistance in Mantle Cell Lymphoma" Cancers 15, no. 2: 510. https://doi.org/10.3390/cancers15020510
APA StyleGranau, A. M., Andersen, P. A., Jakobsen, T., Taouxi, K., Dalila, N., Mogensen, J. B., Kristensen, L. S., Grønbæk, K., & Dimopoulos, K. (2023). Concurrent Inhibition of Akt and ERK Using TIC-10 Can Overcome Venetoclax Resistance in Mantle Cell Lymphoma. Cancers, 15(2), 510. https://doi.org/10.3390/cancers15020510