
Co-Expression of an IL-15 Superagonist Facilitates Self-Enrichment of GD2-Targeted CAR-NK Cells and Mediates Potent Cell Killing in the Absence of IL-2

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Generation of GD2-CAR-Expressing NK-92 Cells
2.3. Generation of Recombinant Derivatives of Anti-Idiotypic Antibody Ganglidiomab
2.4. Analysis of GD2 Expression and Cell Killing Activity
2.5. Analysis of RD-IL15 Expression and Activity
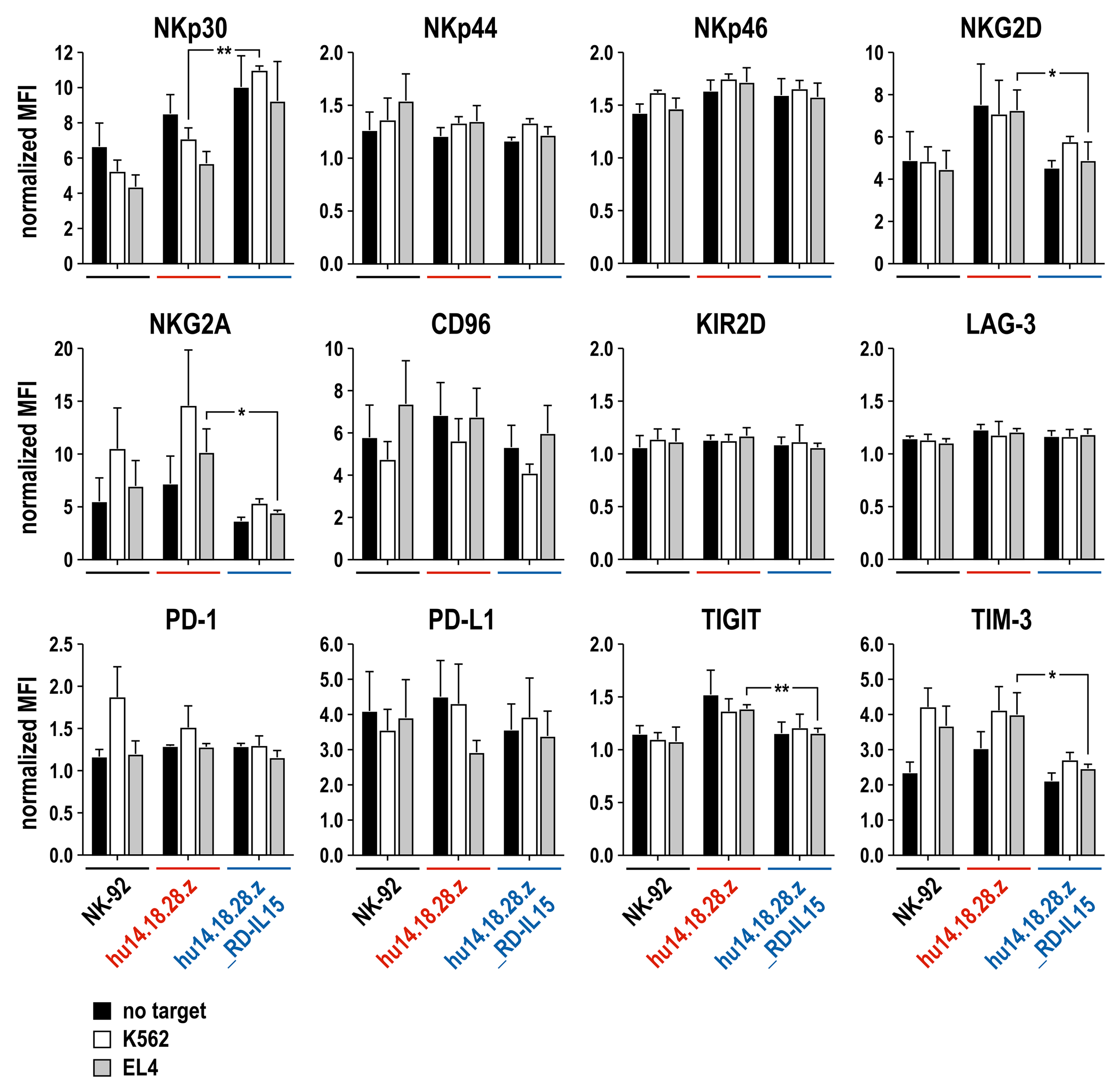
2.6. Surface Marker Analysis
2.7. Transwell Assays
2.8. Statistical Analysis
3. Results
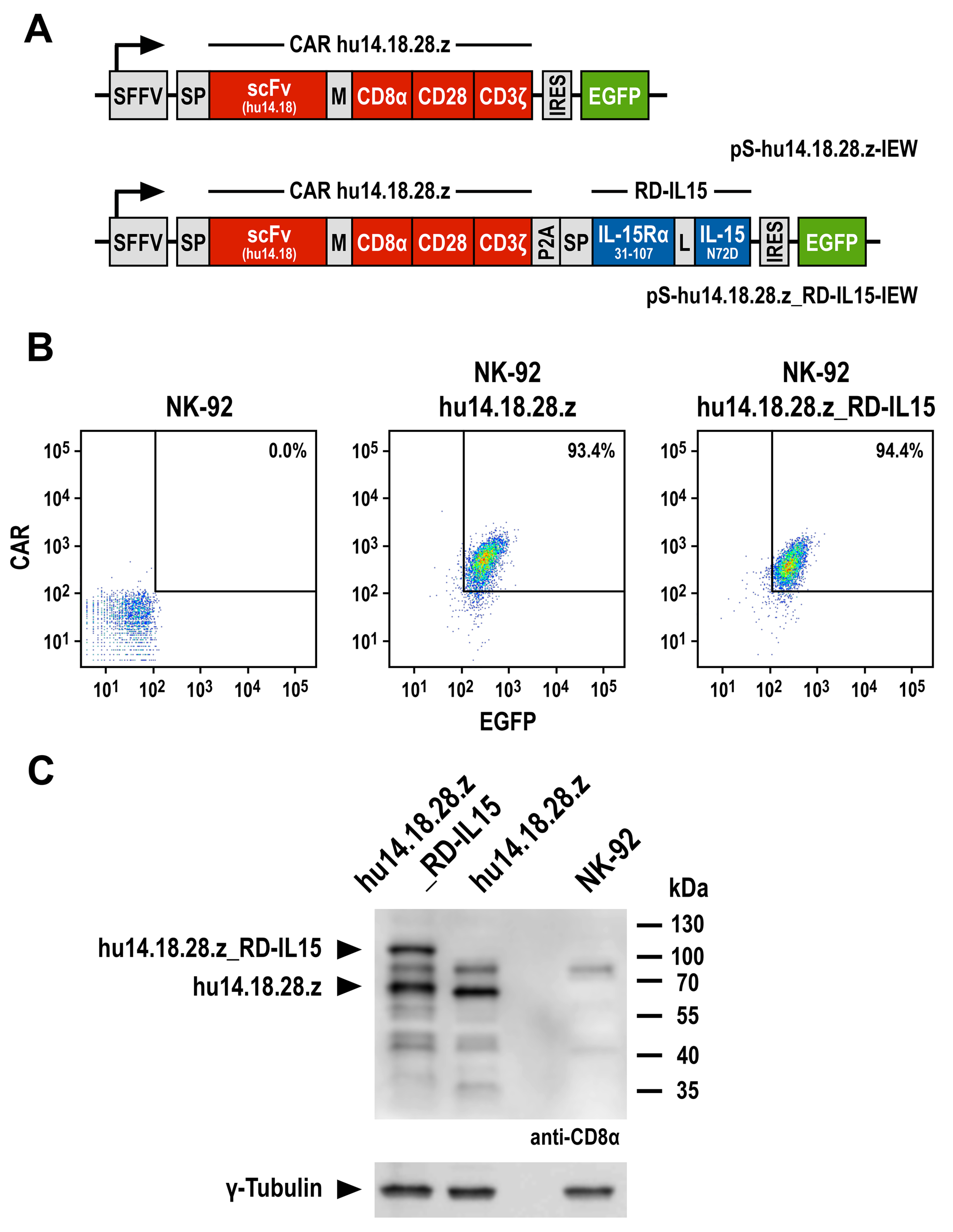
3.1. Generation of GD2-Specific CAR-NK Cells
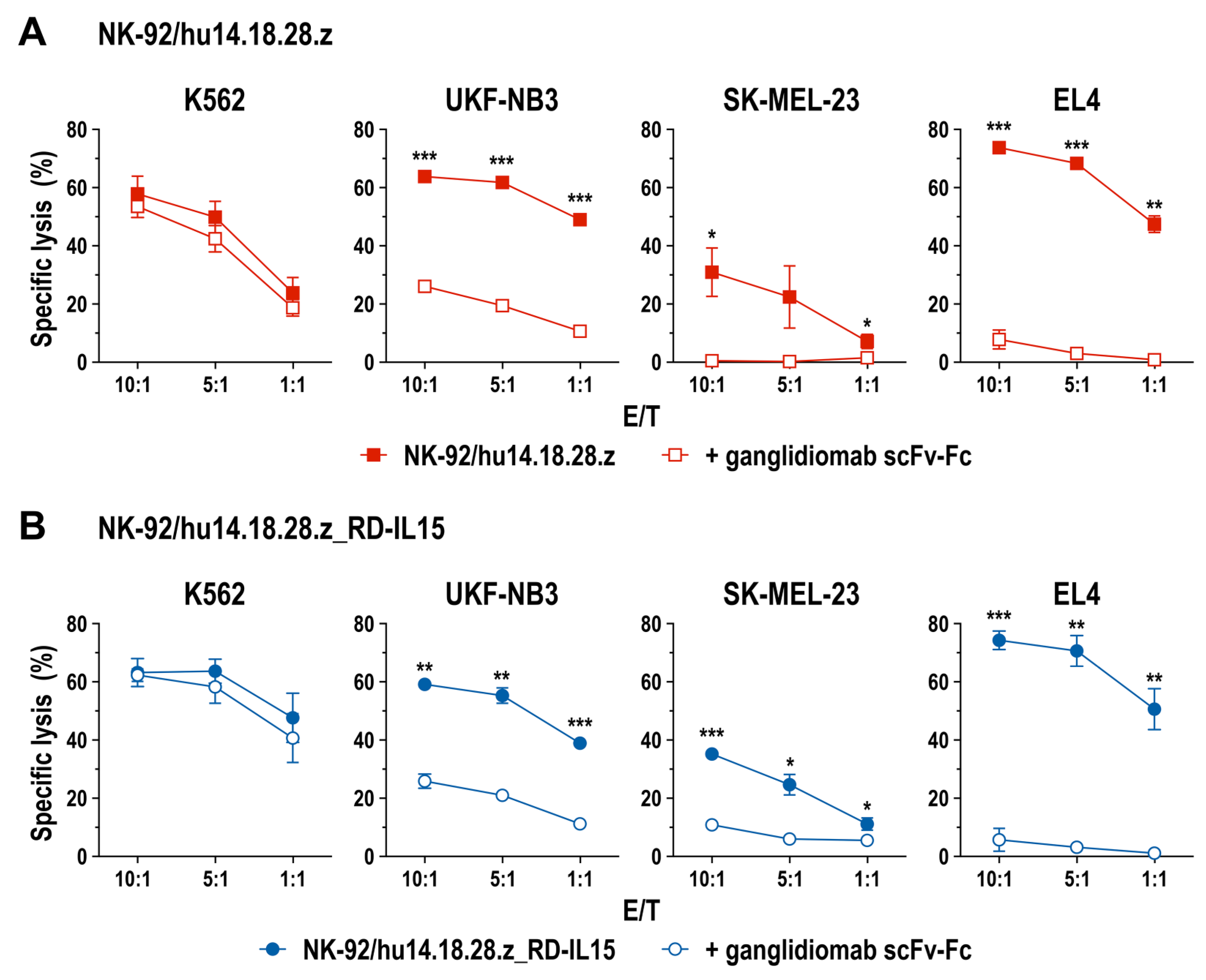
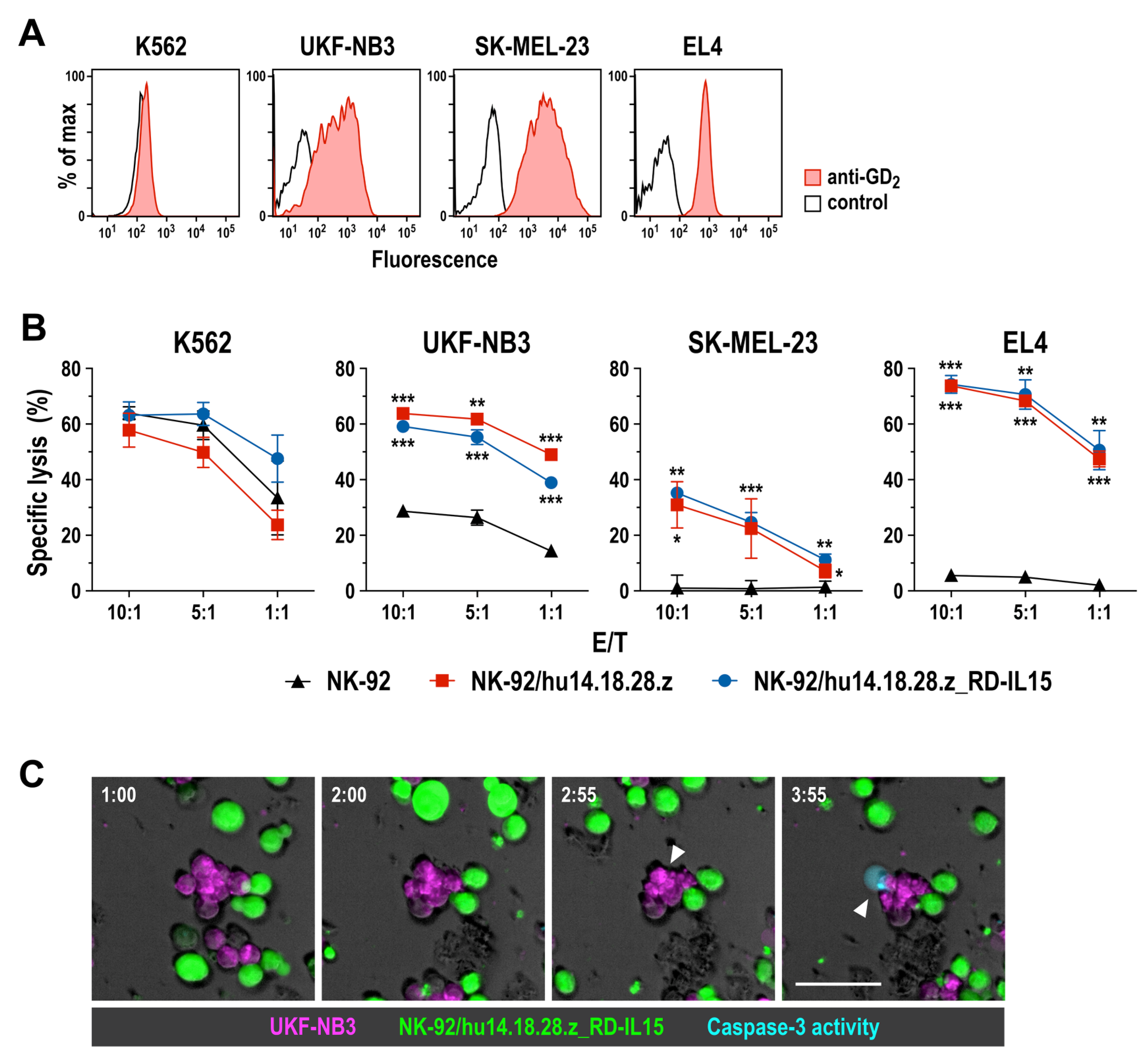
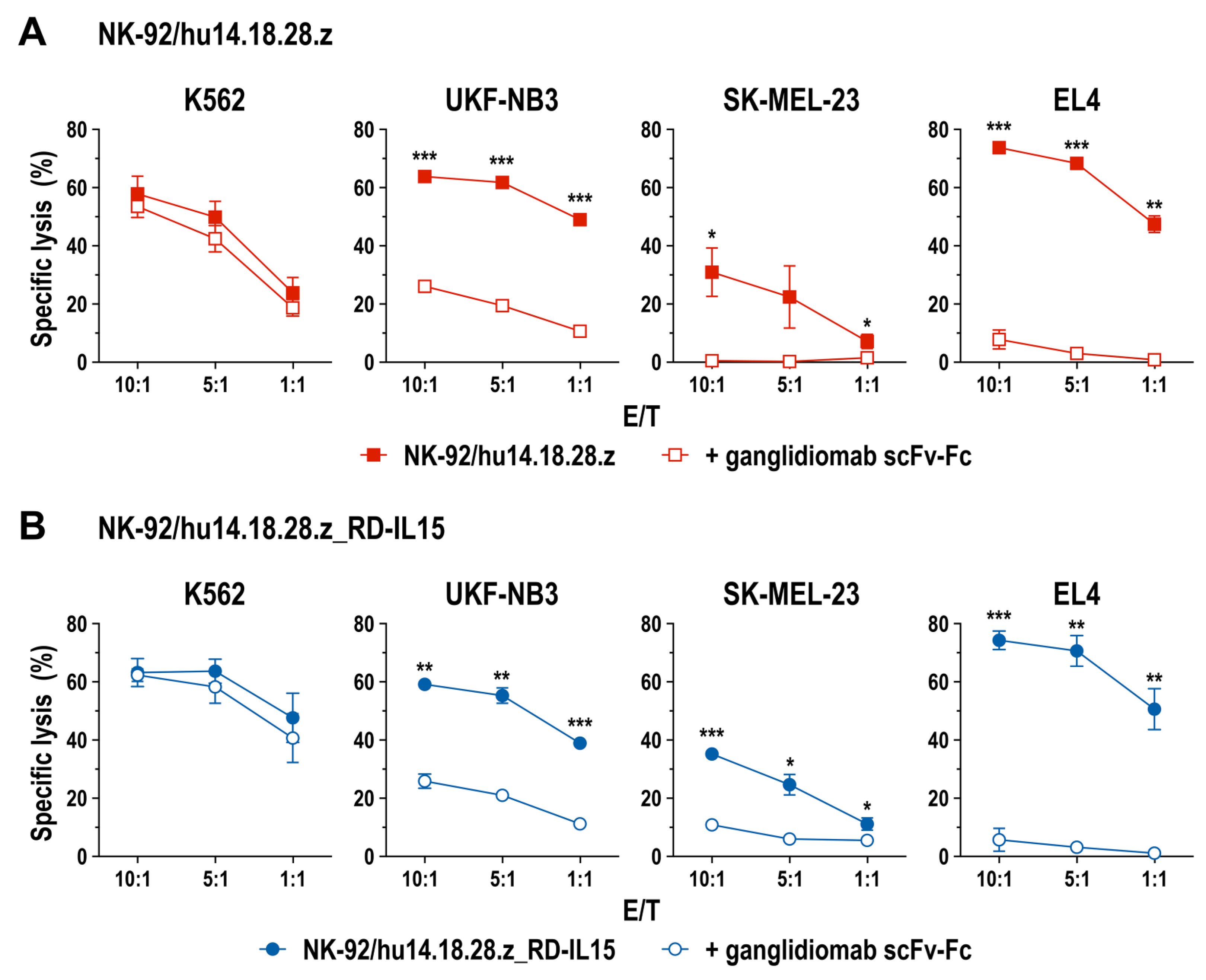
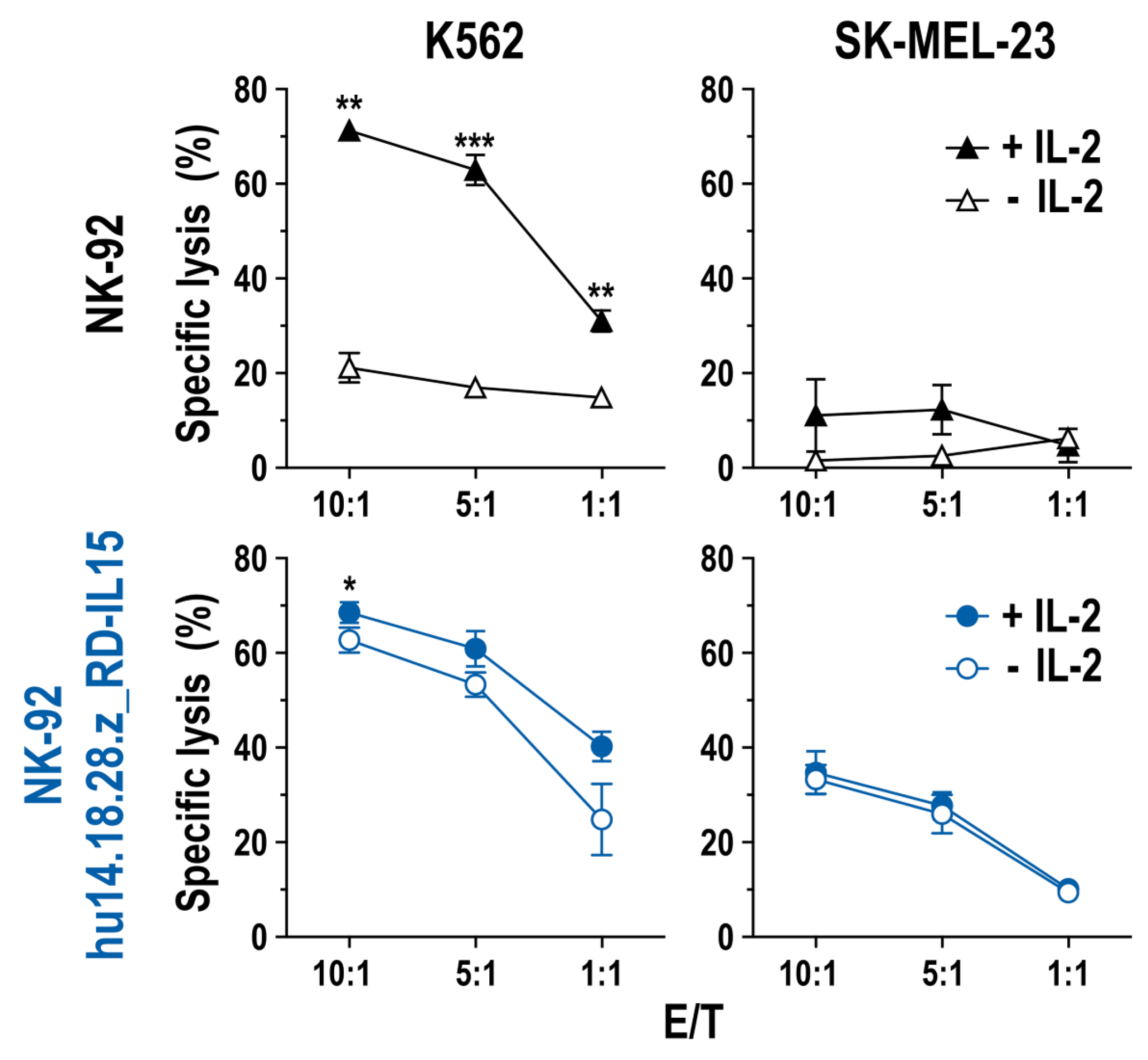
3.2. Cell Killing Activity of GD2-CAR-Engineered NK-92 Cells
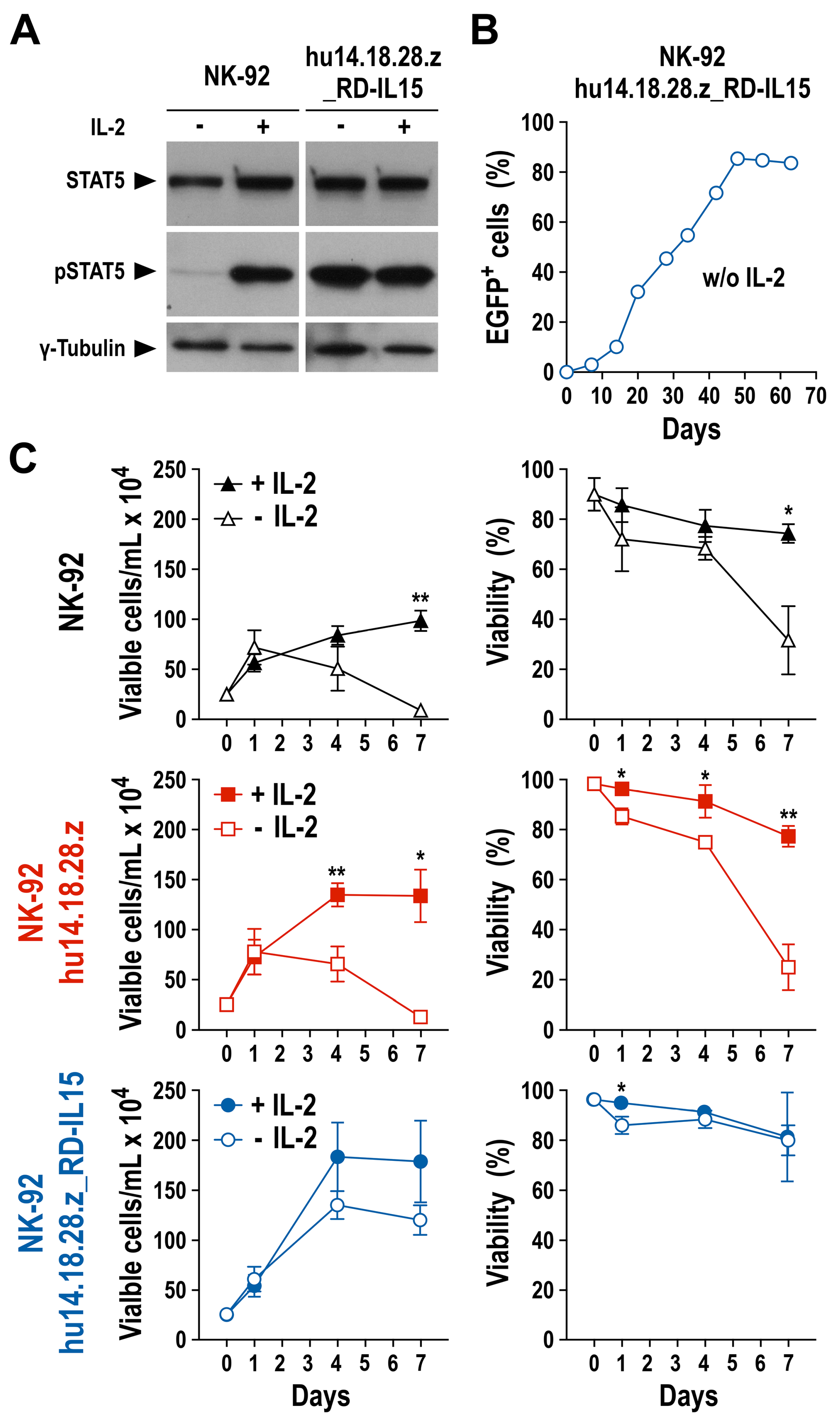
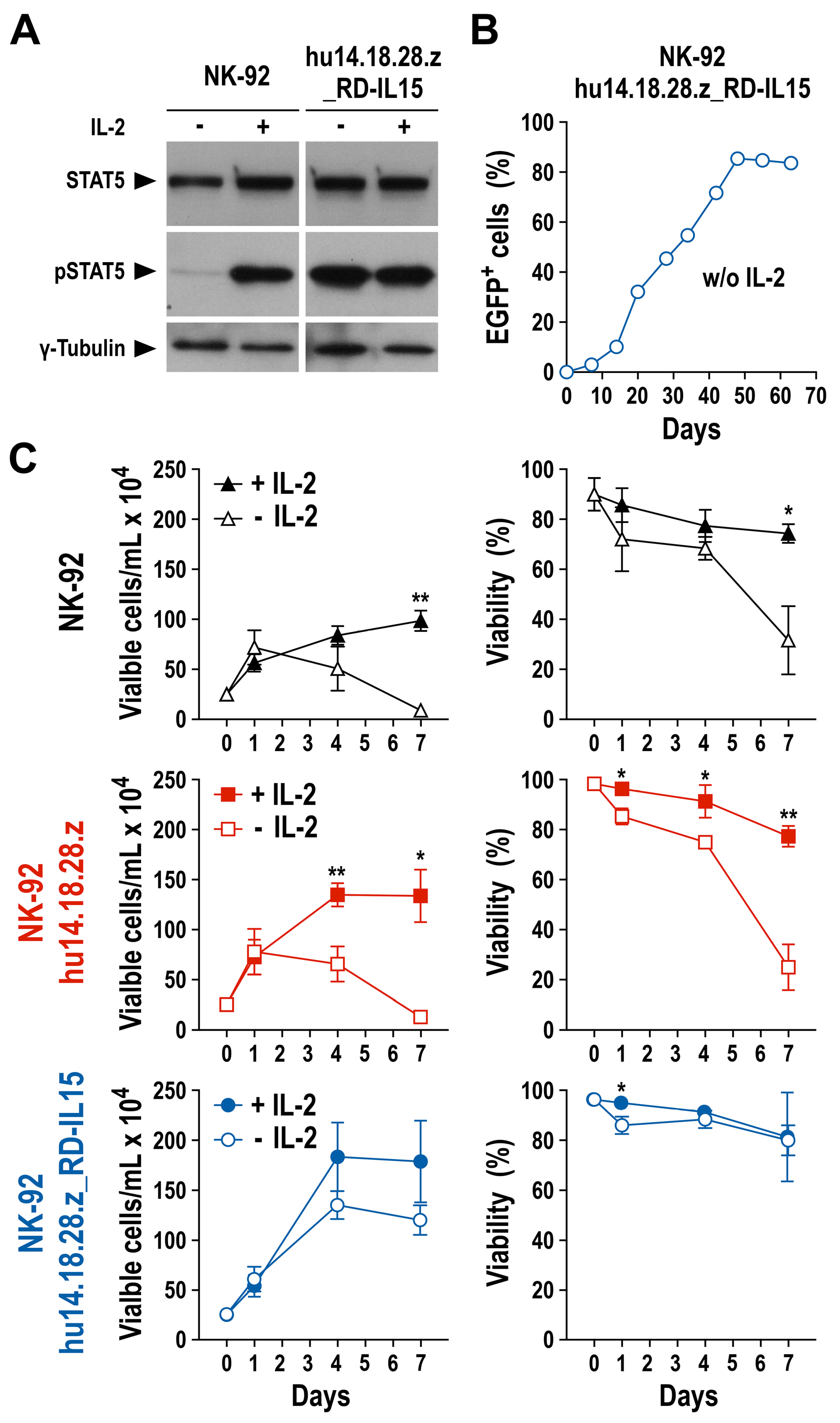
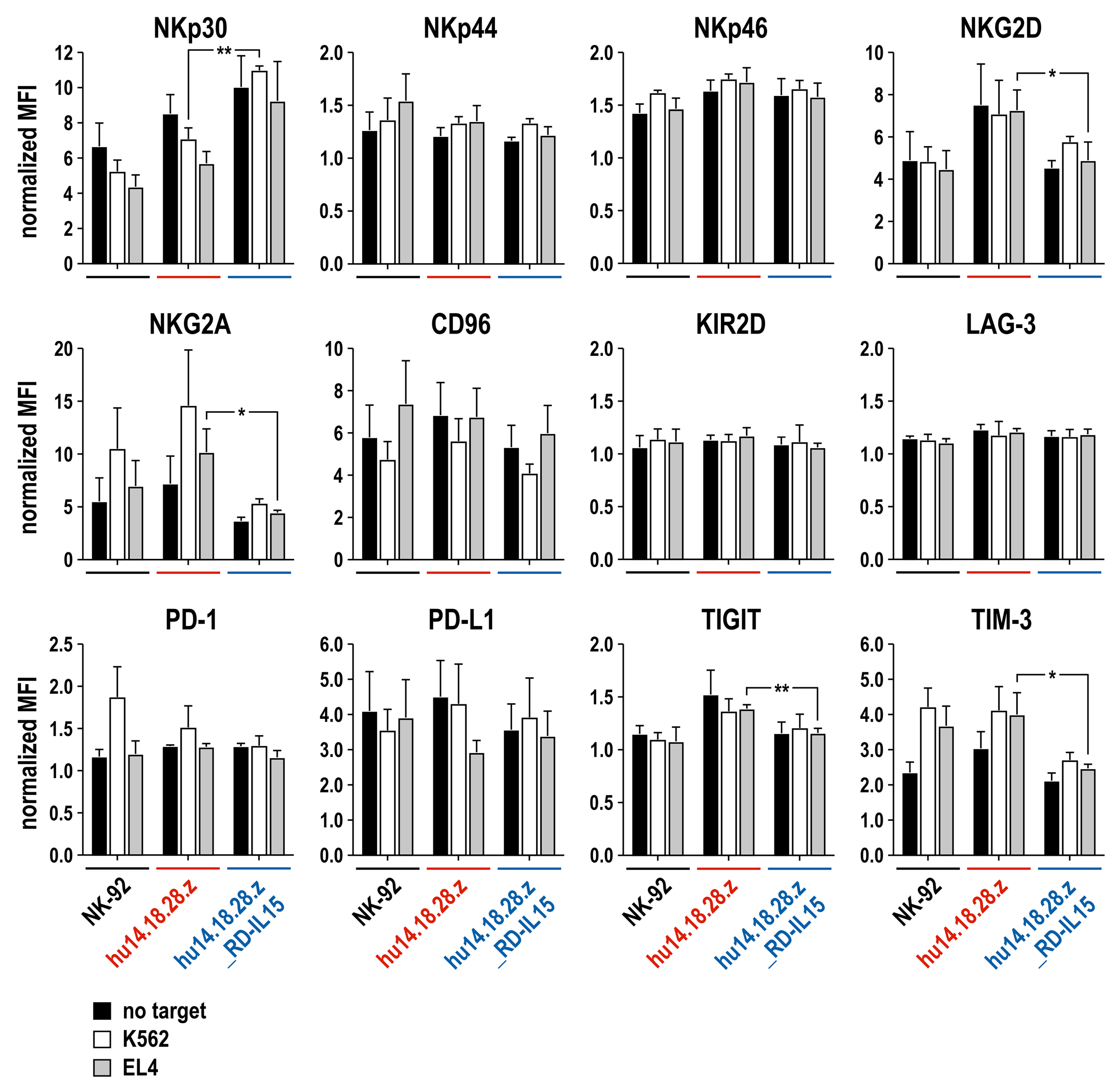
3.3. Growth and Functionality of RD-IL15-Expressing CAR-NK Cells in the Absence of IL-2
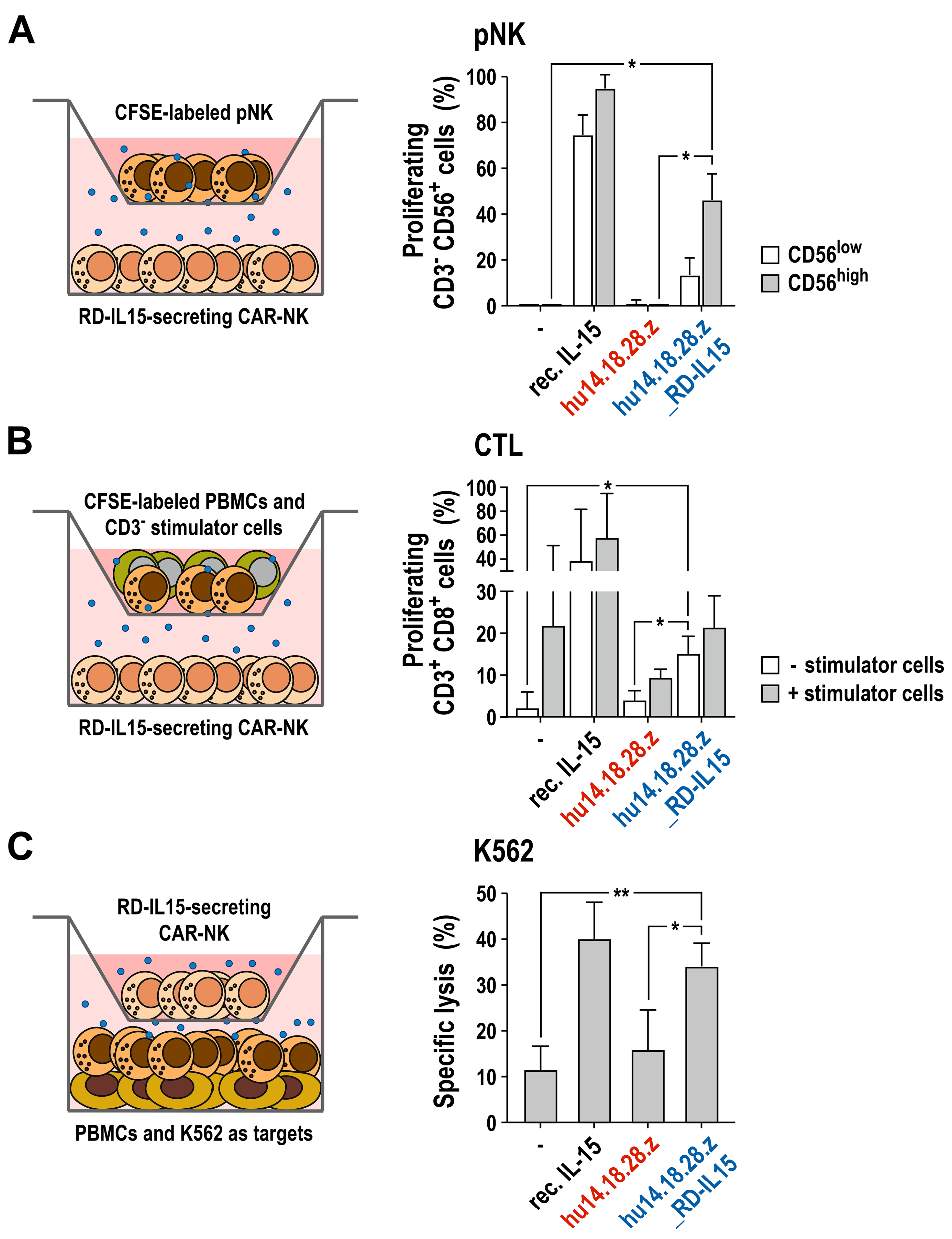
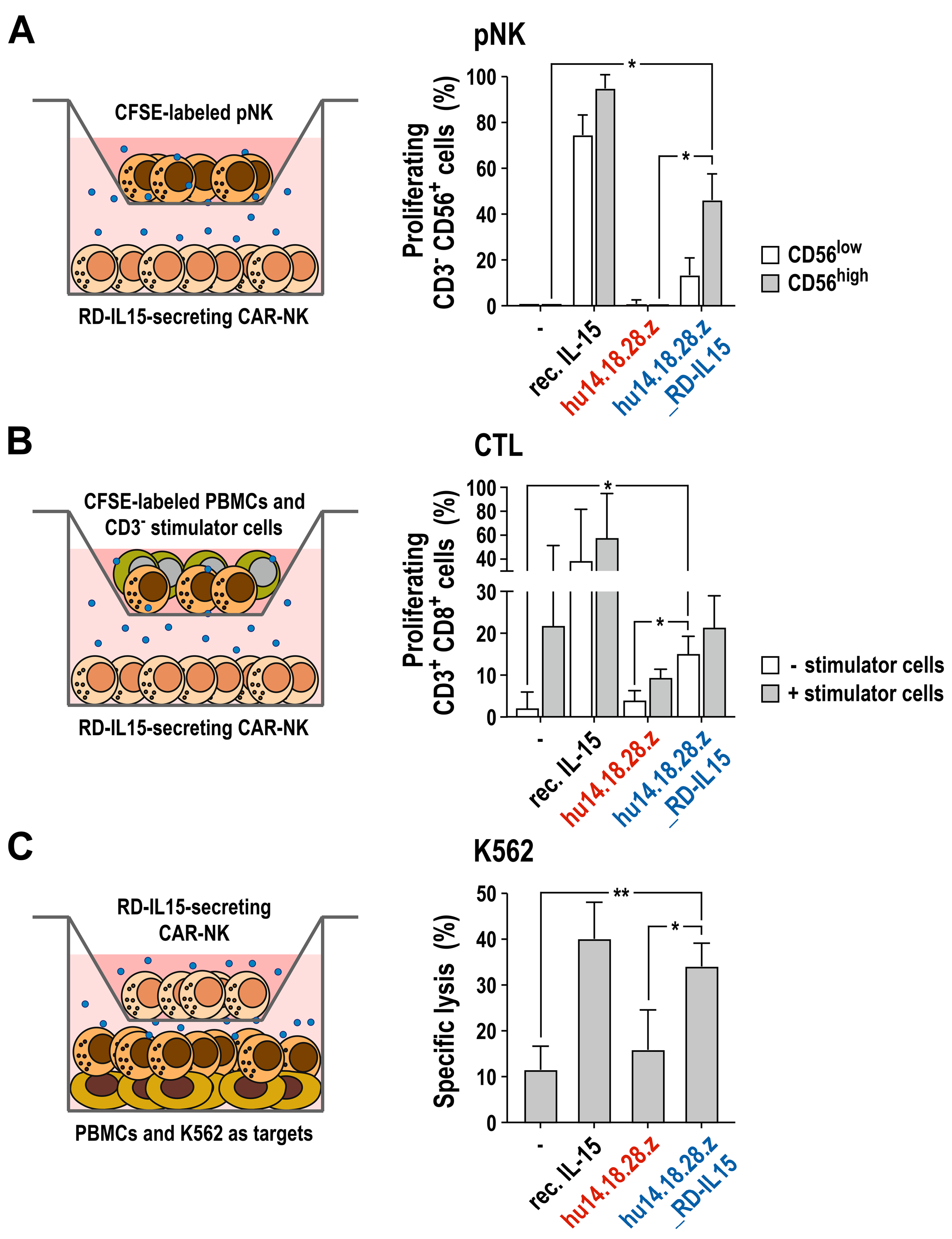
3.4. Activation of Bystander Immune Cells by Secreted RD-IL15
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The emerging landscape of immune cell therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T cell therapy for solid tumors: Bright future or dark reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef]
- Maher, J.; Davies, D.M. CAR based immunotherapy of solid tumours-A clinically based review of target antigens. Biology 2023, 12, 287. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Jin, H.J.; Nam, H.Y.; Bae, Y.K.; Kim, S.Y.; Im, I.R.; Oh, W.; Yang, Y.S.; Choi, S.J.; Kim, S.W. GD2 expression is closely associated with neuronal differentiation of human umbilical cord blood-derived mesenchymal stem cells. Cell. Mol. Life Sci. 2010, 67, 1845–1858. [Google Scholar] [CrossRef]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef]
- Yu, L.; Huang, L.; Lin, D.; Lai, X.; Wu, L.; Liao, X.; Liu, J.; Zeng, Y.; Liang, L.; Zhang, G.; et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J. Cancer Res. Clin. Oncol. 2022, 148, 2643–2652. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Heczey, A.; Xu, X.; Courtney, A.N.; Tian, G.; Barragan, G.A.; Guo, L.; Amador, C.M.; Ghatwai, N.; Rathi, P.; Wood, M.S.; et al. Anti-GD2 CAR-NKT cells in relapsed or refractory neuroblastoma: Updated phase 1 trial interim results. Nat. Med. 2023, 29, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Altvater, B.; Landmeier, S.; Pscherer, S.; Temme, J.; Schweer, K.; Kailayangiri, S.; Campana, D.; Juergens, H.; Pule, M.; Rossig, C. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res. 2009, 15, 4857–4866. [Google Scholar] [CrossRef]
- Esser, R.; Muller, T.; Stefes, D.; Kloess, S.; Seidel, D.; Gillies, S.D.; Aperlo-Iffland, C.; Huston, J.S.; Uherek, C.; Schonfeld, K.; et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J. Cell. Mol. Med. 2012, 16, 569–581. [Google Scholar] [CrossRef]
- Seidel, D.; Shibina, A.; Siebert, N.; Wels, W.S.; Reynolds, C.P.; Huebener, N.; Lode, H.N. Disialoganglioside-specific human natural killer cells are effective against drug-resistant neuroblastoma. Cancer Immunol. Immunother. 2015, 64, 621–634. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Spurny, C.; Jamitzky, S.; Schelhaas, S.; Jacobs, A.H.; Wiek, C.; Roellecke, K.; Hanenberg, H.; Hartmann, W.; et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology 2017, 6, e1250050. [Google Scholar] [CrossRef]
- Pahl, J.; Cerwenka, A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiology 2017, 222, 11–20. [Google Scholar] [CrossRef]
- Maskalenko, N.A.; Zhigarev, D.; Campbell, K.S. Harnessing natural killer cells for cancer immunotherapy: Dispatching the first responders. Nat. Rev. Drug Discov. 2022, 21, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Nowakowska, P.; Romanski, A.; Miller, N.; Odendahl, M.; Bonig, H.; Zhang, C.; Seifried, E.; Wels, W.S.; Tonn, T. Clinical grade manufacturing of genetically modified, CAR-expressing NK-92 cells for the treatment of ErbB2-positive malignancies. Cancer Immunol. Immunother. 2018, 67, 25–38. [Google Scholar] [CrossRef]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Bexte, T.; Gebel, V.; Kalensee, F.; Stolzenberg, E.; Hartmann, J.; Koehl, U.; Schambach, A.; Wels, W.S.; Modlich, U.; et al. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front. Immunol. 2020, 10, 3123. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef]
- Klingemann, H. The NK-92 cell line-30 years later: Its impact on natural killer cell research and treatment of cancer. Cytotherapy 2023, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Burger, M.C.; Forster, M.T.; Romanski, A.; Straßheimer, F.; Macas, J.; Zeiner, P.S.; Steidl, E.; Herkt, S.; Weber, K.J.; Schupp, J.; et al. Intracranial injection of NK cells engineered with a HER2-targeted chimeric antigen receptor in patients with recurrent glioblastoma. Neuro Oncol. 2023, noad087. [Google Scholar] [CrossRef]
- Ma, S.; Caligiuri, M.A.; Yu, J. Harnessing IL-15 signaling to potentiate NK cell-mediated cancer immunotherapy. Trends Immunol. 2022, 43, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Sahm, C.; Schönfeld, K.; Wels, W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Marcus, W.D.; Xu, W.; Lee, H.-i.; Han, K.; Egan, J.O.; Yovandich, J.L.; Rhode, P.R.; Wong, H.C. Novel human interleukin-15 agonists. J. Immunol. 2009, 183, 3598–3607. [Google Scholar] [CrossRef] [PubMed]
- Shiku, H.; Takahashi, T.; Oettgen, H.F. Cell surface antigens of human malignant melanoma. II. Serological typing with immune adherence assays and definition of two new surface antigens. J. Exp. Med. 1976, 144, 873–881. [Google Scholar] [CrossRef]
- Cinatl, J.; Cinatl, J.; Mainke, M.; Weissflog, A.; Rabenau, H.; Kornhuber, B.; Doerr, H.W. In vitro differentiation of human neuroblastoma cells induced by sodium phenylacetate. Cancer Lett. 1993, 70, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar] [PubMed]
- Gillies, S.D.; Lo, K.-M. Humanized Antibody (h14.18) of the Mouse 14.18 Antibody Binding to GD2 and its Fusion with IL-2. International Patent Application WO 2004/055056 A1, 1 July 2004. [Google Scholar]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Demaison, C.; Parsley, K.; Brouns, G.; Scherr, M.; Battmer, K.; Kinnon, C.; Grez, M.; Thrasher, A.J. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum. Gene Ther. 2002, 13, 803–813. [Google Scholar] [CrossRef]
- Lode, H.N.; Schmidt, M.; Seidel, D.; Huebener, N.; Brackrock, D.; Bleeke, M.; Reker, D.; Brandt, S.; Mueller, H.-P.; Helm, C.; et al. Vaccination with anti-idiotype antibody ganglidiomab mediates a GD2-specific anti-neuroblastoma immune response. Cancer Immunol. Immunother. 2013, 62, 999–1010. [Google Scholar] [CrossRef]
- Elpek, K.G.; Rubinstein, M.P.; Bellemare-Pelletier, A.; Goldrath, A.W.; Turley, S.J. Mature natural killer cells with phenotypic and functional alterations accumulate upon sustained stimulation with IL-15/IL-15Ralpha complexes. Proc. Natl. Acad. Sci. USA 2010, 107, 21647–21652. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, A.J.; McElmurry, R.; Chu, S.; Hinderlie, P.; Bendzick, L.; Geller, M.A.; Tolar, J.; Blazar, B.R.; Miller, J.S. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight 2018, 3, e96219. [Google Scholar] [CrossRef]
- Anderson, J.; Majzner, R.G.; Sondel, P.M. Immunotherapy of neuroblastoma: Facts and hopes. Clin. Cancer Res. 2022, 28, 3196–3206. [Google Scholar] [CrossRef]
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T cell therapy for neuroblastoma. Front. Immunol. 2018, 9, 2380. [Google Scholar] [CrossRef]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef]
- Szymczak-Workman, A.L.; Vignali, K.M.; Vignali, D.A. Design and construction of 2A peptide-linked multicistronic vectors. Cold Spring Harb. Protoc. 2012, 2012, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Rudek, L.S.; Zimmermann, K.; Galla, M.; Meyer, J.; Kuehle, J.; Stamopoulou, A.; Brand, D.; Sandalcioglu, I.E.; Neyazi, B.; Moritz, T.; et al. Generation of an NFκB-driven alpharetroviral “All-in-One” vector construct as a potent tool for CAR NK cell therapy. Front. Immunol. 2021, 12, 751138. [Google Scholar] [CrossRef]
- Imamura, M.; Shook, D.; Kamiya, T.; Shimasaki, N.; Chai, S.M.; Coustan-Smith, E.; Imai, C.; Campana, D. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood 2014, 124, 1081–1088. [Google Scholar] [CrossRef]
- Teng, K.Y.; Mansour, A.G.; Zhu, Z.; Li, Z.; Tian, L.; Ma, S.; Xu, B.; Lu, T.; Chen, H.; Hou, D.; et al. Off-the-shelf prostate stem cell antigen-directed chimeric antigen receptor natural killer cell therapy to treat pancreatic cancer. Gastroenterology 2022, 162, 1319–1333. [Google Scholar] [CrossRef] [PubMed]
- Granzin, M.; Wagner, J.; Köhl, U.; Cerwenka, A.; Huppert, V.; Ullrich, E. Shaping of natural killer cell antitumor activity by ex vivo cultivation. Front. Immunol. 2017, 8, 458. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A.; Dubois, S.; Miljkovic, M.D.; Conlon, K.C. IL-15 in the combination immunotherapy of cancer. Front. Immunol. 2020, 11, 868. [Google Scholar] [CrossRef]
- Waldmann, T.A. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: Implications for cancer therapy. Cancer Immunol. Res. 2015, 3, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Knudson, K.M.; Hodge, J.W.; Schlom, J.; Gameiro, S.R. Rationale for IL-15 superagonists in cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Berrien-Elliott, M.M.; Becker-Hapak, M.; Cashen, A.F.; Jacobs, M.; Wong, P.; Foster, M.; McClain, E.; Desai, S.; Pence, P.; Cooley, S.; et al. Systemic IL-15 promotes allogeneic cell rejection in patients treated with natural killer cell adoptive therapy. Blood 2022, 139, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bodden, M.; Häcker, A.; Röder, J.; Kiefer, A.; Zhang, C.; Bhatti, A.; Pfeifer Serrahima, J.; Ullrich, E.; Kühnel, I.; Wels, W.S. Co-Expression of an IL-15 Superagonist Facilitates Self-Enrichment of GD2-Targeted CAR-NK Cells and Mediates Potent Cell Killing in the Absence of IL-2. Cancers 2023, 15, 4310. https://doi.org/10.3390/cancers15174310
Bodden M, Häcker A, Röder J, Kiefer A, Zhang C, Bhatti A, Pfeifer Serrahima J, Ullrich E, Kühnel I, Wels WS. Co-Expression of an IL-15 Superagonist Facilitates Self-Enrichment of GD2-Targeted CAR-NK Cells and Mediates Potent Cell Killing in the Absence of IL-2. Cancers. 2023; 15(17):4310. https://doi.org/10.3390/cancers15174310
Chicago/Turabian StyleBodden, Malena, Aline Häcker, Jasmin Röder, Anne Kiefer, Congcong Zhang, Anita Bhatti, Jordi Pfeifer Serrahima, Evelyn Ullrich, Ines Kühnel, and Winfried S. Wels. 2023. "Co-Expression of an IL-15 Superagonist Facilitates Self-Enrichment of GD2-Targeted CAR-NK Cells and Mediates Potent Cell Killing in the Absence of IL-2" Cancers 15, no. 17: 4310. https://doi.org/10.3390/cancers15174310
APA StyleBodden, M., Häcker, A., Röder, J., Kiefer, A., Zhang, C., Bhatti, A., Pfeifer Serrahima, J., Ullrich, E., Kühnel, I., & Wels, W. S. (2023). Co-Expression of an IL-15 Superagonist Facilitates Self-Enrichment of GD2-Targeted CAR-NK Cells and Mediates Potent Cell Killing in the Absence of IL-2. Cancers, 15(17), 4310. https://doi.org/10.3390/cancers15174310