
Proteomic Profile Distinguishes New Subpopulations of Breast Cancer Patients with Different Survival Outcomes


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Identification of Patient Subpopulations
2.3. Clinical Characteristics of Patient Subpopulations
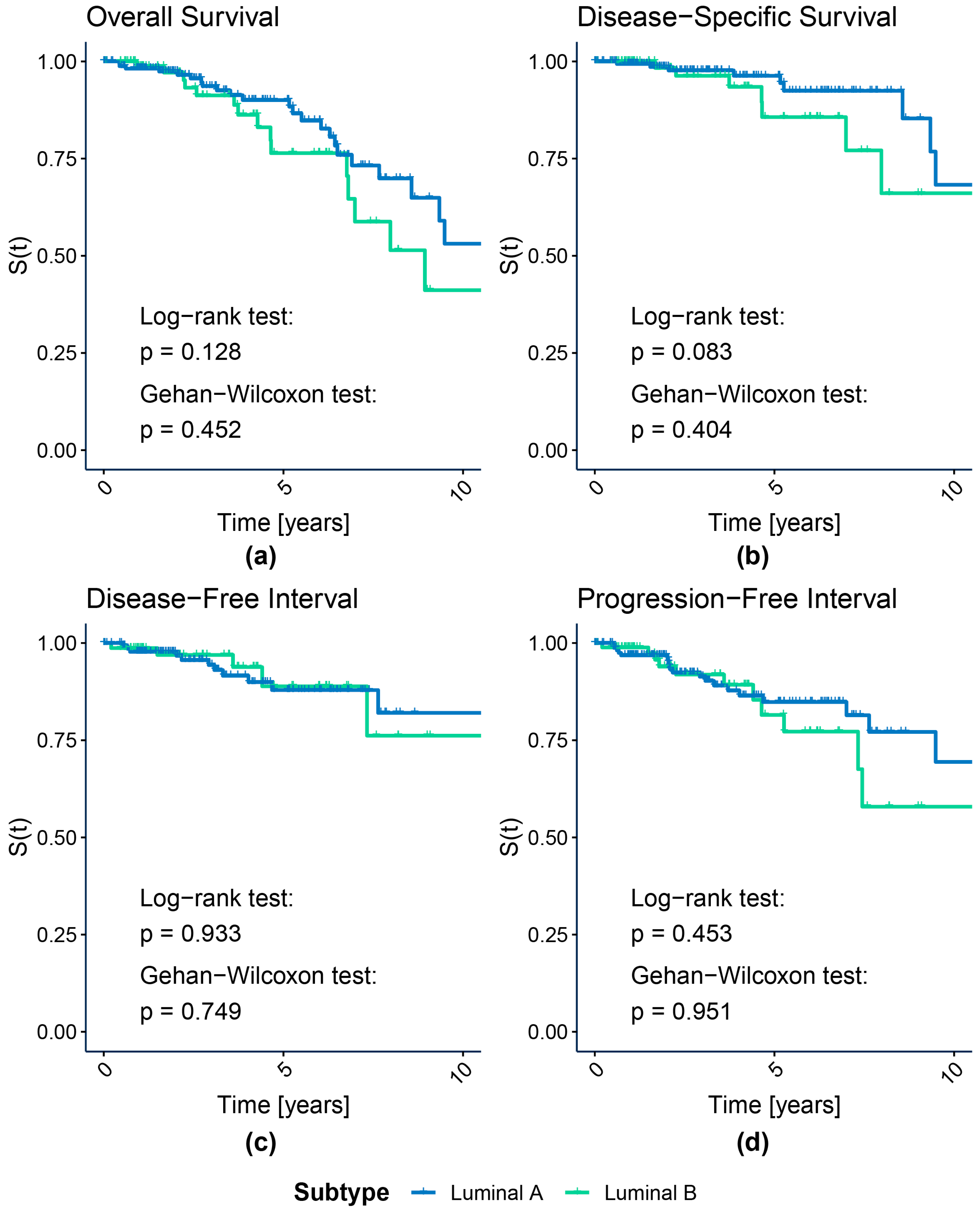
2.3.1. Survival Analysis
2.3.2. Demographic and Clinical Profiles
2.4. Molecular Characteristics of Luminal Patient Subpopulations
3. Results
3.1. Clinical Characteristics of Patient Subpopulations
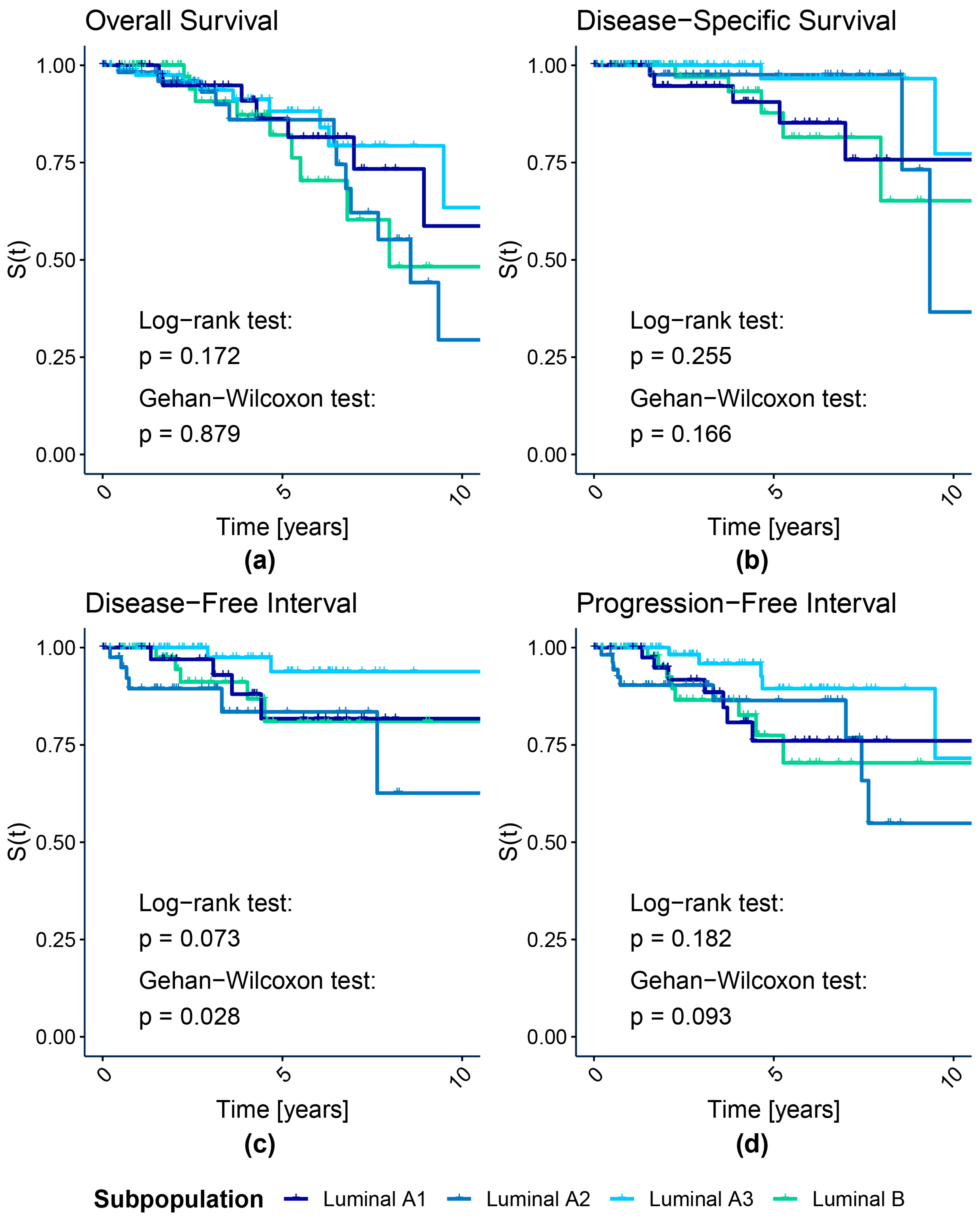
3.1.1. Survival Analysis
3.1.2. Subpopulation Demographic and Clinical Profile
3.2. Molecular Characteristics of Luminal Patient Subpopulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jassem, J.; Shan, A.; Buczek, D. Changing Paradigms in Breast Cancer Treatment. Eur. J. Transl. Clin. Med. 2020, 3, 53–63. [Google Scholar] [CrossRef]
- Szymiczek, A.; Lone, A.; Akbari, M.R. Molecular Intrinsic versus Clinical Subtyping in Breast Cancer: A Comprehensive Review. Clin. Genet. 2020, 99, 613–637. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene Expression Patterns of Breast Carcinomas Distinguish Tumor Subclasses with Clinical Implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated Observation of Breast Tumor Subtypes in Independent Gene Expression Data Sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Neo, S.-Y.; McShane, L.M.; Korn, E.L.; Long, P.M.; Jazaeri, A.; Martiat, P.; Fox, S.B.; Harris, A.L.; Liu, E.T. Breast Cancer Classification and Prognosis Based on Gene Expression Profiles from a Population-Based Study. Proc. Natl. Acad. Sci. USA 2003, 100, 10393–10398. [Google Scholar] [CrossRef]
- Hu, Z.; Fan, C.; Oh, D.S.; Marron, J.S.; He, X.; Qaqish, B.F.; Livasy, C.; Carey, L.A.; Reynolds, E.; Dressler, L.; et al. The Molecular Portraits of Breast Tumors Are Conserved across Microarray Platforms. BMC Genom. 2006, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- El-Rehim, D.M.A.; Ball, G.; Pinder, S.E.; Rakha, E.; Paish, C.; Robertson, J.F.R.; Macmillan, D.; Blamey, R.W.; Ellis, I.O. High-Throughput Protein Expression Analysis Using Tissue Microarray Technology of a Large Well-Characterised Series Identifies Biologically Distinct Classes of Breast Cancer Confirming Recent CDNA Expression Analyses. Int. J. Cancer 2005, 116, 340–350. [Google Scholar] [CrossRef]
- Jönsson, G.; Staaf, J.; Vallon-Christersson, J.; Ringnér, M.; Holm, K.; Hegardt, C.; Gunnarsson, H.; Fagerholm, R.; Strand, C.; Agnarsson, B.A.; et al. Genomic Subtypes of Breast Cancer Identified by Array-Comparative Genomic Hybridization Display Distinct Molecular and Clinical Characteristics. Breast Cancer Res. 2010, 12, R42. [Google Scholar] [CrossRef]
- Guedj, M.; Marisa, L.; de Reynies, A.; Orsetti, B.; Schiappa, R.; Bibeau, F.; MacGrogan, G.; Lerebours, F.; Finetti, P.; Longy, M.; et al. A Refined Molecular Taxonomy of Breast Cancer. Oncogene 2011, 31, 1196–1206. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kiryu, H. MODEC: An Unsupervised Clustering Method Integrating Omics Data for Identifying Cancer Subtypes. Brief. Bioinform. 2022, 23, bbac372. [Google Scholar] [CrossRef]
- Liu, Q.; Cheng, B.; Jin, Y.; Hu, P. Bayesian Tensor Factorization-Drive Breast Cancer Subtyping by Integrating Multi-Omics Data. J. Biomed. Inform. 2022, 125, 103958. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, L.; Zhao, X.; Yang, H.; Sa, J.; Cao, H.; Cui, Y. Cancer Subtyping with Heterogeneous Multi-Omics Data via Hierarchical Multi-Kernel Learning. Brief. Bioinform. 2022, 24, bbac488. [Google Scholar] [CrossRef]
- Sienkiewicz, K.; Chen, J.; Chatrath, A.; Lawson, J.T.; Sheffield, N.C.; Zhang, L.; Ratan, A. Detecting Molecular Subtypes from Multi-Omics Datasets Using SUMO. Cell Rep. Methods 2022, 2, 100152. [Google Scholar] [CrossRef]
- Genomic Data Commons. Genomic Data Commons Data Portal. Available online: https://portal.gdc.cancer.gov (accessed on 2 February 2021).
- Genomic Data Commons. Genomic Data Commons Legacy Archive. Available online: https://portal.gdc.cancer.gov/legacy-archive (accessed on 1 February 2021).
- Hu, J.; He, X.; Baggerly, K.A.; Coombes, K.R.; Hennessy, B.T.J.; Mills, G.B. Non-Parametric Quantification of Protein Lysate Arrays. Bioinformatics 2007, 23, 1986–1994. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Hennessy, B.T.; Meric-Bernstam, F.; Sahin, A.; Liu, W.; Ju, Z.; Carey, M.S.; Myhre, S.; Speers, C.; Deng, L.; et al. Functional Proteomics Can Define Prognosis and Predict Pathologic Complete Response in Patients with Breast Cancer. Clin. Proteom. 2011, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Hotelling, H. Analysis of a Complex of Statistical Variables into Principal Components. J. Educ. Psychol. 1933, 24, 498–520. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Fertig, E.J.; Jaffe, A.E.; Zhang, Y.; Storey, J.D.; Torres, L.C. Sva: Surrogate Variable Analysis. 2021. Available online: https://bioconductor.riken.jp/packages/3.14/bioc/html/sva.html (accessed on 24 March 2021).
- Schwarz, G. Estimating the Dimension of a Model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Marczyk, M.; Jaksik, R.; Polanski, A.; Polanska, J. GaMRed Adaptive Filtering of High-Throughput Biological Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 1, 149–157. [Google Scholar] [CrossRef]
- Tibshirani, R.; Walther, G.; Hastie, T. Estimating the Number of Clusters in a Data Set via the Gap Statistic. J. R. Stat. Soc. Ser. B Stat. Methodol. 2001, 63, 411–423. [Google Scholar] [CrossRef]
- Tobiasz, J.; Polanska, J. How to Compare Various Clustering Outcomes? Metrices to Investigate Breast Cancer Patient Subpopulations Based on Proteomic Profiles. In Bioinformatics and Biomedical Engineering; Springer International Publishing: Berlin/Heidelberg, Germany, 2022; pp. 309–318. [Google Scholar]
- Tobiasz, J.; Hatzis, C.; Polanska, J. Breast Cancer Heterogeneity Investigation: Multiple k-Means Clustering Approach. In Proceedings of the 2019 IEEE 19th International Conference on Bioinformatics and Bioengineering (BIBE), Athens, Greece, 28–30 October 2019. [Google Scholar]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. In Springer Series in Statistics; Springer: New York, NY, USA, 1992; pp. 319–337. [Google Scholar]
- Mantel, N. Evaluation of Survival Data and Two New Rank Order Statistics Arising in Its Consideration. Cancer Chemother. Rep. 1966, 50, 163–170. [Google Scholar]
- Peto, R.; Peto, J. Asymptotically Efficient Rank Invariant Test Procedures. J. R. Stat. Soc. Ser. A 1972, 135, 185. [Google Scholar] [CrossRef]
- Gehan, E.A. A Generalized Wilcoxon Test for Comparing Arbitrarily Singly-Censored Samples. Biometrika 1965, 52, 203. [Google Scholar] [CrossRef]
- Breslow, N. A Generalized Kruskal-Wallis Test for Comparing K Samples Subject to Unequal Patterns of Censorship. Biometrika 1970, 57, 579–594. [Google Scholar] [CrossRef]
- May, S.; Hosmer, D.W., Jr.; Lemeshow, S. Applied Survival Analysis; John Wiley & Sons: Hoboken, NJ, USA, 2014; ISBN 0471754994. [Google Scholar]
- Cox, D.R. Regression Models and Life-Tables. J. R. Stat. Soc. Ser. B (Methodol.) 1972, 34, 187–202. [Google Scholar] [CrossRef]
- Olivier, J.; May, W.L.; Bell, M.L. Relative Effect Sizes for Measures of Risk. Commun. Stat. Theory Methods 2017, 46, 6774–6781. [Google Scholar] [CrossRef]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences; Routledge: Abingdon-on-Thames, UK, 2013. [Google Scholar]
- Yates, F. Contingency Tables Involving Small Numbers and the 2 Test. Suppl. J. R. Stat. Soc. 1934, 1, 217. [Google Scholar] [CrossRef]
- Cramér, H. Mathematical Methods of Statistics (PMS-9), Volume 9; Princeton University Press: Princeton, NJ, USA, 1999; ISBN 0691005478. [Google Scholar]
- Shapiro, S.S.; Wilk, M.B. An Analysis of Variance Test for Normality (Complete Samples). Biometrika 1965, 52, 591. [Google Scholar] [CrossRef]
- Bartlett, M.S. Properties of Sufficiency and Statistical Tests. Proc. R. Soc. Lond. A Math. Phys. Sci. 1937, 160, 268–282. [Google Scholar] [CrossRef]
- Tomczak, M.; Tomczak, E. The Need to Report Effect Size Estimates Revisited. An Overview of Some Recommended Measures of Effect Size. Trends Sport. Sci. 2014, 1, 19–25. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome Pathway Analysis: A High-Performance in-Memory Approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2015, 44, D481–D487. [Google Scholar] [CrossRef]
- Mrukwa, G.; Polanska, J. DiviK: Divisive Intelligent K-Means for Hands-Free Unsupervised Clustering in Big Biological Data. BMC Bioinform. 2022, 23, 538. [Google Scholar] [CrossRef]
- Perou, C.M.; Borresen-Dale, A.-L. Systems Biology and Genomics of Breast Cancer. Cold Spring Harb. Perspect. Biol. 2010, 3, a003293. [Google Scholar] [CrossRef]
- Martin, E.M.; Orlando, K.A.; Yokobori, K.; Wade, P.A. The Estrogen Receptor/GATA3/FOXA1 Transcriptional Network: Lessons Learned from Breast Cancer. Curr. Opin. Struct. Biol. 2021, 71, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.-L.; Pan, Y.-H.; Huang, Q.-Y.; Shi, Y.-B.; Huang, Q.-Y.; Hu, Z.-Z.; Xiong, L.-X. Caveolin-1: A Multifaceted Driver of Breast Cancer Progression and Its Application in Clinical Treatment. Onco Targets Ther. 2019, 12, 1539–1552. [Google Scholar] [CrossRef]
- Ren, L.; Zhou, P.; Wu, H.; Liang, Y.; Xu, R.; Lu, H.; Chen, Q. Caveolin-1 Is a Prognostic Marker and Suppresses the Proliferation of Breast Cancer. Transl. Cancer Res. 2021, 10, 3797–3810. [Google Scholar] [CrossRef]
- Savage, K.; Lambros, M.B.K.; Robertson, D.; Jones, R.L.; Jones, C.; Mackay, A.; James, M.; Hornick, J.L.; Pereira, E.M.; Milanezi, F.; et al. Caveolin 1 Is Overexpressed and Amplified in a Subset of Basal-like and Metaplastic Breast Carcinomas: A Morphologic, Ultrastructural, Immunohistochemical, and IIn Situ/i Hybridization Analysis. Clin. Cancer Res. 2007, 13, 90–101. [Google Scholar] [CrossRef]
- Robinson, T.J.W.; Liu, J.C.; Vizeacoumar, F.; Sun, T.; Maclean, N.; Egan, S.E.; Schimmer, A.D.; Datti, A.; Zacksenhaus, E. RB1 Status in Triple Negative Breast Cancer Cells Dictates Response to Radiation Treatment and Selective Therapeutic Drugs. PLoS ONE 2013, 8, e78641. [Google Scholar] [CrossRef] [PubMed]
- Herschkowitz, J.I.; He, X.; Fan, C.; Perou, C.M. The Functional Loss of the Retinoblastoma Tumour Suppressor Is a Common Event in Basal-like and Luminal B Breast Carcinomas. Breast Cancer Res. 2008, 10, R75. [Google Scholar] [CrossRef] [PubMed]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.-I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/MTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Risso, G.; Blaustein, M.; Pozzi, B.; Mammi, P.; Srebrow, A. Akt/PKB: One Kinase, Many Modifications. Biochem. J. 2015, 468, 203–214. [Google Scholar] [CrossRef]
- Zhang, L.-Y.; Zhang, Y.-Q.; Zeng, Y.-Z.; Zhu, J.-L.; Chen, H.; Wei, X.-L.; Liu, L.-J. TRPC1 Inhibits the Proliferation and Migration of Estrogen Receptor-Positive Breast Cancer and Gives a Better Prognosis by Inhibiting the PI3K/AKT Pathway. Breast Cancer Res. Treat. 2020, 182, 21–33. [Google Scholar] [CrossRef]
- Zheng, F.; Du, F.; Zhao, J.; Wang, X.; Si, Y.; Jin, P.; Qian, H.; Xu, B.; Yuan, P. The Emerging Role of RNA N6-Methyladenosine Methylation in Breast Cancer. Biomark. Res. 2021, 9, 39. [Google Scholar] [CrossRef]
- Stevens, T.A.; Meech, R. BARX2 and Estrogen Receptor- (ESR1) Coordinately Regulate the Production of Alternatively Spliced ESR1 Isoforms and Control Breast Cancer Cell Growth and Invasion. Oncogene 2006, 25, 5426–5435. [Google Scholar] [CrossRef] [PubMed]
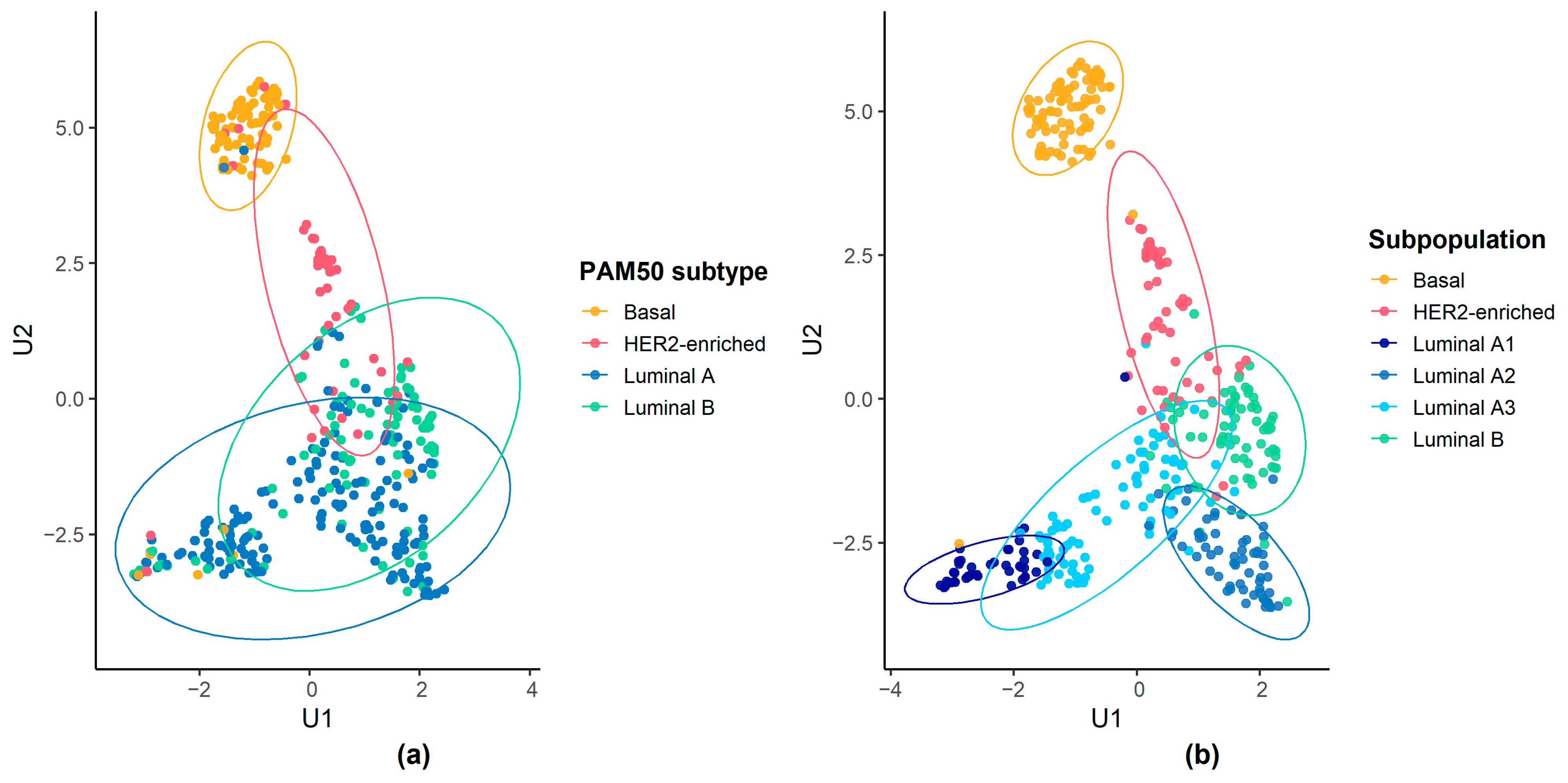


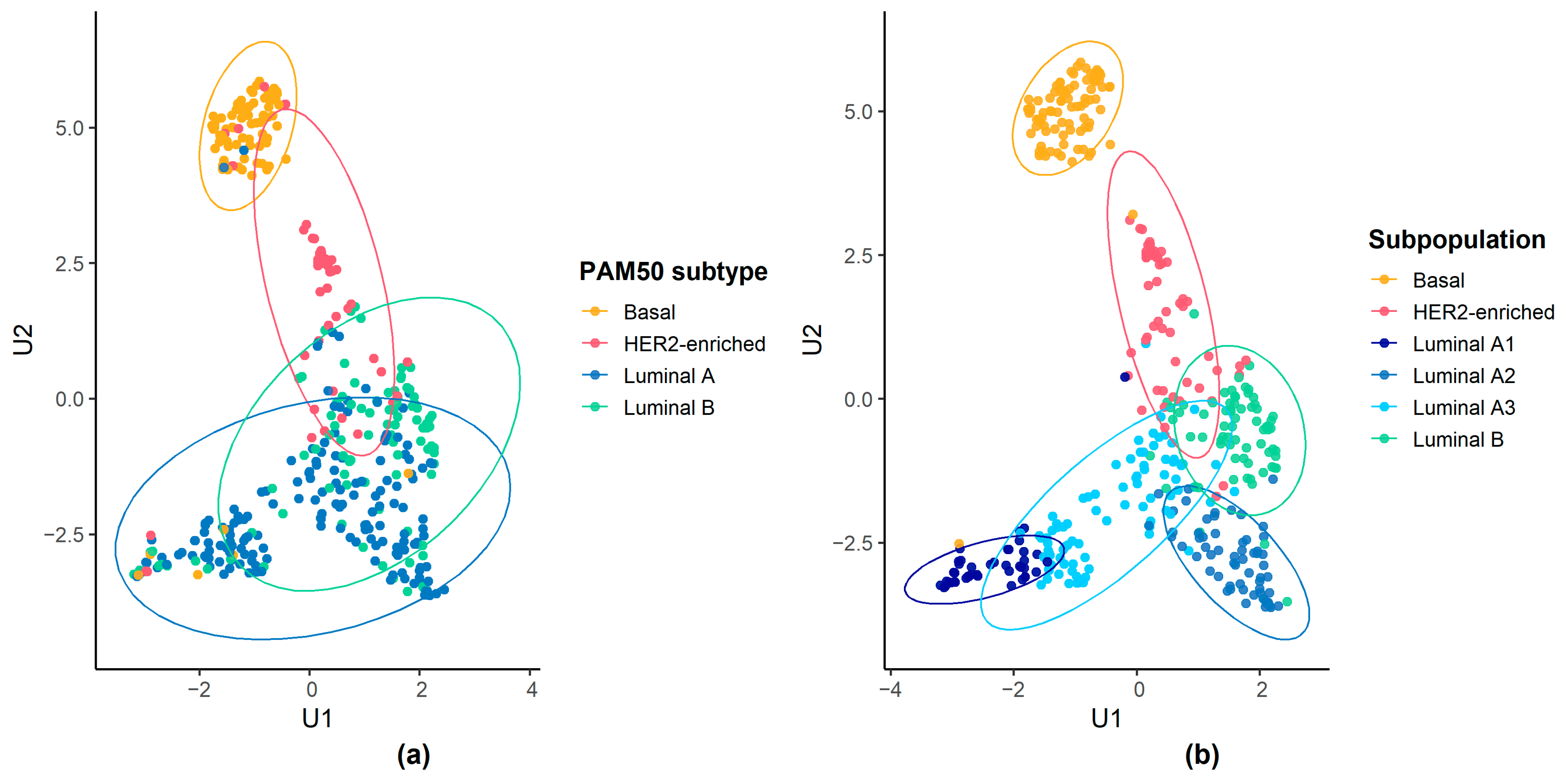{kind=link}
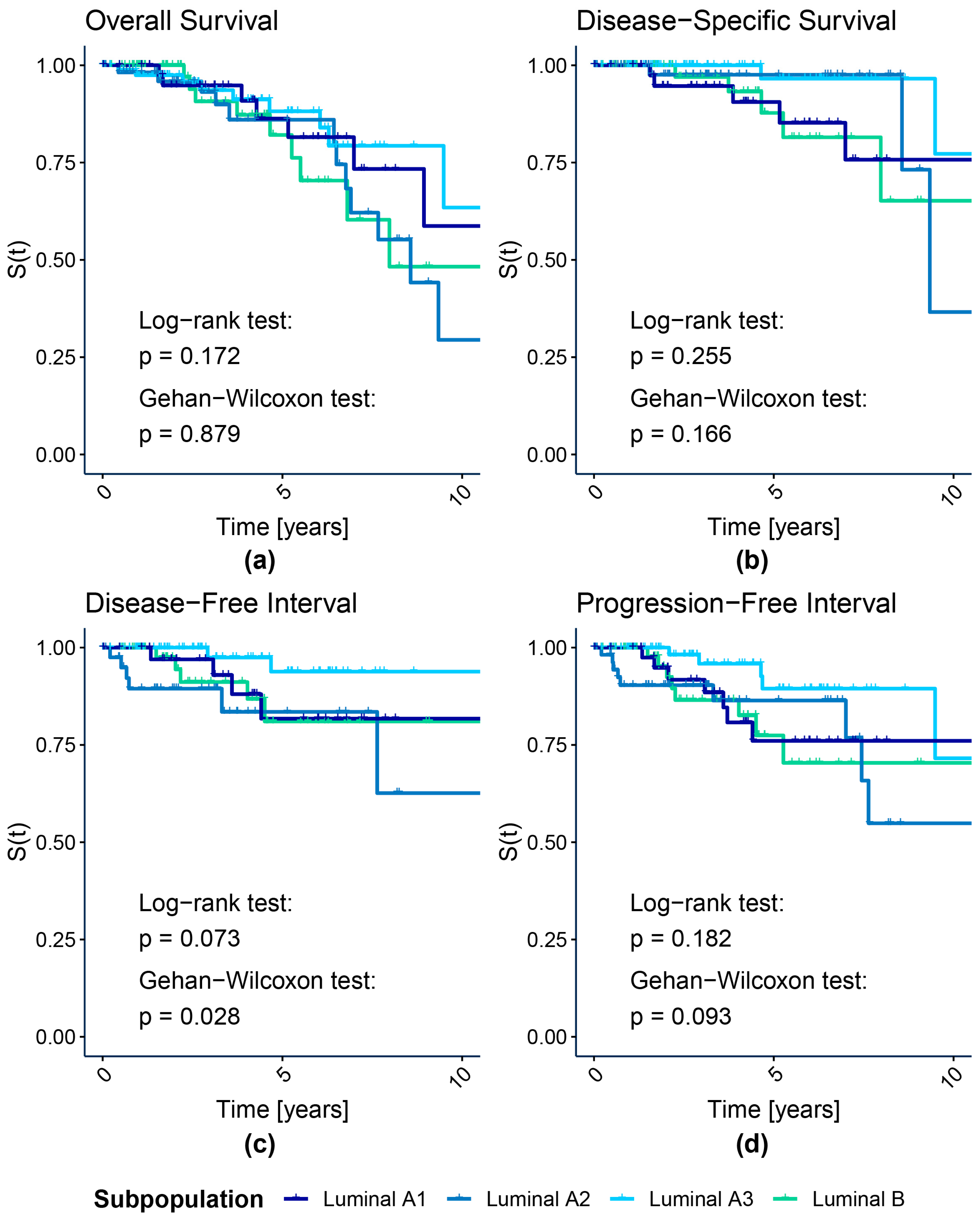{kind=link}
{kind=link}
| PAM50 Subtype | Proteomics-Based Subpopulation | TOTAL | |||||
|---|---|---|---|---|---|---|---|
| Basal | HER2-Enriched | Luminal | |||||
| A1 | A2 | A3 | B | ||||
| Basal | 79 (88.76%) | 0 (0.00%) | 4 (9.09%) | 0 (0.00%) | 2 (2.30%) | 1 (1.39%) | 86 (21.13%) |
| HER2-enriched | 8 (8.99%) | 34 (62.96%) | 2 (4.55%) | 0 (0.00%) | 2 (2.30%) | 4 (5.56%) | 50 (12.29%) |
| Luminal A | 2 (2.25%) | 9 (16.67%) | 27 (61.36%) | 47 (77.05%) | 65 (74.71%) | 23 (31.94%) | 173 (42.51%) |
| Luminal B | 0 (0.00%) | 11 (20.37%) | 11 (25.00%) | 14 (22.95%) | 18 (20.69%) | 44 (61.11%) | 98 (24.08%) |
| TOTAL | 89 (100.00%) | 54 (100.00%) | 44 (100.00%) | 61 (100.00%) | 87 (100.00%) | 72 (100.00%) | 407 (100.00%) |
| Subpopulation | N | Ne | Nc | HR | HR Effect | HR Allocation Adjusted Critical Value | ||
|---|---|---|---|---|---|---|---|---|
| α = 0.1 Small Effect | α = 0.3 Medium Effect | α = 0.5 Large Effect | ||||||
| Overall Survival | ||||||||
| Luminal A1 | 44 | 8 | 36 | 0.657 | Small | HR < 0.773; HR > 1.293 | HR < 0.47; HR > 2.13 | HR < 0.275; HR > 3.636 |
| Luminal A2 | 61 | 14 | 47 | 1.275 | Small | HR < 0.805; HR > 1.242 | HR < 0.517; HR > 1.934 | HR < 0.314; HR > 3.18 |
| Luminal A3 | 87 | 9 | 78 | 0.533 | Medium | HR < 0.831; HR > 1.203 | HR < 0.561; HR > 1.783 | HR < 0.354; HR > 2.828 |
| Luminal B | 72 | 9 | 63 | Reference | ||||
| Disease-Specific Survival | ||||||||
| Luminal A1 | 43 | 5 | 38 | 0.759 | Small | HR < 0.771; HR > 1.297 | HR < 0.466; HR > 2.146 | HR < 0.272; HR > 3.674 |
| Luminal A2 | 58 | 4 | 54 | 0.776 | Small | HR < 0.801; HR > 1.249 | HR < 0.51; HR > 1.961 | HR < 0.309; HR > 3.241 |
| Luminal A3 | 85 | 2 | 83 | 0.213 | Large | HR < 0.83; HR > 1.205 | HR < 0.558; HR > 1.792 | HR < 0.351; HR > 2.847 |
| Luminal B | 72 | 5 | 67 | Reference | ||||
| Disease-Free Interval | ||||||||
| Luminal A1 | 38 | 4 | 34 | 0.927 | No effect | HR < 0.77; HR > 1.298 | HR < 0.465; HR > 2.15 | HR < 0.271; HR > 3.684 |
| Luminal A2 | 46 | 6 | 40 | 1.748 | Small | HR < 0.79; HR > 1.266 | HR < 0.494; HR > 2.025 | HR < 0.295; HR > 3.391 |
| Luminal A3 | 79 | 2 | 77 | 0.250 | Large | HR < 0.833; HR > 1.201 | HR < 0.563; HR > 1.776 | HR < 0.356; HR > 2.81 |
| Luminal B | 64 | 5 | 59 | Reference | ||||
| Progression-Free Interval | ||||||||
| Luminal A1 | 44 | 7 | 37 | 0.839 | No effect | HR < 0.773; HR > 1.293 | HR < 0.47; HR > 2.13 | HR < 0.275; HR > 3.636 |
| Luminal A2 | 61 | 9 | 52 | 1.101 | No effect | HR < 0.805; HR > 1.242 | HR < 0.517; HR > 1.934 | HR < 0.314; HR > 3.18 |
| Luminal A3 | 87 | 5 | 82 | 0.359 | Medium | HR < 0.831; HR > 1.203 | HR < 0.561; HR > 1.783 | HR < 0.354; HR > 2.828 |
| Luminal B | 72 | 8 | 64 | Reference | ||||
| PAM50 Subtype | N | Ne | Nc | HR | HR Effect | HR Allocation Adjusted Critical Value | ||
|---|---|---|---|---|---|---|---|---|
| α = 0.1 Small Effect | α = 0.3 Medium Effect | α = 0.5 Large Effect | ||||||
| Overall Survival | ||||||||
| Luminal A | 173 | 26 | 147 | 0.612 | Small | HR < 0.852; HR > 1.174 | HR < 0.598; HR > 1.671 | HR < 0.39; HR > 2.566 |
| Luminal B | 98 | 16 | 82 | Reference | ||||
| Disease-Specific Survival | ||||||||
| Luminal A | 169 | 10 | 159 | 0.437 | Medium | HR < 0.852; HR > 1.174 | HR < 0.598; HR > 1.672 | HR < 0.389; HR > 2.568 |
| Luminal B | 96 | 8 | 88 | Reference | ||||
| Disease-Free Interval | ||||||||
| Luminal A | 147 | 11 | 136 | 1.046 | No effect | HR < 0.852; HR > 1.173 | HR < 0.6; HR > 1.668 | HR < 0.391; HR > 2.558 |
| Luminal B | 82 | 5 | 77 | Reference | ||||
| Progression-Free Interval | ||||||||
| Luminal A | 173 | 20 | 153 | 0.752 | Small | HR < 0.852; HR > 1.174 | HR < 0.598; HR > 1.671 | HR < 0.39; HR > 2.566 |
| Luminal B | 98 | 11 | 87 | Reference | ||||
| Feature | χ2 | p-Value | Cramér’s V | Cramér’s V Effect Threshold | ||
|---|---|---|---|---|---|---|
| Small | Medium | Large | ||||
| Proteomics-based subpopulations | ||||||
| Race | 13.42 | 0.0368 | 0.1712 | 0.0707 | 0.2121 | 0.3536 |
| Ethnicity | 0.23 | 0.9718 | 0.0346 | 0.1 | 0.3 | 0.5 |
| AJCC Stage | 18.61 | 0.0287 | 0.1536 | 0.0577 | 0.1732 | 0.2887 |
| AJCC Tumor | 19.34 | 0.0225 | 0.1566 | |||
| AJCC Node | 13.23 | 0.1526 | 0.1292 | |||
| AJCC Tumor Binarized | 13.86 | 0.0031 | 0.2295 | 0.1 | 0.3 | 0.5 |
| AJCC Node Binarized | 3.75 | 0.2900 | 0.1191 | |||
| AJCC Metastasis | 2.23 | 0.5254 | 0.0922 | |||
| Radiotherapy | 4.42 | 0.2193 | 0.1294 | |||
| Chemotherapy | 12.37 | 0.0062 | 0.2165 | |||
| Hormone Therapy | 2.11 | 0.5500 | 0.0894 | |||
| PAM50-based subtypes | ||||||
| Race | 3.74 | 0.1543 | 0.1269 | 0.1 | 0.3 | 0.5 |
| Ethnicity | 1.26 | 0.2610 | 0.0793 | |||
| AJCC Stage | 9.19 | 0.0269 | 0.1848 | |||
| AJCC Tumor | 14.40 | 0.0024 | 0.2309 | |||
| AJCC Node | 0.91 | 0.8228 | 0.0580 | |||
| AJCC Tumor Binarized | 13.25 | 0.0003 | 0.2215 | |||
| AJCC Node Binarized | 0.67 | 0.4133 | 0.0497 | |||
| AJCC Metastasis | 1.42 | 0.2335 | 0.0725 | |||
| Radiotherapy | 0.05 | 0.8295 | 0.0131 | |||
| Chemotherapy | 1.45 | 0.2280 | 0.0732 | |||
| Hormone Therapy | 0.09 | 0.7613 | 0.0185 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tobiasz, J.; Polanska, J. Proteomic Profile Distinguishes New Subpopulations of Breast Cancer Patients with Different Survival Outcomes. Cancers 2023, 15, 4230. https://doi.org/10.3390/cancers15174230
Tobiasz J, Polanska J. Proteomic Profile Distinguishes New Subpopulations of Breast Cancer Patients with Different Survival Outcomes. Cancers. 2023; 15(17):4230. https://doi.org/10.3390/cancers15174230
Chicago/Turabian StyleTobiasz, Joanna, and Joanna Polanska. 2023. "Proteomic Profile Distinguishes New Subpopulations of Breast Cancer Patients with Different Survival Outcomes" Cancers 15, no. 17: 4230. https://doi.org/10.3390/cancers15174230
APA StyleTobiasz, J., & Polanska, J. (2023). Proteomic Profile Distinguishes New Subpopulations of Breast Cancer Patients with Different Survival Outcomes. Cancers, 15(17), 4230. https://doi.org/10.3390/cancers15174230