
Absolute Quantification of Pan-Cancer Plasma Proteomes Reveals Unique Signature in Multiple Myeloma

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
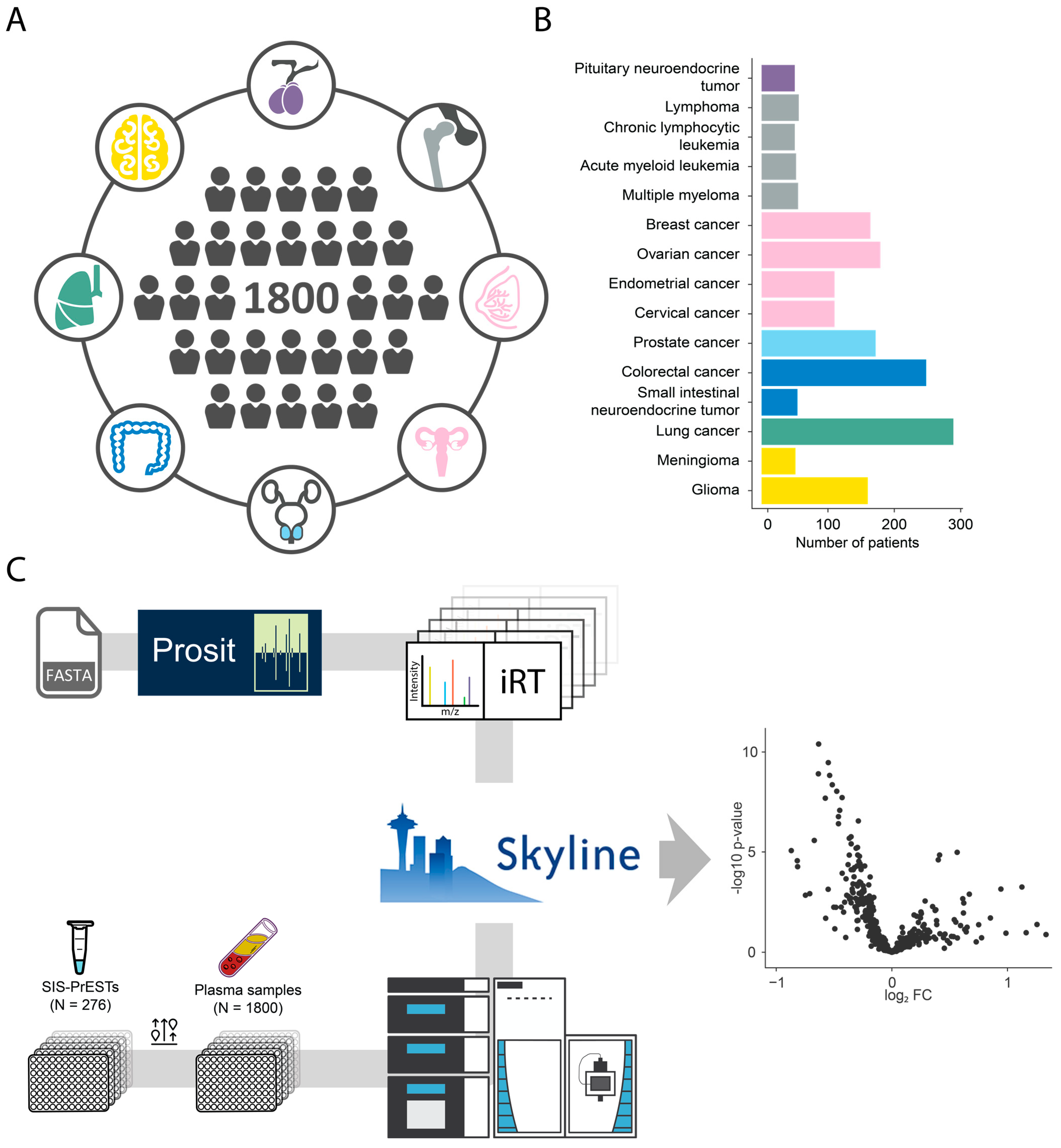
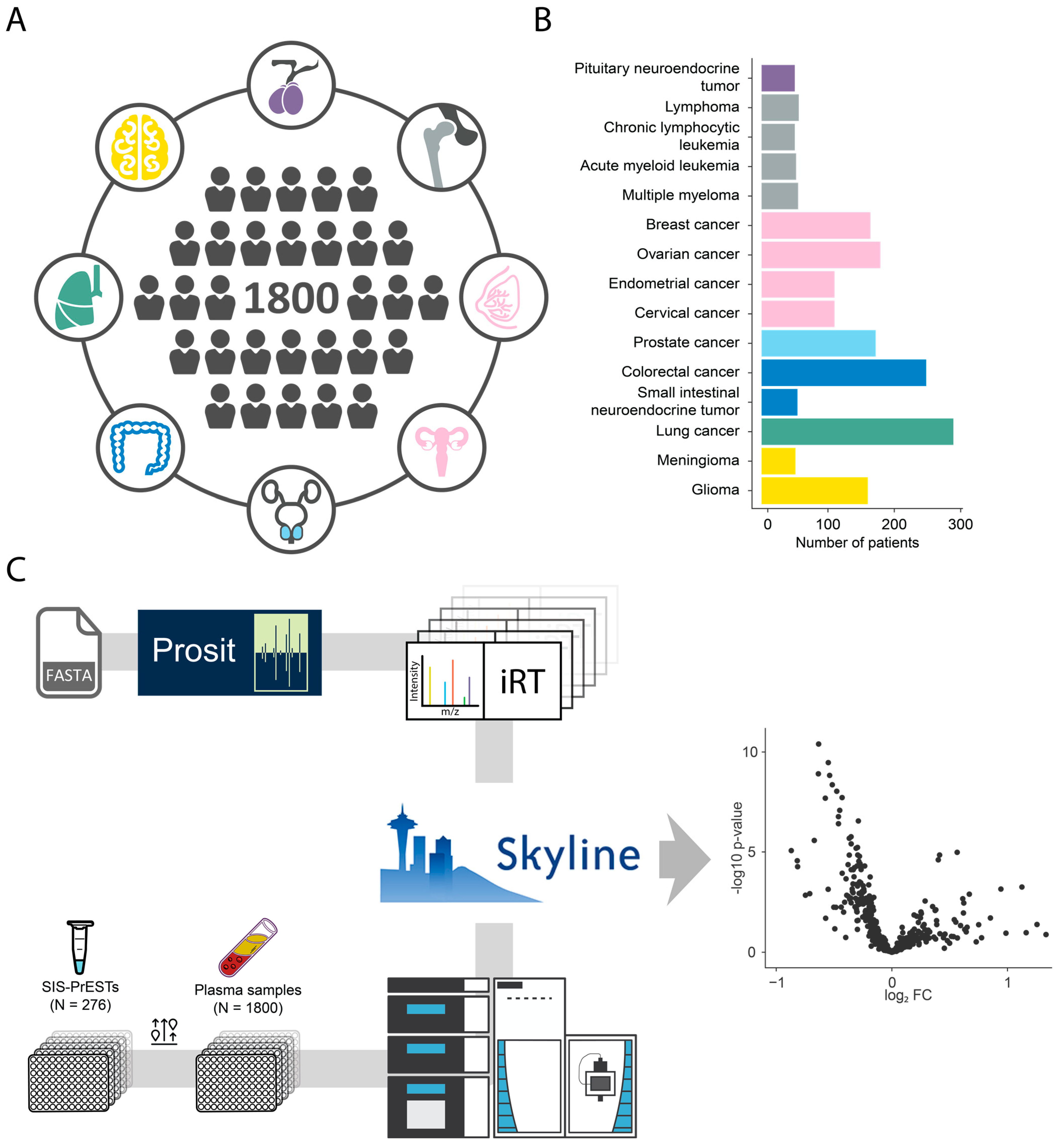
2.1. Cohort and Analytical Strategy
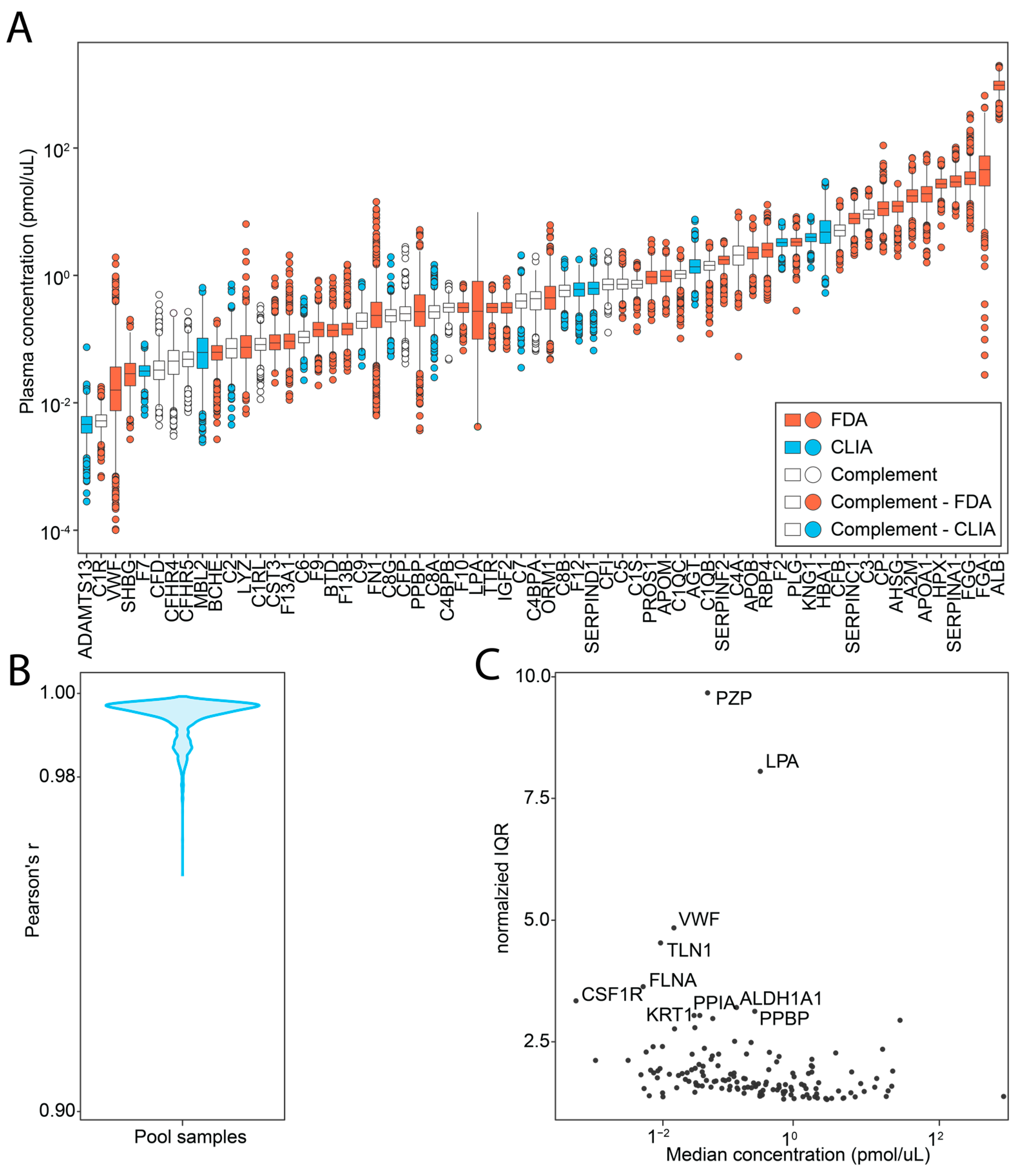
2.2. Investigated Targets and Analytical Performance
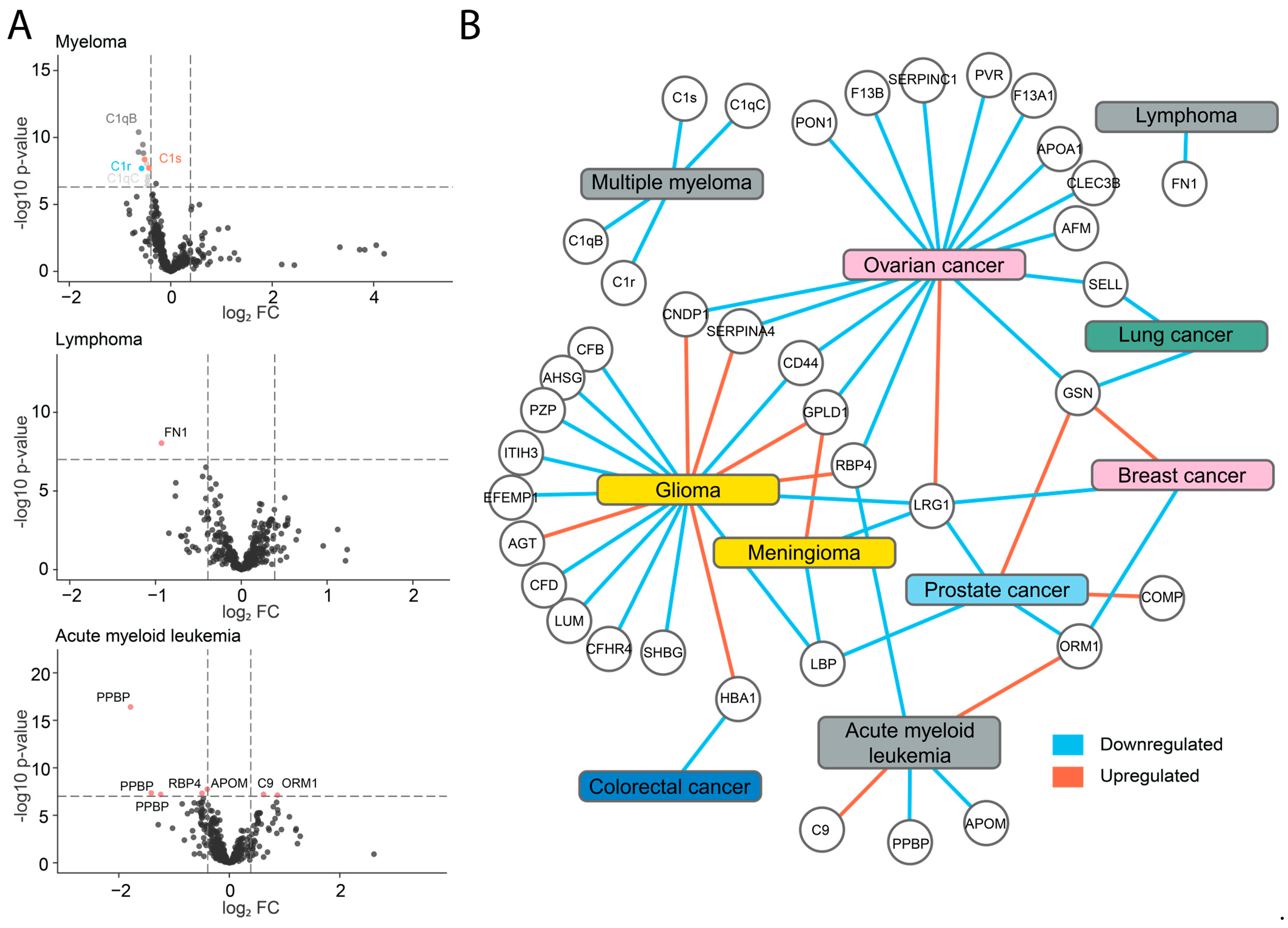
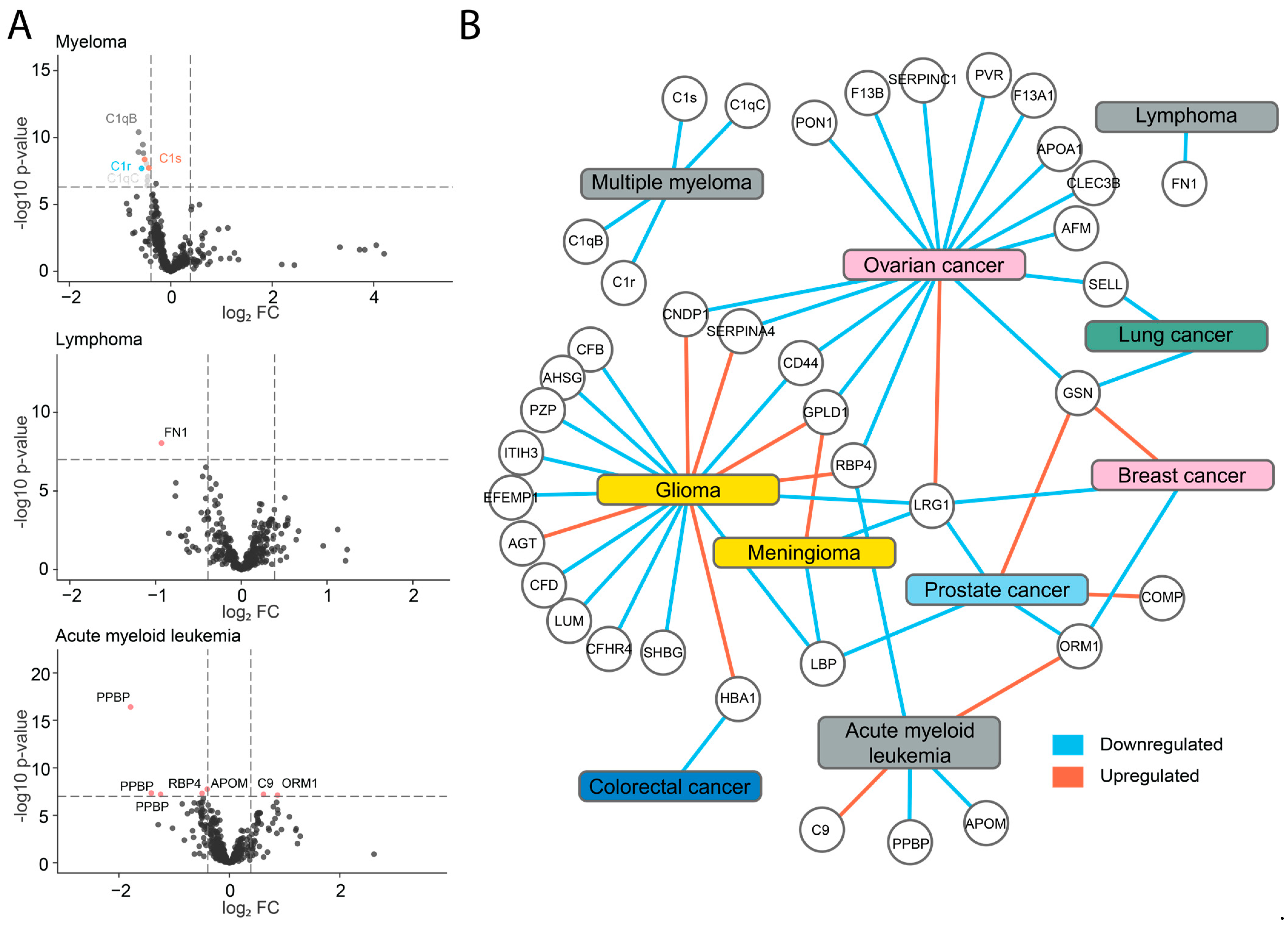
2.3. Identification of Potential Biomarkers
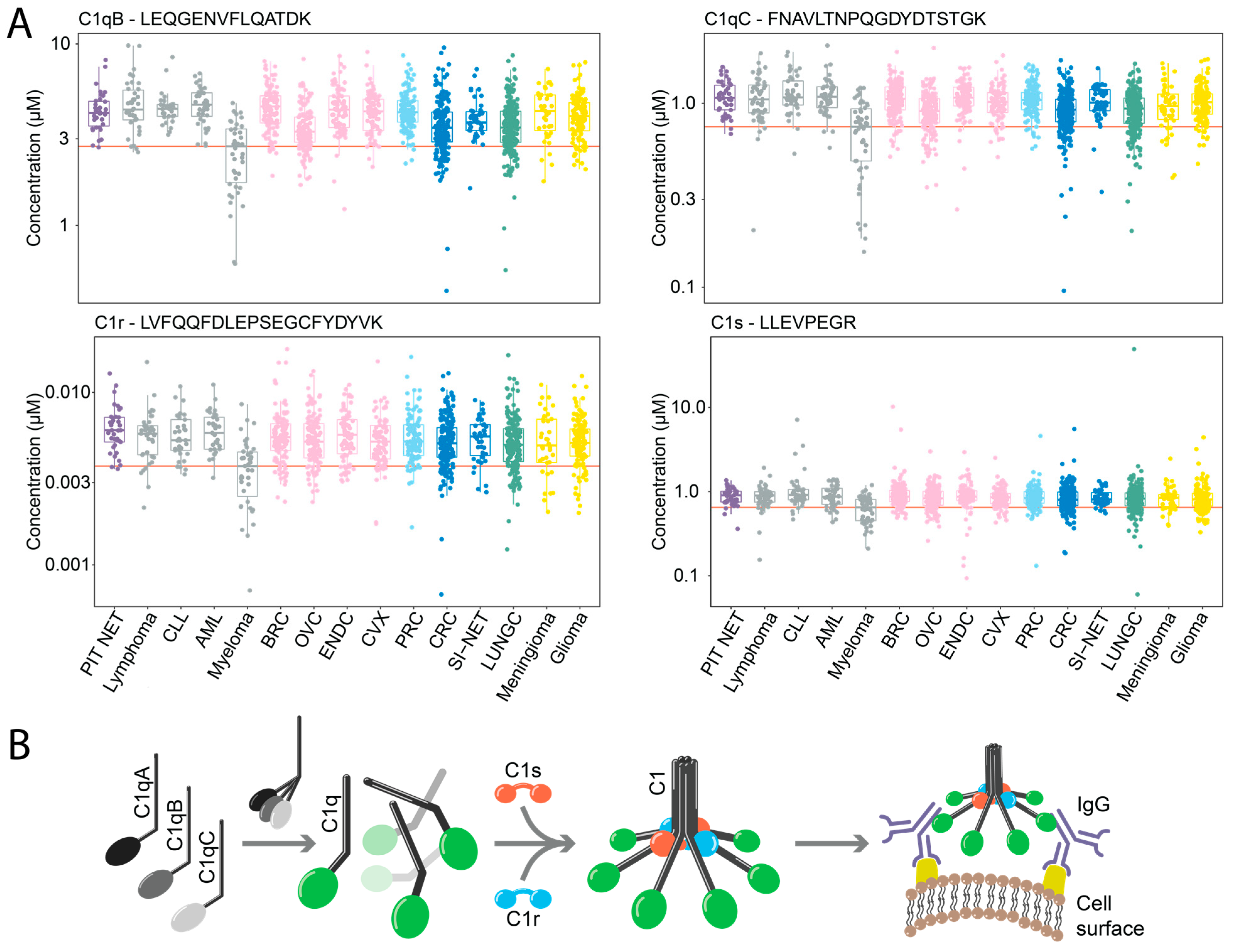
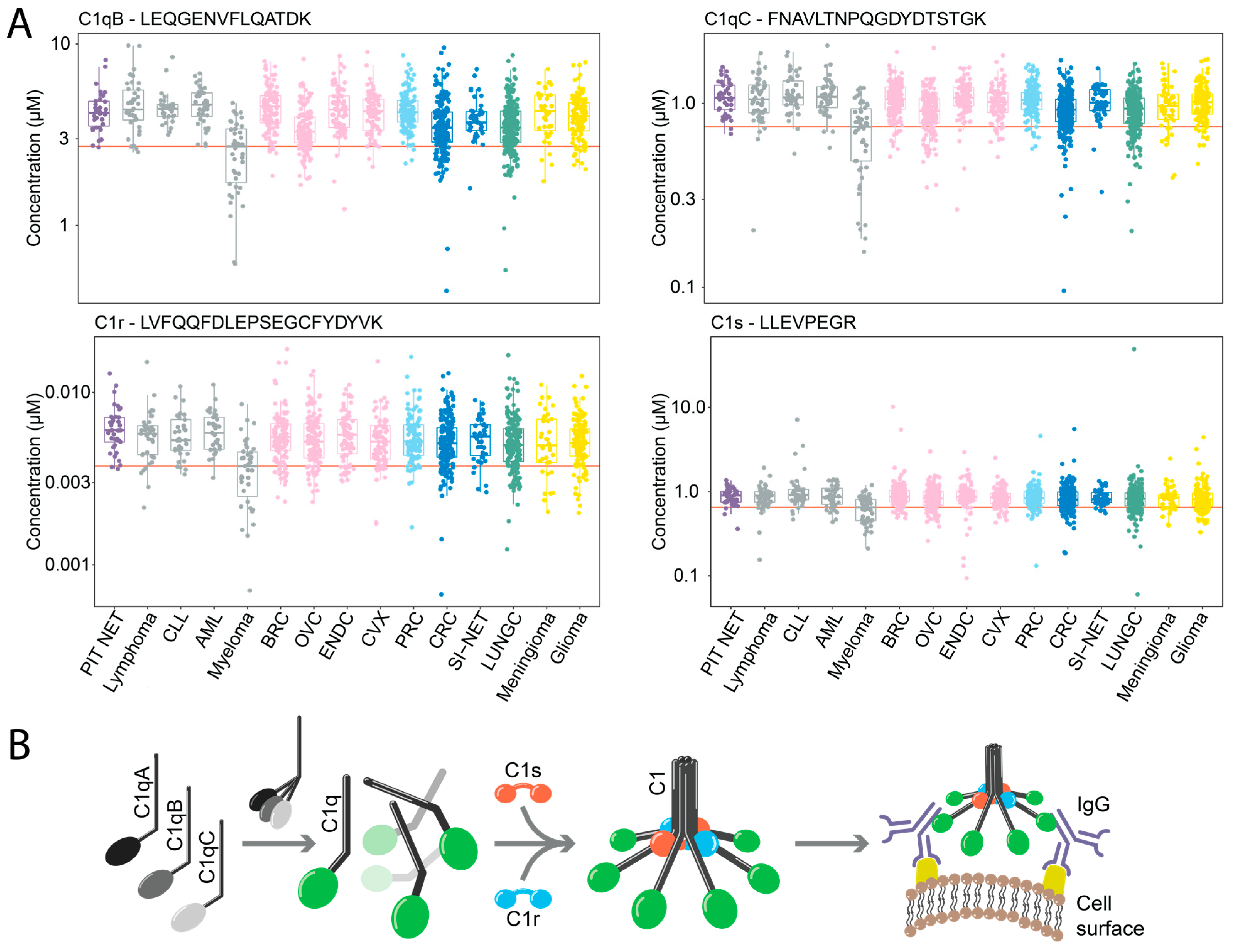
2.4. Downregulation of Components of the C1 Complex in Multiple Myeloma
2.5. Decreased JCHAIN and CD5L Plasma Levels Distinguish Multiple Myeloma
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Cohort
4.3. Sample Preparation
4.4. Mass Spectrometry Analysis
4.5. Absolute Quantification
4.6. Label-Free Data Extraction
4.7. Disease Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Craft, B.; Swatloski, T.; Goldman, M.; Ma, S.; Haussler, D.; Zhu, J. Exploring TCGA Pan-Cancer Data at the UCSC Cancer Genomics Browser. Sci. Rep. 2013, 3, 2652. [Google Scholar] [CrossRef]
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [PubMed]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2010, 31, 1023–1031. [Google Scholar] [CrossRef]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e18. [Google Scholar] [CrossRef]
- Ludwig, J.A.; Weinstein, J.N. Biomarkers in Cancer Staging, Prognosis and Treatment Selection. Nat. Rev. Cancer 2005, 5, 845–856. [Google Scholar] [CrossRef]
- Haber, D.A.; Velculescu, V.E. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef]
- Anderson, N.L.; Polanski, M.; Pieper, R.; Gatlin, T.; Tirumalai, R.S.; Conrads, T.P.; Veenstra, T.D.; Adkins, J.N.; Pounds, J.G.; Fagan, R.; et al. The human plasma proteome. Mol. Cell Proteom. 2004, 3, 311–326. [Google Scholar] [CrossRef]
- Ankerst, D.P. di Societa Italiana di ITOU2006 Sensitivity and Specificity of Prostate-Specific Antigen for Prostate Cancer Detection with High Rates of Biopsy Verification—Abstract—Europe PMC. Available online: https://europepmc.org/article/med/17269614 (accessed on 10 March 2022).
- Larson, M.H.; Pan, W.; Kim, H.J.; Mauntz, R.E.; Stuart, S.M.; Pimentel, M.; Zhou, Y.; Knudsgaard, P.; Demas, V.; Aravanis, A.M.; et al. A comprehensive characterization of the cell-free transcriptome reveals tissue- and subtype-specific biomarkers for cancer detection. Nat. Commun. 2021, 12, 2357. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.J.; Byrns, S.; Pennington, S.R.; Holmes, D.T.; Anderson, N.L.; Agreste, T.M.; Duffy, M.A. Clinical translation of MS-based, quantitative plasma proteomics: Status, challenges, requirements, and potential. Expert Rev. Proteom. 2016, 13, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Ferrari, M.; Petricoin, E. Clinical proteomics: Written in blood. Nature 2003, 425, 905. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Targeted proteomics. Nat. Methods 2012, 10, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Zeiler, M.; Straube, W.L.; Lundberg, E.; Uhlén, M.; Mann, M. A Protein Epitope Signature Tag (PrEST) library allows SILAC-based absolute quantification and multiplexed determination of protein copy numbers in cell lines. Mol. Cell Proteom. 2012, 11, O111.009613. [Google Scholar] [CrossRef]
- Nakayasu, E.S.; Gritsenko, M.; Piehowski, P.D.; Gao, Y.; Orton, D.J.; Schepmoes, A.A.; Fillmore, T.L.; Frohnert, B.I.; Rewers, M.; Krischer, J.P.; et al. Tutorial: Best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat. Protoc. 2021, 16, 3737–3760. [Google Scholar] [CrossRef]
- Kotol, D.; Hober, A.; Strandberg, L.; Svensson, A.-S.; Uhlén, M.; Edfors, F. Targeted proteomics analysis of plasma proteins using recombinant protein standards for addition only workflows. BioTechniques 2021, 71, 473–483. [Google Scholar] [CrossRef]
- Uhlén, M.; Karlsson, M.J.; Hober, A.; Svensson, A.-S.; Scheffel, J.; Kotol, D.; Zhong, W.; Tebani, A.; Strandberg, L.; Edfors, F.; et al. The human secretome. Sci. Signal. 2019, 12, eaaz0274. [Google Scholar] [CrossRef]
- Lassman, M.E.; McLaughlin, T.M.; Zhou, H.; Pan, Y.; Marcovina, S.M.; Laterza, O.; Roddy, T.P. Simultaneous quantitation and size characterization of apolipoprotein(a) by ultra-performance liquid chromatography/mass spectrometry. Rapid Commun. Mass. Spectrom. 2014, 28, 1101–1106. [Google Scholar] [CrossRef]
- Cai, D.; Liang, J.; Cai, X.-D.; Yang, Y.; Liu, G.; Zhou, F.; He, D. Identification of six hub genes and analysis of their correlation with drug sensitivity in acute myeloid leukemia through bioinformatics. Transl. Cancer Res. 2021, 10, 126–140. [Google Scholar] [CrossRef]
- Liang, L.; Li, J.; Fu, H.; Liu, X.; Liu, P. Identification of High Serum Apolipoprotein A1 as a Favorable Prognostic Indicator in Patients with Multiple Myeloma. J. Cancer 2019, 10, 4852–4859. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [PubMed]
- Johansen, F.E.; Braathen, R.; Brandtzaeg, P. Role of J chain in secretory immunoglobulin formation. Scand J. Immunol. 2000, 52, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.C.; Roux, K.H.; Shulman, M.J. On the structure of polymeric IgM. Eur. J. Immunol. 1988, 18, 1001–1008. [Google Scholar] [CrossRef]
- Wiersma, E.J.; Collins, C.; Fazel, S.; Shulman, M.J. Structural and functional analysis of J chain-deficient IgM. J. Immunol. 1998, 160, 5979–5989. [Google Scholar] [CrossRef]
- Hiramoto, E.; Tsutsumi, A.; Suzuki, R.; Matsuoka, S.; Arai, S.; Kikkawa, M.; Miyazaki, T. The IgM pentamer is an asymmetric pentagon with an open groove that binds the AIM protein. Sci. Adv. 2018, 4, eaau1199. [Google Scholar] [CrossRef]
- Arai, S.; Miyazaki, T. Impacts of the apoptosis inhibitor of macrophage (AIM) on obesity-associated inflammatory diseases. Semin Immunopathol. 2013, 36, 3–12. [Google Scholar] [CrossRef]
- Miyazaki, T.; Yamazaki, T.; Sugisawa, R.; Gershwin, M.E.; Arai, S. AIM associated with the IgM pentamer: Attackers on stand-by at aircraft carrier. Cell Mol. Immunol. 2018, 15, 563–574. [Google Scholar] [CrossRef]
- Oskam, N.; den Boer, M.A.; Lukassen, M.V.; Ooijevaar-de Heer, P.; Veth, T.S.; van Mierlo, G.; Lai, S.-H.; Derksen, N.I.L.; Yin, V.C.; Streutker, M.; et al. CD5L is a canonical component of circulatory IgM. bioRxiv 2023. [Google Scholar] [CrossRef]
- Corona, A.; Blobe, G.C. The role of the extracellular matrix protein TGFBI in cancer. Cell. Signal. 2021, 84, 110028. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yosef, N.; Gaublomme, J.; Wu, C.; Lee, Y.; Clish, C.B.; Kaminski, J.; Xiao, S.; Zu Horste, G.M.; Pawlak, M.; et al. CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell 2015, 163, 1413–1427. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Sze, C.-I.; Lin, S.-R.; Lee, M.-H.; He, R.-Y.; Schultz, L.; Chang, J.-Y.; Chen, S.-J.; Boackle, R.J.; Hsu, L.-J.; et al. Complement C1q Activates Tumor Suppressor WWOX to Induce Apoptosis in Prostate Cancer Cells. PLoS ONE 2019, 4, e5755. [Google Scholar] [CrossRef] [PubMed]
- Bandini, S.; Macagno, M.; Hysi, A.; Lanzardo, S.; Conti, L.; Bello, A.; Riccardo, F.; Ruiu, R.; Merighi, I.F.; Forni, G.; et al. The non-inflammatory role of C1q during Her2/neu-driven mammary carcinogenesis. OncoImmunology 2016, 5, e1253653. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Sultan, S.H.A.; Murugaiah, V.; Pathan, A.A.; Alhamlan, F.S.; Karteris, E.; Kishore, U. Human C1q Induces Apoptosis in an Ovarian Cancer Cell Line via Tumor Necrosis Factor Pathway. Front. Immunol. 2016, 7, 599. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Huang, J.; Ma, H.; Li, S.; Gao, X.; Liu, Y.; Shen, J.; Liao, A. Is complement C1q a potential marker for tumor burden and immunodeficiency in multiple myeloma? Leuk. Lymphoma 2019, 60, 1812–1818. [Google Scholar] [CrossRef]
- Barratt, J.; Weitz, I. Complement Factor D as a Strategic Target for Regulating the Alternative Complement Pathway. Front. Immunol. 2021, 12, 712572. [Google Scholar] [CrossRef]
- Flier, J.S.; Cook, K.S.; Usher, P.; Spiegelman, B.M. Severely Impaired Adipsin Expression in Genetic and Acquired Obesity. Science 1987, 237, 405–408. [Google Scholar] [CrossRef]
- Nezhad, P.R.; Riihilä, P.; Knuutila, J.S.; Viiklepp, K.; Peltonen, S.; Kallajoki, M.; Meri, S.; Nissinen, L.; Kähäri, V.-M. Complement Factor D Is a Novel Biomarker and Putative Therapeutic Target in Cutaneous Squamous Cell Carcinoma. Cancers 2022, 14, 305. [Google Scholar] [CrossRef]
- Gheorghe, S.R.; Crăciun, A.M. Matrix Gla protein in tumoral pathology. Clujul. Med. 2016, 89, 319–321. [Google Scholar] [CrossRef]
- Bokisch, V.A.; Müller-Eberhard, H.J. Anaphylatoxin inactivator of human plasma: Its isolation and characterization as a carboxypeptidase. J. Clin. Investig. 1970, 49, 2427–2436. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.W.; Mueller-Ortiz, S.L.; Wetsel, R.A. Carboxypeptidase N: A pleiotropic regulator of inflammation. Mol. Immunol. 2004, 40, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Skidgel, R.A.; Erdös, E.G. Structure and function of human plasma carboxypeptidase N, the anaphylatoxin inactivator. Int. Immunopharmacol. 2007, 7, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Cumová, J.; Jedličková, L.; Potěšil, D.; Sedo, O.; Stejskal, K.; Potáčová, A.; Zdráhal, Z.; Hájek, R. Comparative plasma proteomic analysis of patients with multiple myeloma treated with bortezomib-based regimens. Klin. Onkol. 2012, 25, 17–25. [Google Scholar]
- Cui, R.; Wang, C.; Li, T.; Hua, J.; Zhao, T.; Ren, L.; Wang, Y.; Li, Y. Carboxypeptidase N1 is anticipated to be a synergy metrics for chemotherapy effectiveness and prognostic significance in invasive breast cancer. Cancer Cell Int. 2021, 21, 571. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Searle, B.C.; Swearingen, K.E.; Barnes, C.A.; Schmidt, T.; Gessulat, S.; Kuster, B.; Wilhelm, M. Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A.; et al. Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509–518. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Searle, B.C.; Pino, L.K.; Egertson, J.D.; Ting, Y.S.; Lawrence, R.T.; MacLean, B.X.; Villén, J.; MacCoss, M.J. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 2018, 9, 5128. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotol, D.; Woessmann, J.; Hober, A.; Álvez, M.B.; Tran Minh, K.H.; Pontén, F.; Fagerberg, L.; Uhlén, M.; Edfors, F. Absolute Quantification of Pan-Cancer Plasma Proteomes Reveals Unique Signature in Multiple Myeloma. Cancers 2023, 15, 4764. https://doi.org/10.3390/cancers15194764
Kotol D, Woessmann J, Hober A, Álvez MB, Tran Minh KH, Pontén F, Fagerberg L, Uhlén M, Edfors F. Absolute Quantification of Pan-Cancer Plasma Proteomes Reveals Unique Signature in Multiple Myeloma. Cancers. 2023; 15(19):4764. https://doi.org/10.3390/cancers15194764
Chicago/Turabian StyleKotol, David, Jakob Woessmann, Andreas Hober, María Bueno Álvez, Khue Hua Tran Minh, Fredrik Pontén, Linn Fagerberg, Mathias Uhlén, and Fredrik Edfors. 2023. "Absolute Quantification of Pan-Cancer Plasma Proteomes Reveals Unique Signature in Multiple Myeloma" Cancers 15, no. 19: 4764. https://doi.org/10.3390/cancers15194764
APA StyleKotol, D., Woessmann, J., Hober, A., Álvez, M. B., Tran Minh, K. H., Pontén, F., Fagerberg, L., Uhlén, M., & Edfors, F. (2023). Absolute Quantification of Pan-Cancer Plasma Proteomes Reveals Unique Signature in Multiple Myeloma. Cancers, 15(19), 4764. https://doi.org/10.3390/cancers15194764