
NOTCH and AKT Signalling Interact to Drive Mammary Tumour Heterogeneity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Mouse Cohorts
2.2. Tumour Phenotyping
2.3. Gene Expression Analysis Using Quantitative Real-Time rtPCR
2.4. Western Blotting
2.5. Statistics
3. Results
3.1. Notch1 and Notch2 Are Differentially Expressed in Mammary Tumours
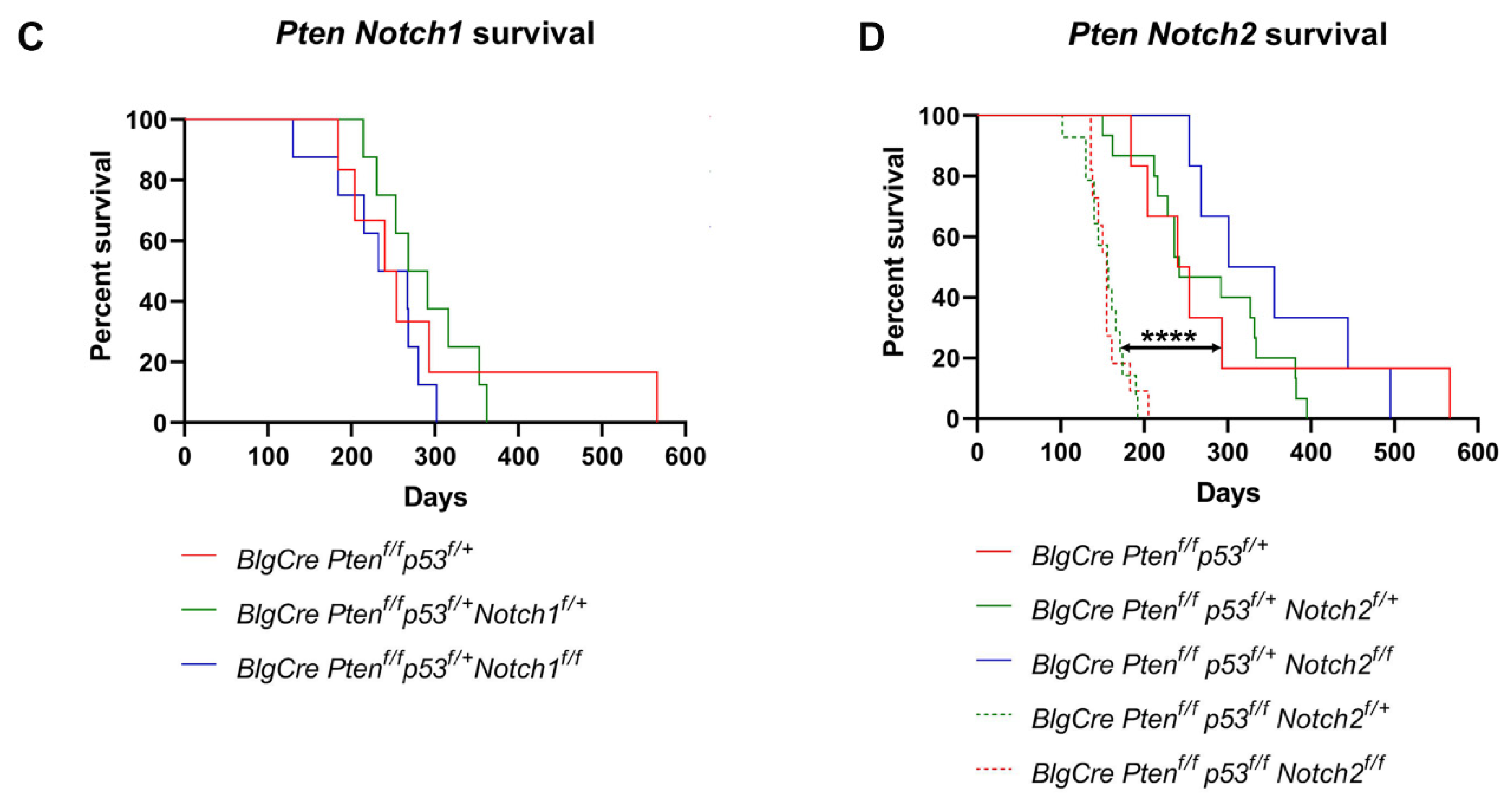
3.2. Notch1/2 Deletion Does Not Alter Tumour Onset
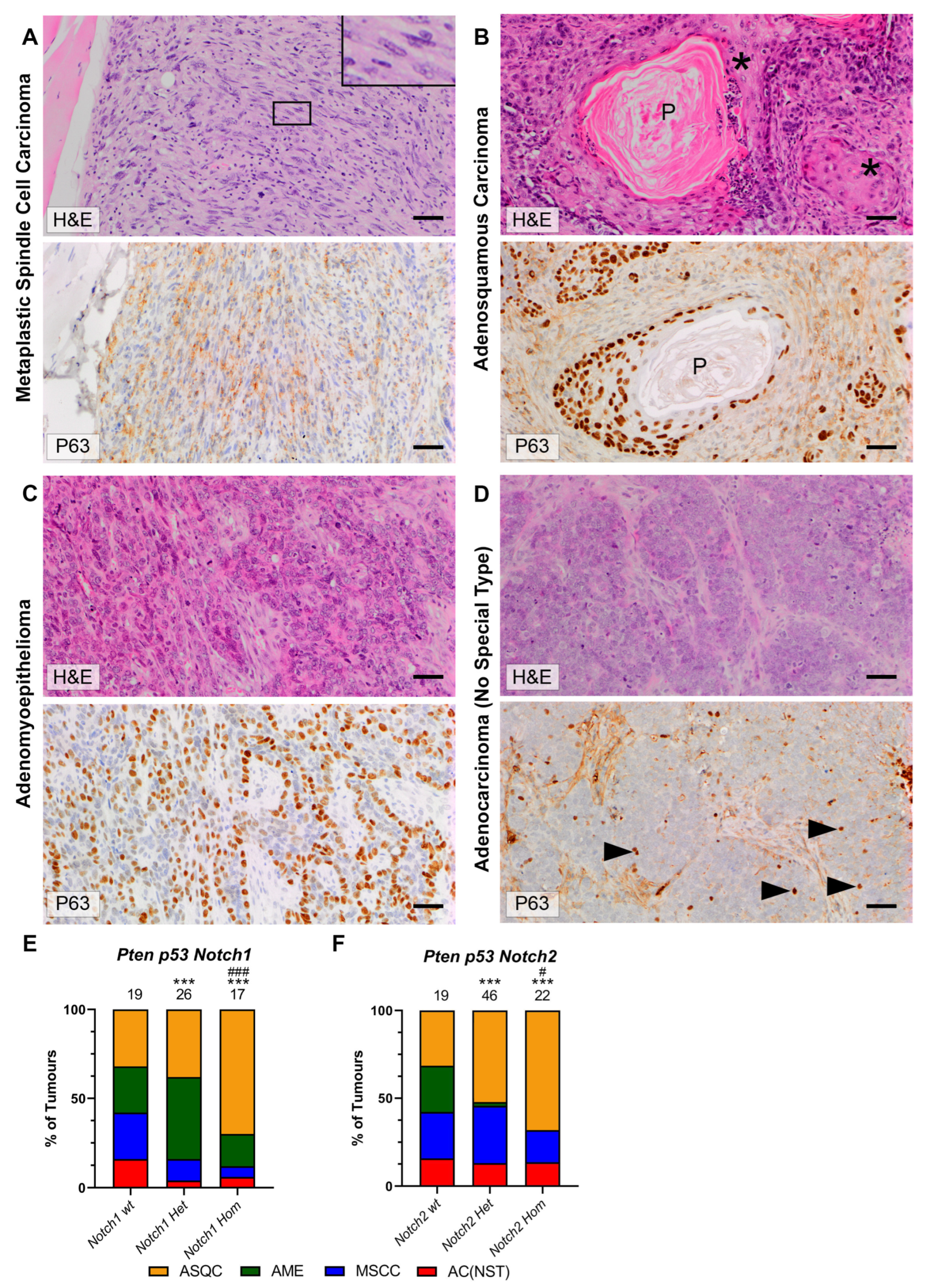
3.3. Notch Deletion Increases the Proportion of AME and ASQC Tumours
3.4. Expression of Lineage-Associated Genes Is Associated with Histotype Rather than Notch1/2 Status
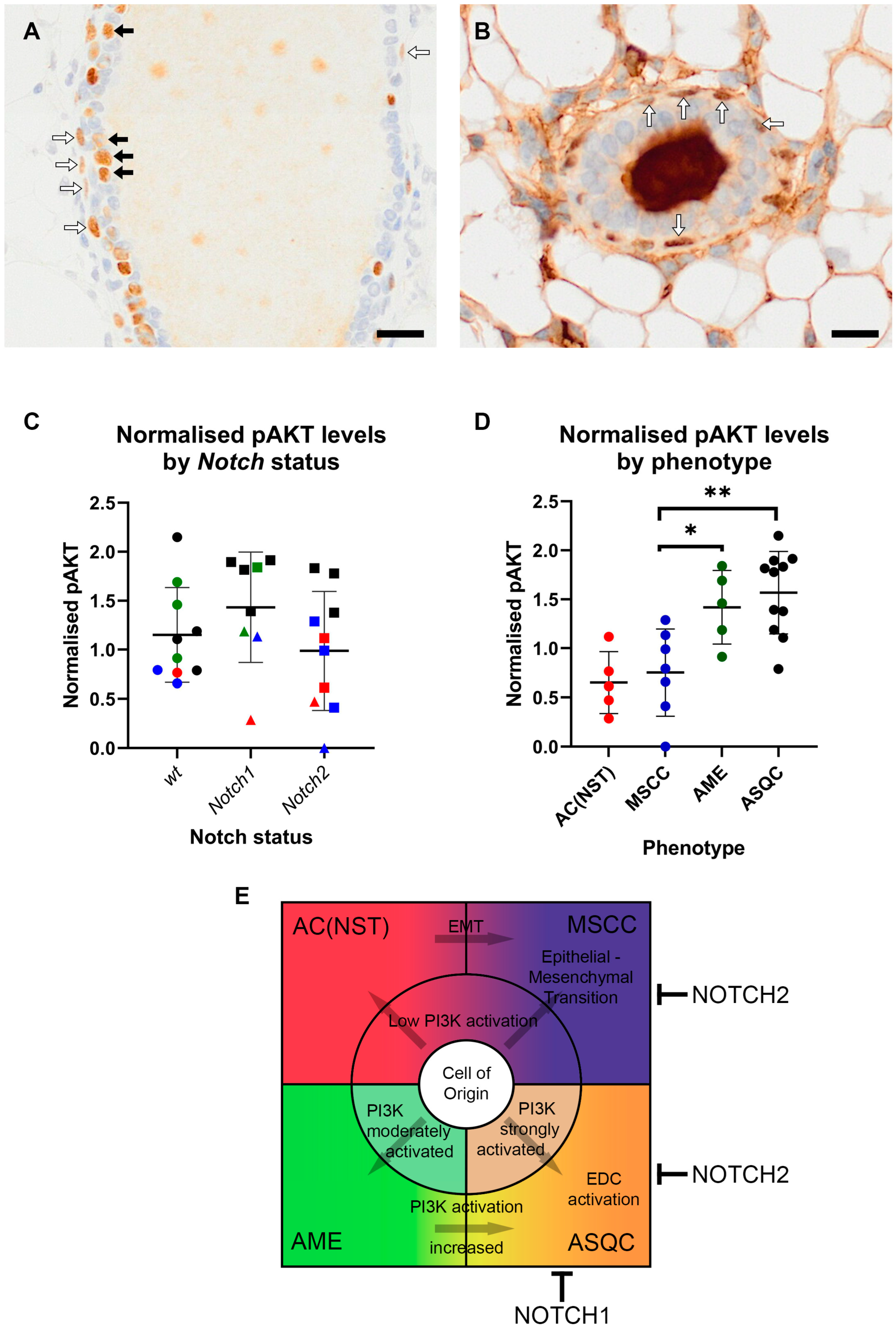
3.5. AKT Signalling Is Upregulated in AME and ASQC Tumours
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Melchor, L.; Molyneux, G.; Mackay, A.; Magnay, F.A.; Atienza, M.; Kendrick, H.; Nava-Rodrigues, D.; Lopez-Garcia, M.A.; Milanezi, F.; Greenow, K.; et al. Identification of cellular and genetic drivers of breast cancer heterogeneity in genetically engineered mouse tumour models. J. Pathol. 2014, 233, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, G.; Geyer, F.C.; Magnay, F.A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; Mackay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, L.D.; Melchor, L.; Greenow, K.R.; Kendrick, H.; Tornillo, G.; Bradford, J.; Giles, P.; Smalley, M.J. Reproductive history determines Erbb2 locus amplification, WNT signalling and tumour phenotype in a murine breast cancer model. Dis. Model. Mech. 2021, 14, dmm048736. [Google Scholar] [CrossRef]
- WHO Classification of Tumours: Breast Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2019.
- Aiello, N.M.; Stanger, B.Z. Echoes of the embryo: Using the developmental biology toolkit to study cancer. Dis. Model. Mech. 2016, 9, 105–114. [Google Scholar] [CrossRef]
- Bianchi, S.; Dotti, M.T.; Federico, A. Physiology and pathology of notch signalling system. J. Cell Physiol. 2006, 207, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Lilja, A.M.; Rodilla, V.; Huyghe, M.; Hannezo, E.; Landragin, C.; Renaud, O.; Leroy, O.; Rulands, S.; Simons, B.D.; Fre, S. Clonal analysis of Notch1-expressing cells reveals the existence of unipotent stem cells that retain long-term plasticity in the embryonic mammary gland. Nat. Cell Biol. 2018, 20, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Lafkas, D.; Rodilla, V.; Huyghe, M.; Mourao, L.; Kiaris, H.; Fre, S. Notch3 marks clonogenic mammary luminal progenitor cells in vivo. J. Cell Biol. 2013, 203, 47–56. [Google Scholar] [CrossRef]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef]
- Speiser, J.; Foreman, K.; Drinka, E.; Godellas, C.; Perez, C.; Salhadar, A.; Ersahin, C.; Rajan, P. Notch-1 and Notch-4 biomarker expression in triple-negative breast cancer. Int. J. Surg. Pathol. 2012, 20, 139–145. [Google Scholar] [CrossRef]
- Lamy, M.; Ferreira, A.; Dias, J.S.; Braga, S.; Silva, G.; Barbas, A. Notch-out for breast cancer therapies. New Biotechnol. 2017, 39, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, S.; Clarke, R.B.; Brennan, K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006, 66, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.P.; Edvardsen, H.; Kaushiva, A.; Arhancet, J.P.; Howe, T.M.; Kohaar, I.; Porter-Gill, P.; Shah, A.; Landmark-Hoyvik, H.; Fossa, S.D.; et al. NOTCH2 in breast cancer: Association of SNP rs11249433 with gene expression in ER-positive breast tumors without TP53 mutations. Mol. Cancer 2010, 9, 113. [Google Scholar] [CrossRef]
- Kendrick, H.; Regan, J.L.; Magnay, F.A.; Grigoriadis, A.; Mitsopoulos, C.; Zvelebil, M.; Smalley, M.J. Transcriptome analysis of mammary epithelial subpopulations identifies novel determinants of lineage commitment and cell fate. BMC Genom. 2008, 9, 591. [Google Scholar] [CrossRef]
- Smalley, M.J. Isolation, culture and analysis of mouse mammary epithelial cells. Methods Mol. Biol. 2010, 633, 139–170. [Google Scholar] [CrossRef]
- Cumming, G.; Fidler, F.; Vaux, D.L. Error bars in experimental biology. J. Cell Biol. 2007, 177, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar] [CrossRef]
- Yallowitz, A.R.; Alexandrova, E.M.; Talos, F.; Xu, S.; Marchenko, N.D.; Moll, U.M. p63 is a prosurvival factor in the adult mammary gland during post-lactational involution, affecting PI-MECs and ErbB2 tumorigenesis. Cell Death Differ. 2014, 21, 645–654. [Google Scholar] [CrossRef]
- Tadeu, A.M.; Horsley, V. Notch signaling represses p63 expression in the developing surface ectoderm. Development 2013, 140, 3777–3786. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Barton, C.E.; Pietenpol, J.A. Delta Np63 alpha expression is regulated by the phosphoinositide 3-kinase pathway. J. Biol. Chem. 2003, 278, 51408–51414. [Google Scholar] [CrossRef]
- Koren, S.; Reavie, L.; Couto, J.P.; De Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O.; et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.R.; Land, S.R.; Saad, R.S.; Fisher, B.; Wickerham, D.L.; Wang, M.; Costantino, J.P.; Wolmark, N. Pathologic variables predictive of breast events in patients with ductal carcinoma in situ. Am. J. Clin. Pathol. 2007, 128, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Rasotto, R.; Berlato, D.; Goldschmidt, M.H.; Zappulli, V. Prognostic Significance of Canine Mammary Tumor Histologic Subtypes: An Observational Cohort Study of 229 Cases. Vet. Pathol. 2017, 54, 571–578. [Google Scholar] [CrossRef]
- Grego-Bessa, J.; Diez, J.; Timmerman, L.; de la Pompa, J.L. Notch and epithelial-mesenchyme transition in development and tumor progression: Another turn of the screw. Cell Cycle 2004, 3, 718–721. [Google Scholar] [CrossRef]
- Cavazza, A.; Miccio, A.; Romano, O.; Petiti, L.; Malagoli Tagliazucchi, G.; Peano, C.; Severgnini, M.; Rizzi, E.; De Bellis, G.; Bicciato, S.; et al. Dynamic Transcriptional and Epigenetic Regulation of Human Epidermal Keratinocyte Differentiation. Stem Cell Rep. 2016, 6, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Jarde, T.; Lloyd-Lewis, B.; Thomas, M.; Kendrick, H.; Melchor, L.; Bougaret, L.; Watson, P.D.; Ewan, K.; Smalley, M.J.; Dale, T.C. Wnt and Neuregulin1/ErbB signalling extends 3D culture of hormone responsive mammary organoids. Nat. Commun. 2016, 7, 13207. [Google Scholar] [CrossRef]
- Koster, M.I.; Dai, D.; Marinari, B.; Sano, Y.; Costanzo, A.; Karin, M.; Roop, D.R. p63 induces key target genes required for epidermal morphogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 3255–3260. [Google Scholar] [CrossRef]
- Koster, M.I.; Marinari, B.; Payne, A.S.; Kantaputra, P.N.; Costanzo, A.; Roop, D.R. DeltaNp63 knockdown mice: A mouse model for AEC syndrome. Am. J. Med. Genet. A 2009, 149, 1942–1947. [Google Scholar] [CrossRef]
- Romano, R.A.; Smalley, K.; Magraw, C.; Serna, V.A.; Kurita, T.; Raghavan, S.; Sinha, S. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 2012, 139, 772–782. [Google Scholar] [CrossRef]
- Mikkola, M.L. p63 in skin appendage development. Cell Cycle 2007, 6, 285–290. [Google Scholar] [CrossRef]
- Yoh, K.; Prywes, R. Pathway Regulation of p63, a Director of Epithelial Cell Fate. Front. Endocrinol. 2015, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Damonte, P.; Gregg, J.P.; Borowsky, A.D.; Keister, B.A.; Cardiff, R.D. EMT tumorigenesis in the mouse mammary gland. Lab. Investig. 2007, 87, 1218–1226. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ordonez, L.; Tornillo, G.; Kendrick, H.; Hay, T.; Smalley, M.J. NOTCH and AKT Signalling Interact to Drive Mammary Tumour Heterogeneity. Cancers 2023, 15, 4324. https://doi.org/10.3390/cancers15174324
Ordonez L, Tornillo G, Kendrick H, Hay T, Smalley MJ. NOTCH and AKT Signalling Interact to Drive Mammary Tumour Heterogeneity. Cancers. 2023; 15(17):4324. https://doi.org/10.3390/cancers15174324
Chicago/Turabian StyleOrdonez, Liliana, Giusy Tornillo, Howard Kendrick, Trevor Hay, and Matthew John Smalley. 2023. "NOTCH and AKT Signalling Interact to Drive Mammary Tumour Heterogeneity" Cancers 15, no. 17: 4324. https://doi.org/10.3390/cancers15174324
APA StyleOrdonez, L., Tornillo, G., Kendrick, H., Hay, T., & Smalley, M. J. (2023). NOTCH and AKT Signalling Interact to Drive Mammary Tumour Heterogeneity. Cancers, 15(17), 4324. https://doi.org/10.3390/cancers15174324