
TCF12 Activates TGFB2 Expression to Promote the Malignant Progression of Melanoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Reagents and Antibodies
2.3. RNA Interference
2.4. Cell Proliferation and Colony Formation Assays
2.5. Migration and Invasion Assays
2.6. Immunoblotting
2.7. Quantitative Real-Time PCR
2.8. Immunohistochemical (IHC) Staining
2.9. Animal Experiments
2.10. RNA-Sequencing Analysis
2.11. Chromatin Immunoprecipitation (ChIP)
2.12. Luciferase Reporter Assay
2.13. Statistical Analysis
3. Results
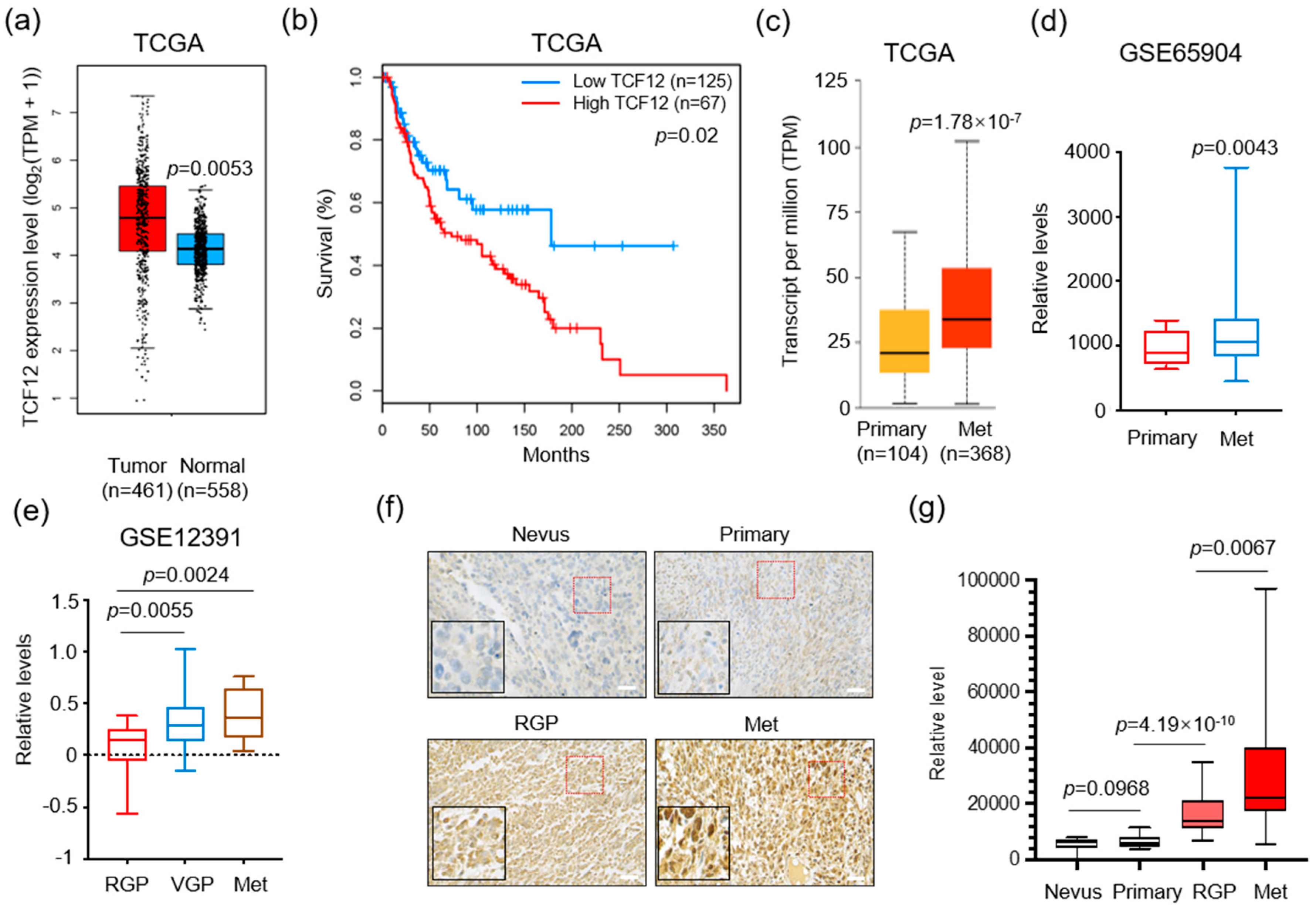
3.1. TCF12 Expression Is Positively Correlated with Poor Prognosis in Melanoma Patients
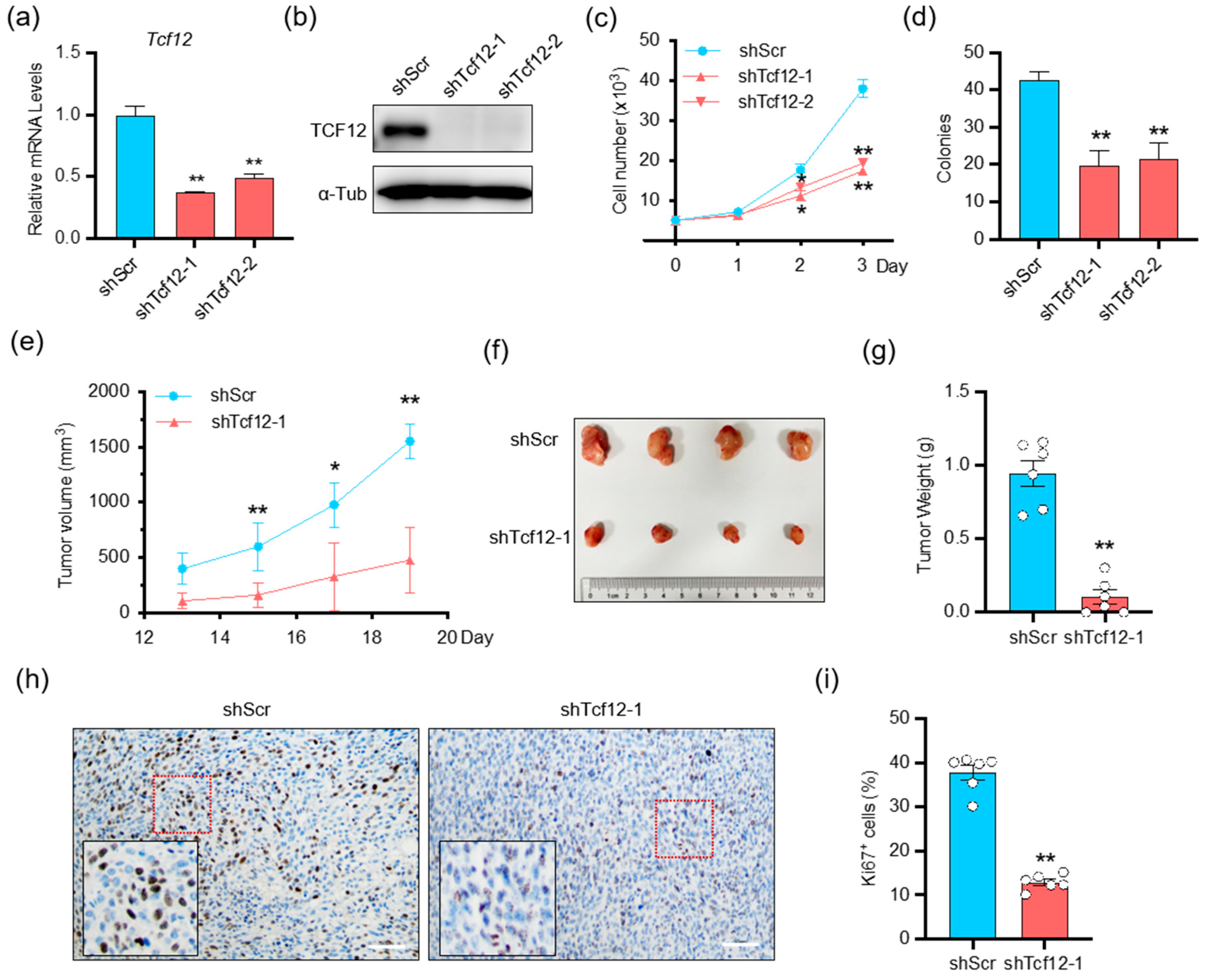
3.2. TCF12 Enhances Melanoma Cell Proliferation In Vitro and Tumorigenicity In Vivo
3.3. TCF12 Promotes Melanoma Cell Migration, Invasion In Vitro and Metastasis In Vivo
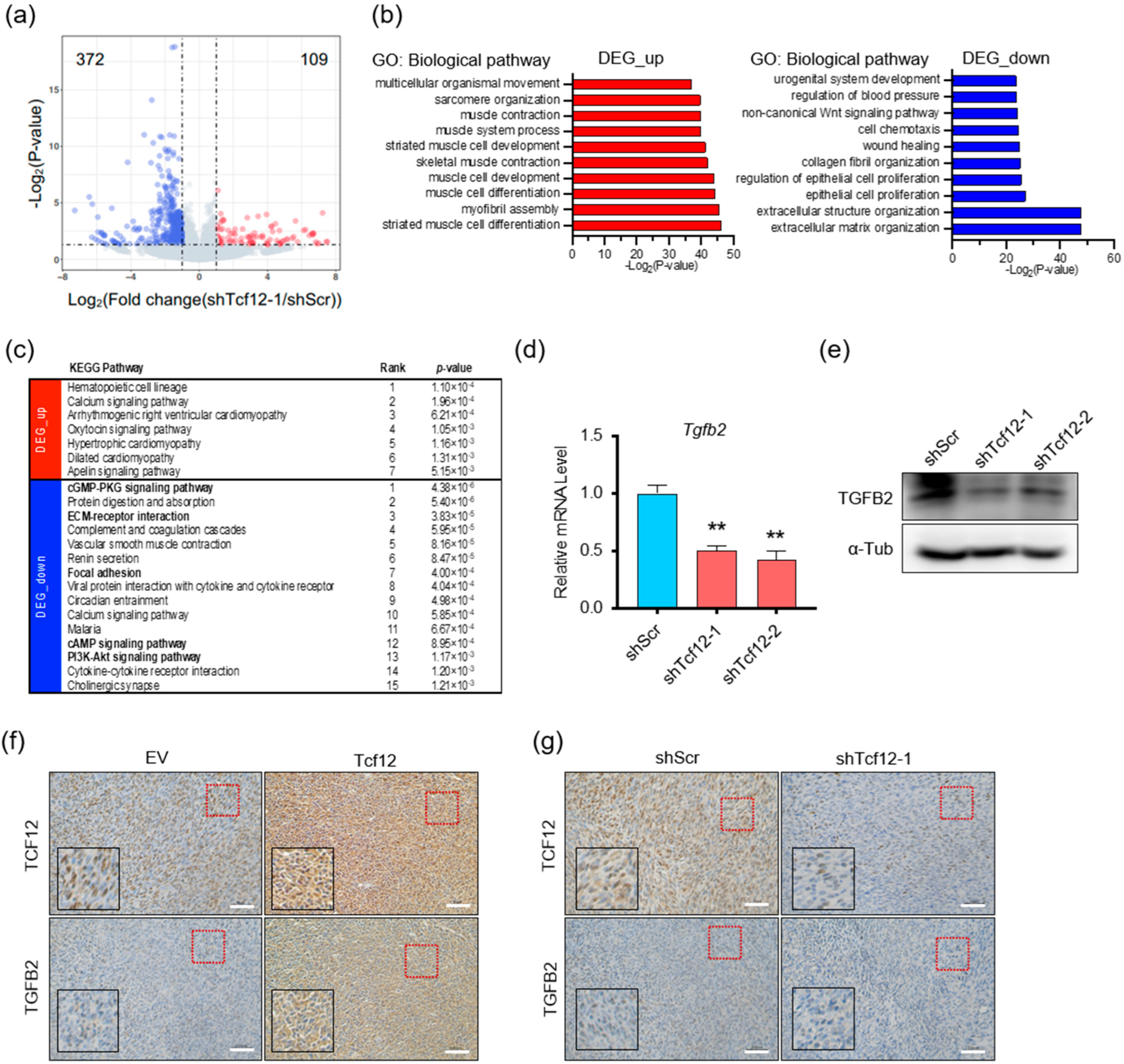
3.4. TGFB2 Is a Direct Downstream Target Gene of TCF12
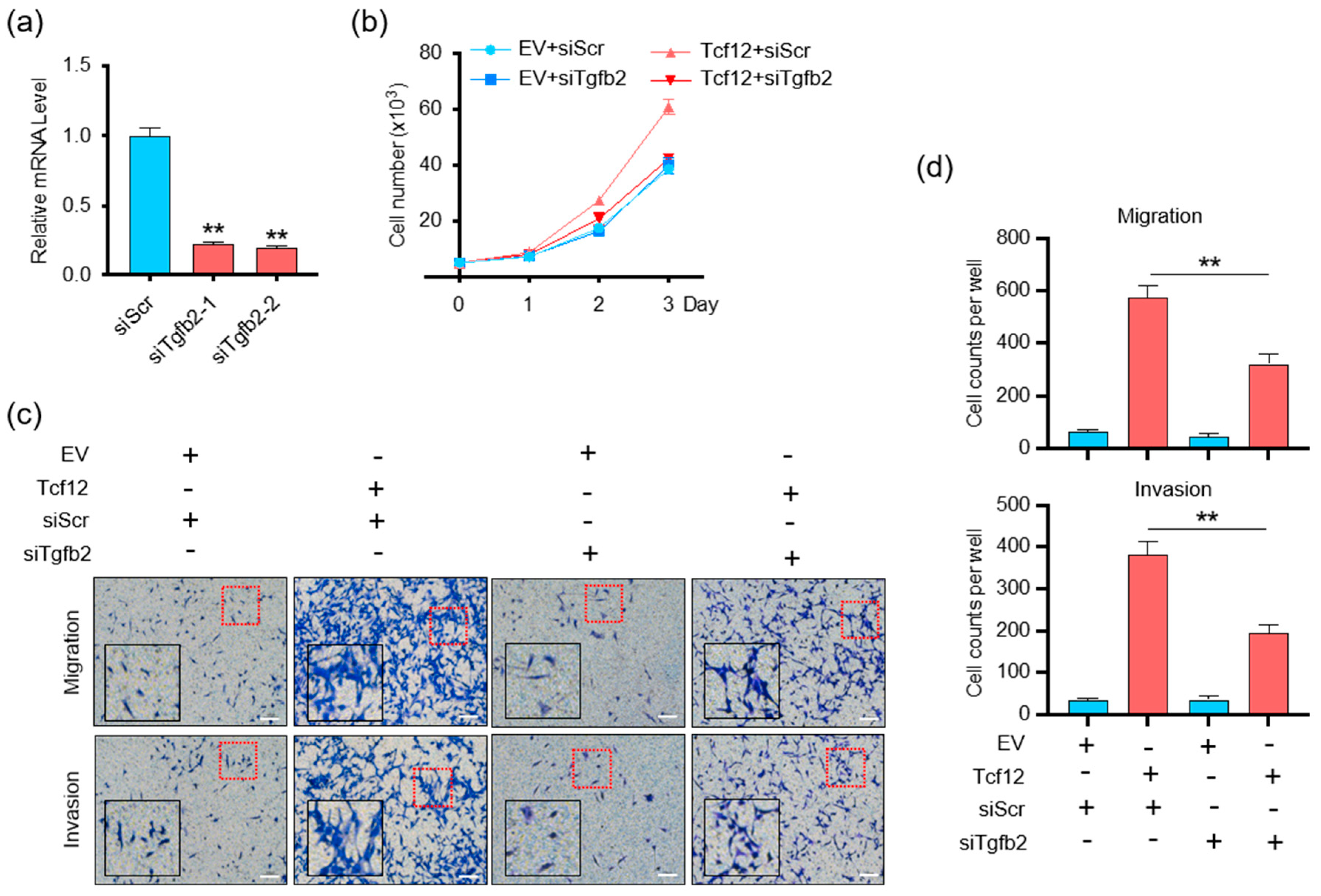
3.5. TGFB2 Is Essential for TCF12-Induced Cell Proliferation, Migration and Invasion In Vitro
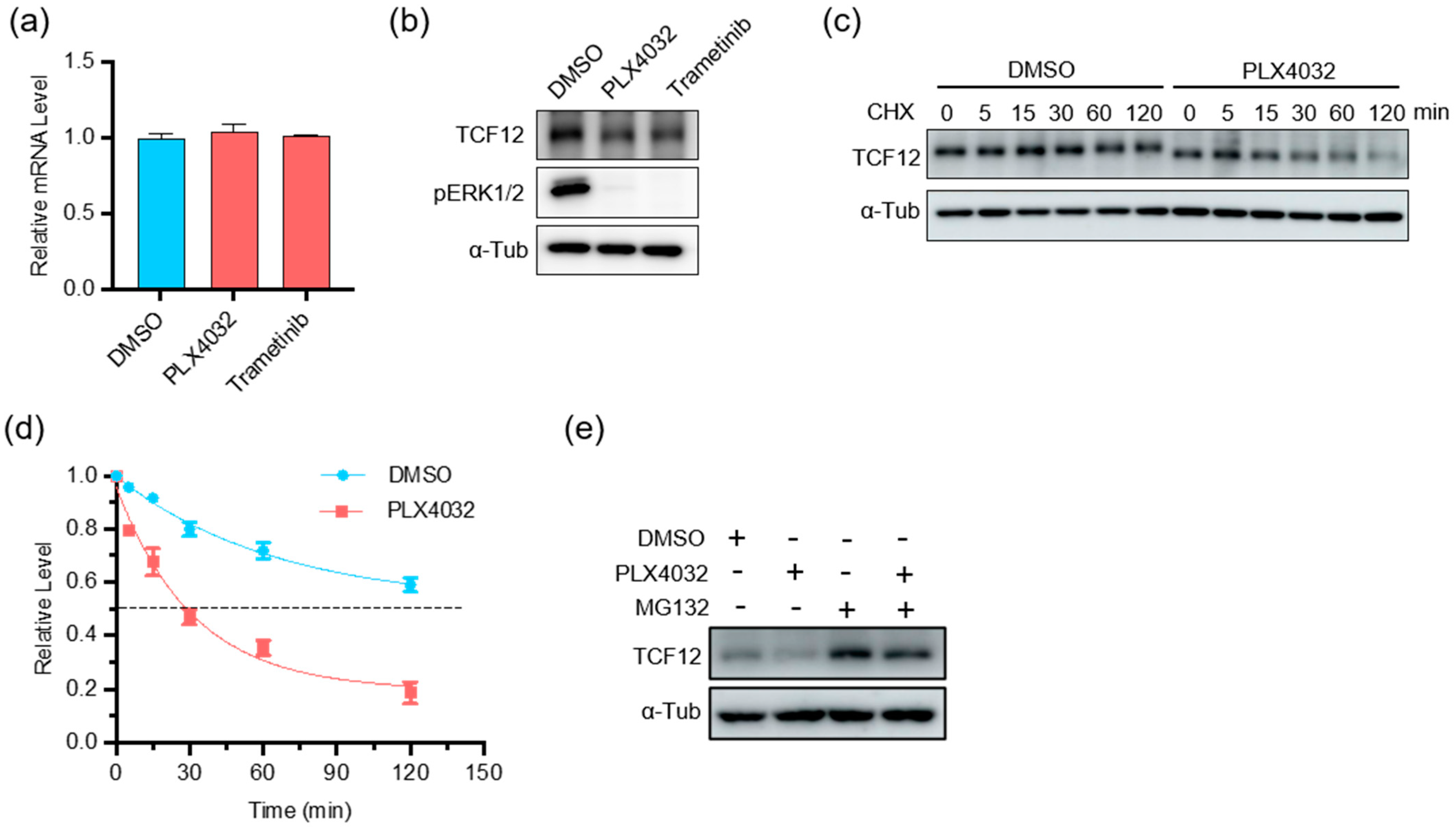
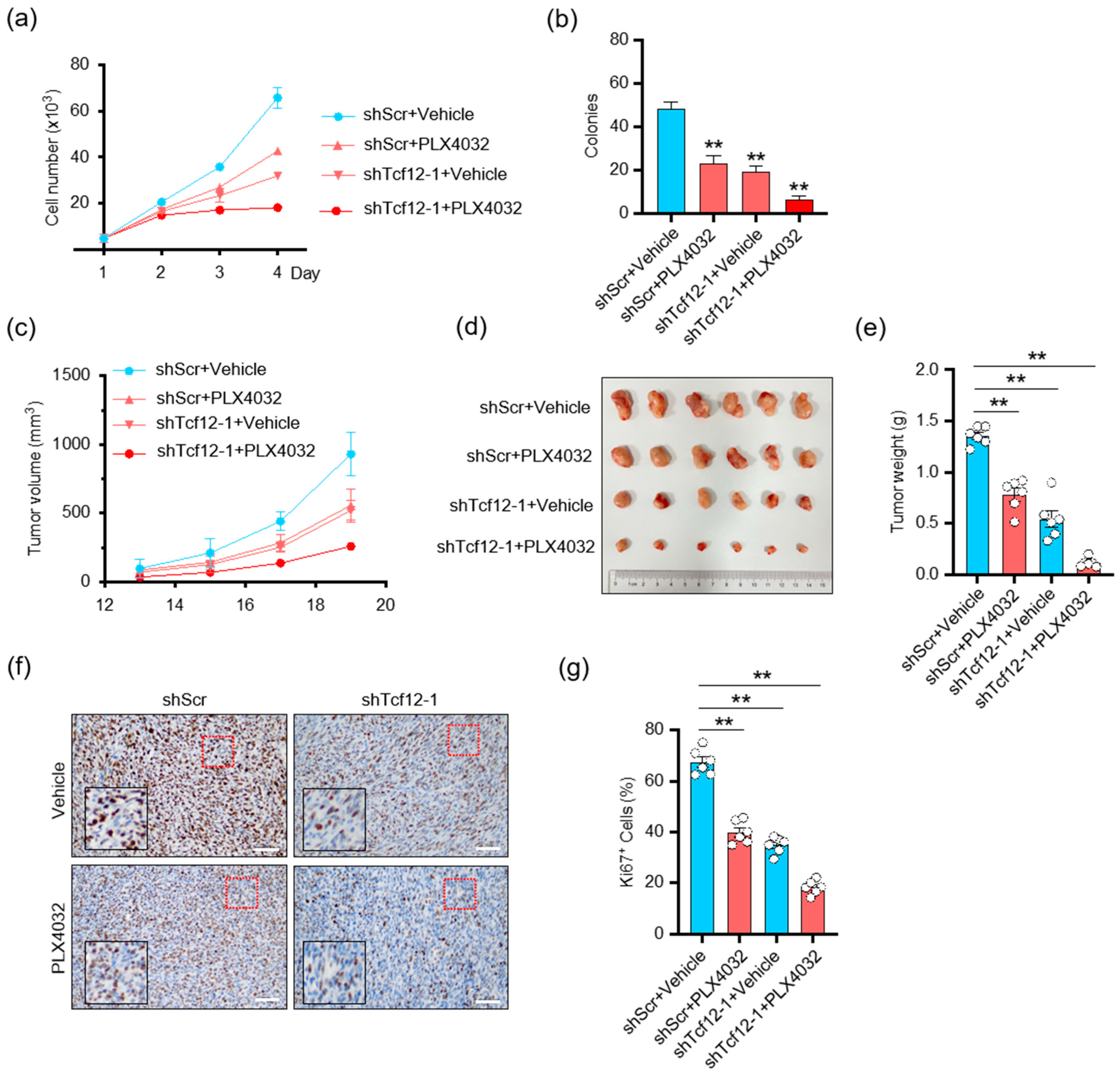
3.6. Depletion of TCF12 Sensitizes Melanoma to BRAF Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centeno, P.P.; Pavet, V.; Marais, R. The journey from melanocytes to melanoma. Nat. Rev. Cancer 2023, 23, 372–390. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Nascentes Melo, L.M.; Kumar, S.; Riess, V.; Szylo, K.J.; Eisenburger, R.; Schadendorf, D.; Ubellacker, J.M.; Tasdogan, A. Advancements in melanoma cancer metastasis models. Pigment Cell Melanoma Res. 2023, 36, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Sullivan, R.J.; Yaeger, R. Molecular Pathways and Mechanisms of BRAF in Cancer Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 4618–4628. [Google Scholar] [CrossRef]
- Ziogas, D.C.; Theocharopoulos, C.; Koutouratsas, T.; Haanen, J.; Gogas, H. Mechanisms of resistance to immune checkpoint inhibitors in melanoma: What we have to overcome? Cancer Treat. Rev. 2023, 113, 102499. [Google Scholar] [CrossRef] [PubMed]
- Merlino, G.; Herlyn, M.; Fisher, D.E.; Bastian, B.C.; Flaherty, K.T.; Davies, M.A.; Wargo, J.A.; Curiel-Lewandrowski, C.; Weber, M.J.; Leachman, S.A.; et al. The state of melanoma: Challenges and opportunities. Pigment Cell Melanoma Res. 2016, 29, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Bashash, D.; Zandi, Z.; Kashani, B.; Pourbagheri-Sigaroodi, A.; Salari, S.; Ghaffari, S.H. Resistance to immunotherapy in human malignancies: Mechanisms, research progresses, challenges, and opportunities. J. Cell. Physiol. 2022, 237, 346–372. [Google Scholar] [CrossRef]
- Ganesh, K.; Massagué, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N.; et al. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef]
- Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef]
- Ngeow, K.C.; Friedrichsen, H.J.; Li, L.; Zeng, Z.; Andrews, S.; Volpon, L.; Brunsdon, H.; Berridge, G.; Picaud, S.; Fischer, R.; et al. BRAF/MAPK and GSK3 signaling converges to control MITF nuclear export. Proc. Natl. Acad. Sci. USA 2018, 115, E8668–E8677. [Google Scholar] [CrossRef] [PubMed]
- Vachtenheim, J.; Ondrušová, L. Microphthalmia-associated transcription factor expression levels in melanoma cells contribute to cell invasion and proliferation. Exp. Dermatol. 2015, 24, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Balsa, E.; Perry, E.A.; Liang, J.; Tavares, C.D.; Vazquez, F.; Widlund, H.R.; Puigserver, P. H3K27me3-mediated PGC1α gene silencing promotes melanoma invasion through WNT5A and YAP. J. Clin. Investig. 2020, 130, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Lim, J.H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.R.; Puigserver, P. A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016, 537, 422–426. [Google Scholar] [CrossRef]
- Di Rocco, G.; Pennuto, M.; Illi, B.; Canu, N.; Filocamo, G.; Trani, E.; Rinaldi, A.M.; Possenti, R.; Mandolesi, G.; Sirinian, M.I.; et al. Interplay of the E box, the cyclic AMP response element, and HTF4/HEB in transcriptional regulation of the neurospecific, neurotrophin-inducible vgf gene. Mol. Cell Biol. 1997, 17, 1244–1253. [Google Scholar] [CrossRef]
- Li, Y.; Brauer, P.M.; Singh, J.; Xhiku, S.; Yoganathan, K.; Zúñiga-Pflücker, J.C.; Anderson, M.K. Targeted Disruption of TCF12 Reveals HEB as Essential in Human Mesodermal Specification and Hematopoiesis. Stem Cell Rep. 2017, 9, 779–795. [Google Scholar] [CrossRef]
- Parker, M.H.; Perry RL, S.; Fauteux, M.C.; Berkes, C.A.; Rudnicki, M.A. MyoD synergizes with the E-protein HEB beta to induce myogenic differentiation. Mol. Cell. Biol. 2006, 26, 5771–5783. [Google Scholar] [CrossRef]
- Yi, S.; Yu, M.; Yang, S.; Miron, R.J.; Zhang, Y. Tcf12, A Member of Basic Helix-Loop-Helix Transcription Factors, Mediates Bone Marrow Mesenchymal Stem Cell Osteogenic Differentiation In Vitro and In Vivo. Stem Cells 2017, 35, 386–397. [Google Scholar] [CrossRef]
- Yoon, S.J.; Foley, J.W.; Baker, J.C. HEB associates with PRC2 and SMAD2/3 to regulate developmental fates. Nat. Commun. 2015, 6, 6546. [Google Scholar] [CrossRef]
- Lee, C.-C.; Chen, W.-S.; Chen, C.-C.; Chen, L.-L.; Lin, Y.-S.; Fan, C.-S.; Huang, T.-S. TCF12 protein functions as transcriptional repressor of E-cadherin, and its overexpression is correlated with metastasis of colorectal cancer. J. Biol. Chem. 2012, 287, 2798–2809. [Google Scholar] [CrossRef]
- Wang, L.; Tang, Y.; Wu, H.; Shan, G. TCF12 activates MAGT1 expression to regulate the malignant progression of pancreatic carcinoma cells. Oncol. Lett. 2022, 23, 62. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, L.; Jiang, Z.; Ge, C.; Zhao, F.; Jiang, J.; Tian, H.; Chen, T.; Xie, H.; Cui, Y.; et al. TCF12 promotes the tumorigenesis and metastasis of hepatocellular carcinoma via upregulation of CXCR4 expression. Theranostics 2019, 9, 5810–5827. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Bian, T.; Zhang, Y.; Su, M.; Liu, Y. TCF12 overexpression as a poor prognostic factor in ovarian cancer. Pathol. Res. Pract. 2019, 215, 152527. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhao, M.; Yang, Y.; Xu, R.; Tong, L.; Liang, J.; Zhang, X.; Sun, Y.; Fan, Y. Reversal of epithelial-mesenchymal transition and inhibition of tumor stemness of breast cancer cells through advanced combined chemotherapy. Acta Biomater. 2022, 152, 380–392. [Google Scholar] [CrossRef]
- Tai, G.; Fu, H.; Bai, H.; Liu, H.; Li, L.; Song, T. Long non-coding RNA GLIDR accelerates the tumorigenesis of lung adenocarcinoma by miR-1270/TCF12 axis. Cell Cycle (Georget. Tex.) 2021, 20, 1653–1662. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Plantefaber, L.C.; Hynes, R.O. Changes in integrin receptors on oncogenically transformed cells. Cell 1989, 56, 281–290. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Fagin, J.A.; Krishnamoorthy, G.P.; Landa, I. Pathogenesis of cancers derived from thyroid follicular cells. Nat. Rev. Cancer 2023. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Delyon, J.; Vallet, A.; Bernard-Cacciarella, M.; Kuzniak, I.; Reger de Moura, C.; Louveau, B.; Jouenne, F.; Mourah, S.; Lebbé, C.; Dumaz, N. TERT Expression Induces Resistance to BRAF and MEK Inhibitors in BRAF-Mutated Melanoma In Vitro. Cancers 2023, 15, 2888. [Google Scholar] [CrossRef] [PubMed]
- Marranci, A.; Prantera, A.; Masotti, S.; De Paolo, R.; Baldanzi, C.; Podda, M.S.; Mero, S.; Vitiello, M.; Franchin, C.; Laezza, M.; et al. PARP1 negatively regulates MAPK signaling by impairing BRAF-X1 translation. J. Hematol. Oncol. 2023, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Mahumud, R.A.; Shahjalal, M. The Emerging Burden of Genetic Instability and Mutation in Melanoma: Role of Molecular Mechanisms. Cancers 2022, 14, 6202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef]
- Baba, A.B.; Rah, B.; Bhat, G.R.; Mushtaq, I.; Parveen, S.; Hassan, R.; Hameed Zargar, M.; Afroze, D. Transforming Growth Factor-Beta (TGF-β) Signaling in Cancer-A Betrayal Within. Front. Pharmacol. 2022, 13, 791272. [Google Scholar] [CrossRef]
- Krasagakis, K.; Krüger-Krasagakes, S.; Fimmel, S.; Eberle, J.; Thölke, D.; von der Ohe, M.; Mansmann, U.; Orfanos, C.E. Desensitization of melanoma cells to autocrine TGF-beta isoforms. J. Cell Physiol. 1999, 178, 179–187. [Google Scholar] [CrossRef]
- Javelaud, D.; Mohammad, K.S.; McKenna, C.R.; Fournier, P.; Luciani, F.; Niewolna, M.; André, J.; Delmas, V.; Larue, L.; Guise, T.A.; et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 2007, 67, 2317–2324. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Alexaki, V.I.; Mauviel, A. Transforming growth factor-beta in cutaneous melanoma. Pigment Cell Melanoma Res. 2008, 21, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Berking, C.; Takemoto, R.; Schaider, H.; Showe, L.; Satyamoorthy, K.; Robbins, P.; Herlyn, M. Transforming growth factor-beta1 increases survival of human melanoma through stroma remodeling. Cancer Res. 2001, 61, 8306–8316. [Google Scholar] [PubMed]
- Cosgarea, I.; McConnell, A.T.; Ewen, T.; Tang, D.; Hill, D.S.; Anagnostou, M.; Elias, M.; Ellis, R.A.; Murray, A.; Spender, L.C.; et al. Melanoma secretion of transforming growth factor-β2 leads to loss of epidermal AMBRA1 threatening epidermal integrity and facilitating tumour ulceration. Br. J. Dermatol. 2022, 186, 694–704. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Zhou, J.; Chai, X.; Ping, Z.; Zhao, Y.; Xu, X.; Luo, C.; Sheng, J. TCF12 Activates TGFB2 Expression to Promote the Malignant Progression of Melanoma. Cancers 2023, 15, 4505. https://doi.org/10.3390/cancers15184505
Tian Y, Zhou J, Chai X, Ping Z, Zhao Y, Xu X, Luo C, Sheng J. TCF12 Activates TGFB2 Expression to Promote the Malignant Progression of Melanoma. Cancers. 2023; 15(18):4505. https://doi.org/10.3390/cancers15184505
Chicago/Turabian StyleTian, Youjia, Jiang Zhou, Xinxin Chai, Zejun Ping, Yurong Zhao, Xin Xu, Chi Luo, and Jinghao Sheng. 2023. "TCF12 Activates TGFB2 Expression to Promote the Malignant Progression of Melanoma" Cancers 15, no. 18: 4505. https://doi.org/10.3390/cancers15184505
APA StyleTian, Y., Zhou, J., Chai, X., Ping, Z., Zhao, Y., Xu, X., Luo, C., & Sheng, J. (2023). TCF12 Activates TGFB2 Expression to Promote the Malignant Progression of Melanoma. Cancers, 15(18), 4505. https://doi.org/10.3390/cancers15184505