
Oncolytic Viruses and Immune Checkpoint Inhibitors: The “Hot” New Power Couple


Abstract
:Simple Summary
Abstract
1. Introduction
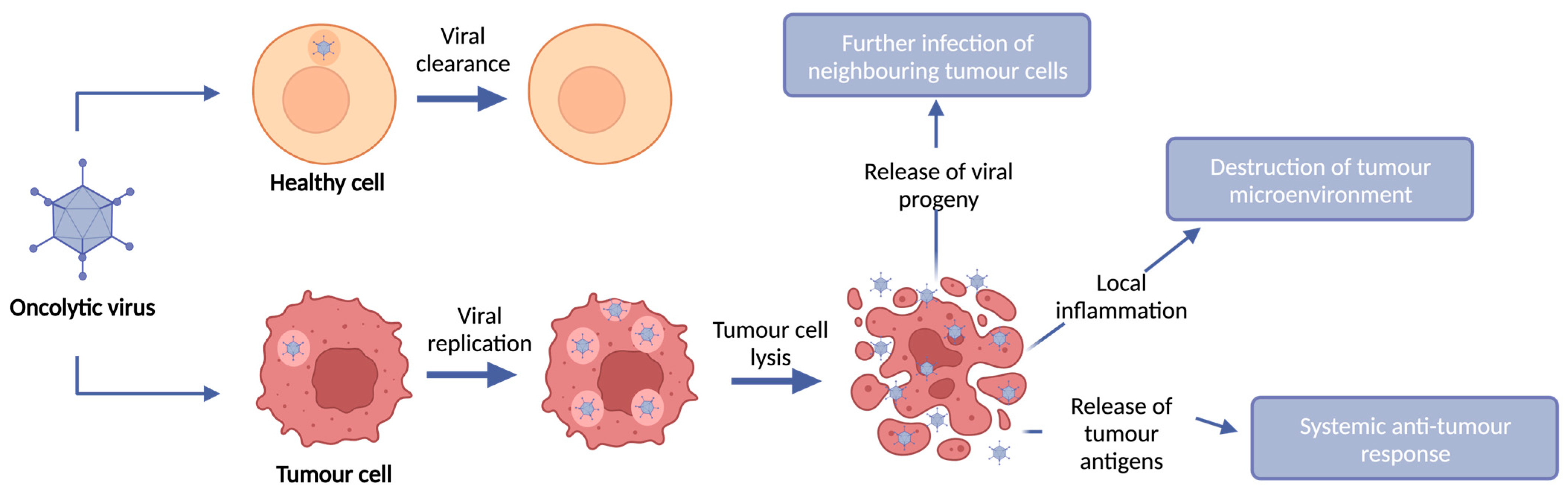
2. Oncolytic Viruses
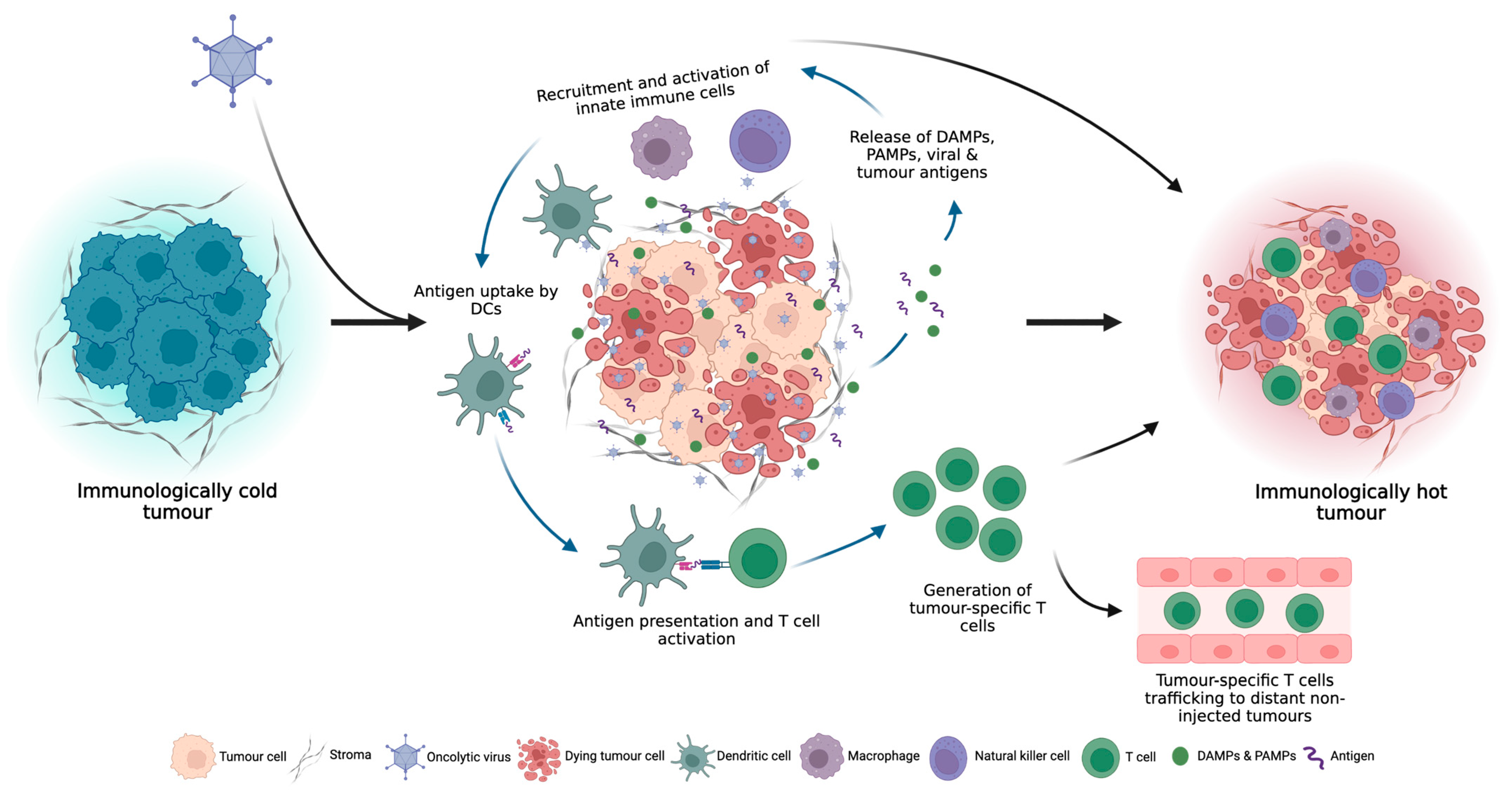
Turning Cold Tumours Hot: The OV Immune Response
3. Oncolytic Virus Monotherapy
4. Combined OV and ICI Therapy
4.1. Neoadjuvant Therapies
4.1.1. Markers of Response

{kind=link}
{kind=link}
| Virus | OV | ICI | Indication | Key Findings | Ref. |
|---|---|---|---|---|---|
| Ad | CG0070 (IVS) | PD-1: Pembrolizumab | NMIBC Phase II | Disease control: 82% 6-month CR; 81% 9-month CR; 68% 12-month CR | |
| DNX-2401 (IT) | GBM Phase II | Survival: 52.7% 12-month survival; 12.5 months median OS; 3 patients alive > 45 months Disease control: ORR 10.4%; 42.9% SD; 4.8% CR; 7.1% PR; | [59] | ||
| EnAd (IT) | PD-1: Nivolumab | mCRC Phase I | Survival: median OS 15.4 months (5 months placebo) Disease control: median PFS 2.8 months Immune response: 85% demonstrated increased CD8+ TILs; 77% increased CD4+ TILs; 62% increased PD-L1+ TILs | [64,65,66] | |
| ONCOS-102 (IT) | PD-1: Pembrolizumab | Melanoma progressing post-PD-1 blockade Pilot | Disease control: 35% ORR; 64% SD; 27% demonstrated CR in injected tumour 53% demonstrated reduction in ≥1 non-injected tumour Immune response: increased CD4+ & CD8+ TILs | [62] | |
| HSV | T-VEC (IT) | PD-1: Pembrolizumab | Melanoma Phase Ib | Disease control: 82% demonstrated >50% reduction of injected tumours; 43% in non-injected tumours Immune response: 67% demonstrated increased CD8+ TILs; demonstrated increased systemic proliferating CD8+ T cells | [67] |
| Melanoma Phase III T-VEC + Pemb vs Pemb | Disease control: T + P: 17.9% CR; 48.6% ORR (CR/PR); 14.3 months PFS P: 11.6% CR; 41.3% ORR; 8.5 months PFS | [60] | |||
| Sarcoma Phase II | Disease control: 21% PR; 47% SD; median PFS 17.1 months Immune response responders saw increased CD8+ TILs and CD8+ aggregates at tumour edge; non-responders saw no increase in CD8+ TILs or aggregates | [63] | |||
| CTLA-4: Ipilimumab | Melanoma Phase II TVEC + Ipi vs Ipi | Disease control: T + I: 13% CR; 26% PR; 39% ORR (CR/PR); 8.2 months median PFS; 52% non-injected visceral tumour reduction I: 7% CR; 11% PR; 18% ORR; 6.4 months median PFS; 23% non-injected visceral tumour reduction | [61] | ||
| HF10 (IT) | CTLA-4: Ipilimumab | Melanoma Phase II | Survival: Median OS 26 months Disease control: median PFS 19 months; 68% SD Immune response: increased CD8+ and decreased CD4+ TILs | [34] | |
| VV | JX-594 (IT) | CTLA-4: Tremelumab PD-L1: Durvalumab | ICI refractory CRC Phase I/II | Survival: J + D: Median OS 7.5 months J + D + T: Median OS 5.2 months Disease control: J + D: median PFS 2.3 months; 12.5% DCR J + D + T: median PFS 2.1 months; 16.7% DCR Immune response: Increased proliferating CD3+ TILs after OV treatment and again after ICI treatment; increased M1 macrophages in tumours | [68] |
4.1.2. Adverse Events
4.2. OVs Encoding ICIs
| OV | Target | ICI Format | Indication | Key Findings | Ref. |
|---|---|---|---|---|---|
| Ad5 | CTLA-4 mouse | IgG2 | Melanoma NSCLC SCLC | Subcutaneous mouse xenograft model with intravenous OV injection: Disease control: significant 72% reduction in tumour growth compared to untreated tumours Subcutaneous mouse xenograft model with intra-tumoural OV injection: Disease control: significant 3-fold decrease in tumour growth compared to untreated tumours | [71] |
| Ad5/3 | CTLA-4 human | IgG2 | NSCLC Prostate | Subcutaneous T-cell-deficient mouse xenograft model with intra-tumoural OV injection: Disease control: significantly decreased tumour growth compared to untreated Immune response: 43-fold increase in tumour anti-CTLA-4 antibody concentrations compared to systemic plasma In vitro human T cell activation assay: PBMCs from advanced solid cancer patients cultured in the presence of supernatant from OV-infected cells saw increase in T cell IL-2 and IFN-γ production | [72] |
| HSV-1 | CTLA-4 & GM-CSF mouse | scFv fused to mouse IgG1 | Lymphoma | Bilateral subcutaneous mouse xenograft model with single-sided intra-tumoural OV injection: Disease control: decreased tumour growth in both injected and non-injected tumours (not significant) | [73] |
| IAV | CTLA-4 mouse | scFV | Melanoma | Bilateral subcutaneous mouse xenograft model with single-sided intra-tumoural OV injection: Disease control: significantly decreased tumour growth in both injected and non-injected tumours and prolonged survival compared to parental virus | [74] |
| IAV | CTLA-4 mouse | scFV | HCC | Spontaneous homograft model with intra-tumoural OV injection: Survival: prolonged survival compared to parental OV Disease control: significantly decreased tumour compared to parental OV | [85] |
| NDV | CTLA-4 mouse | scFV | Melanoma | Intradermal mouse tumour model with intra-tumoural OV injection: Survival: prolonged survival compared to systemic CTLA-4 treatment plus parental NDV Disease control: comparable tumour growth inhibition | [93] |
| MV | CTLA-4 mouse | scFV-IgG1 Fc fusion | Melanoma | Subcutaneous synergic mouse tumour model with intra-tumoural OV injection: Disease control: significantly decreased tumour growth compared to parental virus and untreated Immune response: significant increase in tumour T cell infiltration and a decrease in Treg infiltration compared to parental OV and untreated; increased splenocyte IFN-γ release upon re-stimulation with tumour cells in vitro compared to parental OV and untreated | [91] |
| Ad68 | PD-1 | IgG4 | Colorectal | Bilateral subcutaneous humanised PD-1 transgenic mouse tumour model with single-sided intra-tumoural OV injection: Survival: prolonged survival compared to parental OV and untreated Disease control: significantly decreased tumour growth compared to parental OV and untreated, with successful tumour rejection upon rechallenge Immune response: significantly increased systemic CD8+ T cell and effector and central memory T cell proportions; significantly decreased PD-1+ CD4+ and CD8+ T cell proportions | [75] |
| HSV-1 | PD-1 mouse | scFv | HCC | Bilateral subcutaneous synergic mouse tumour model with single-sided intra-tumoural OV injection: Disease control: significantly decreased tumour growth in both injected and non-injected tumours and greater long-term tumour growth inhibition compared to parental OV and untreated; successful tumour rejection upon rechallenge Immune response: significantly increased activated CD4+ and CD8+ cell tumour infiltration compared to parental OV; however, also saw significantly greater MDSC infiltration compared to parental OV | [76] |
| HSV-1 | PD-1 human | scFv | HCC | Orthotopic HCC xenograft tumour model with intravenous OV injection in humanised PD-1 transgenic mice: Survival: and increased overall survival compared to parental OV and untreated mice Disease control: significantly decreased tumour growth compared to parental OV and untreated mice, with all anti-PD-1 OV treated mice tumour free at 12 weeks Bilateral subcutaneous mouse xenograft tumour model with single-sided intra-tumoural OV injection in humanised PD-1 transgenic mice: Disease control: significantly decreased tumour growth in both injected and non-injected tumours compared to parental OV and untreated Immune response: anti-PD-1 OV treated tumours demonstrated significantly reduced proportions of exhausted CD8+ T cell populations and increased effector memory CD8+ T cell populations compared to parental OV and untreated | [77] |
| HSV-1 | PD-1 human | scFv | Melanoma | Bilateral subcutaneous mouse xenograft tumour model with intra-tumoural OV injection in humanised PD-1 transgenic mice: Disease control: significantly decreased tumour growth compared to untreated and parental OV Immune response: significantly increased tumour CD4+ and CD8+ T cell infiltration compared to untreated; RNA-seq analysis demonstrated significant enrichment in anti-viral, IFN and antigen presentation and processing pathways compared to untreated | [78] |
| HSV-1 | PD-1 human | scFV | GBM | Orthoptic GBM synergic mouse tumour model with intra-tumoural OV injection: Survival: increased median survival time compared to untreated (significant) and parental OV (not significant) Disease control: successful tumour rejection following rechallenge | [89] |
| HSV-2 | PD-1 human | IgG | Melanoma | Subcutaneous mouse xenograft tumour model with intra-tumoural OV injection in humanised PD-1 transgenic mice: Survival: prolonged survival compared to untreated; improved tumour-free survival compared to parental OV and untreated Disease control: significantly decreased tumour growth compared to untreated; successful tumour rejection following rechallenge Immune response: increased systemic percentages of CD4+, CD8+ and CD3+ T cells and significant increase in T cell activation markers compared to parental OV and untreated; significant reduction in Tregs and MDSCs compared to untreated | [79] |
| VV | PD-1 mouse | IgG & scFV | Fibrosarcoma Melanoma | Subcutaneous synergic mouse tumour model with intra-tumoural OV injection: Survival: prolonged survival (IgG significant; scFV not significant) compared to parental OV and untreated Disease control: significantly decreased tumour growth compared to parental OV and untreated Immune response: IgG-OV significantly increased tumour infiltration of CD4+ and CD8+ T cells, the proportion of activated CD8+ T cells, and the CD8+/Foxp3+ T cell ratio compared to systemic anti-PD-L1 treatment, but to a lesser extent than parental OV alone | [86] |
| NDV | PD-1 and PD-L1 mouse & IL-2 | scFV | Melanoma | Unilateral subcutaneous synergic mouse tumour model with intra-tumoural OV injection: Survival: prolonged survival compared to parental OV Disease control: significantly decreased tumour growth compared to parental OV Bilateral subcutaneous synergic mouse tumour model with single-sided intra-tumoural OV injection: Survival: when combined with systemic anti-CTLA-4 treatment, PD-1 and PD-L1 OV demonstrated significantly prolonged survival compared to parental OV Disease control: when combined with systemic anti-CTLA-4 treatment, PD-1 and PD-L1 OV demonstrated significantly inhibited tumour growth in non-injected tumours compared to parental OV | [87] |
| MV | PD-1 & PD-L1 mouse | scFV-IgG1 Fc fusion | Melanoma | Subcutaneous synergic mouse tumour model with intra-tumoural OV injection: Survival: significantly prolonged survival compared to parental OV and untreated Disease control: significantly decreased tumour growth compared to parental OV and untreated; successful tumour rejection following rechallenge Immune response: significantly increased activated CD8+ T cell and reduced Foxp3+ Treg tumour infiltration; higher effector memory T cell: central memory T cell ratio for PD-1 (significant) and PD-L1 (not significant) OVs compared to untreated | [84,91] |
| Ad5/24 | PD-L1 mouse | scFV | Colorectal | Bilateral subcutaneous synergic mouse tumour model with intra-tumoural OV injection: Survival: significantly prolonged survival compared to parental OV and untreated Disease control: significantly decreased tumour growth compared to parental OV and untreated Immune response: significantly increased tumour CD8+ T cell infiltration compared to parental OV | [88] |
| Chimeric poxvirus | PD-L1 human | scFv | Breast cancer Gastric cancer PDAC | Orthotopic synergic mouse breast cancer model with intra-tumoural or intravenous OV injection: Survival: significantly prolonged survival compared to untreated Disease control: significantly decreased tumour growth compared to untreated Orthotopic mouse breast cancer xenograft model with intra-tumoural OV injection: Survival: significantly prolonged survival compared to untreated Disease control: significantly decreased tumour growth compared to untreated Peritoneal mouse GC and PDAC xenograft tumour model with intraperitoneal OV injection: Survival: significantly prolonged survival compared to untreated Disease control: significantly decreased tumour growth compared to untreated | [80,81,82,90] |
| VSV | PD-L1 human | scFV | Lung carcinoma | Subcutaneous mouse hPD-L1 knock-in synergic tumour model with intra-tumoural OV injection: Survival: significantly prolonged survival compared to untreated Disease control: significantly decreased tumour growth compared to untreated successful tumour rejection following rechallenge Immune response: significant systemic increase in total number of CD8+ effector memory and CD8/CD4+ central memory T cells | [83] |
| VV | PD-L1 & GM-CSF human | Soluble PD-1 ED fused to IgG1 Fc | Melanoma | Bilateral subcutaneous synergic mouse tumour models with intra-tumoural OV injection: Survival: significantly decreased prolonged survival upon tumour rechallenge compared to untreated and parental OV Disease control: decreased tumour growth in 3 solid tumour models; significantly decreased tumour growth and prolonged survival upon tumour rechallenge compared to untreated and parental OV Immune response: significantly increased CD45+, DC, CD4+ and CD8+ T cell, and decreased MDSC and Treg tumour infiltration in injected tumours; untreated distant tumours also demonstrated increased infiltration and activation of lymphocytes and other immune cells | [92] |
Additional Targets
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dyck, L.; Mills, K.H.G. Immune Checkpoints and Their Inhibition in Cancer and Infectious Diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef]
- Maleki Vareki, S. High and Low Mutational Burden Tumors versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. J. ImmunoTher. Cancer 2018, 6, 4–8. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing Oncolytic Virotherapy in Cancer Treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sun, Z.-J. Turning Cold Tumors into Hot Tumors by Improving T-Cell Infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.K.; Hong, J.; Yun, C.O. Oncolytic Viruses and Immune Checkpoint Inhibitors: Preclinical Developments to Clinical Trials. Int. J. Mol. Sci. 2020, 21, 8627. [Google Scholar] [CrossRef]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of Virus-Mediated Immunogenic Cancer Cell Death and the Consequences for Oncolytic Virus-Based Immunotherapy of Cancer. Cell Death Dis. 2020, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating Oncolytic Viruses in Combination Cancer Immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Hotte, S.J.; Lorence, R.M.; Hirte, H.W.; Polawski, S.R.; Bamat, M.K.; O’Neil, J.D.; Roberts, M.S.; Groene, W.S.; Major, P.P. An Optimized Clinical Regimen for the Oncolytic Virus PV701. Clin. Cancer Res. 2007, 13, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Heinzerling, L.; Künzi, V.; Oberholzer, P.A.; Kündig, T.; Naim, H.; Dummer, R. Oncolytic Measles Virus in Cutaneous T-Cell Lymphomas Mounts Antitumor Immune Responses in Vivo and Targets Interferon-Resistant Tumor Cells. Blood 2005, 106, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, Y.A.; Wang, E.; Chen, N.; Danner, R.L.; Munson, P.J.; Marincola, F.M.; Szalay, A.A. Eradication of Solid Human Breast Tumors in Nude Mice with an Intravenously Injected Light-Emitting Oncolytic Vaccinia Virus. Cancer Res. 2007, 67, 10038–10046. [Google Scholar] [CrossRef]
- Felt, S.A.; Grdzelishvili, V.Z. Recent Advances in Vesicular Stomatitis Virus-Based Oncolytic Virotherapy: A 5-Year Update. J. Gen. Virol. 2017, 98, 2895–2911. [Google Scholar] [CrossRef] [PubMed]
- Kemp, V.; Hoeben, R.C.; van den Wollenberg, D.J.M. Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus. Viruses 2015, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Geisler, A.; Hazini, A.; Heimann, L.; Kurreck, J.; Fechner, H. Coxsackievirus B3—Its Potential as an Oncolytic Virus. Viruses 2021, 13, 718. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic Viruses: A New Class of Immunotherapy Drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Cunliffe, T.G.; Bates, E.A.; Parker, A.L. Hitting the Target but Missing the Point: Recent Progress towards Adenovirus-Based Precision Virotherapies. Cancers 2020, 12, 3327. [Google Scholar] [CrossRef]
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with Oncolytic Viruses: Progress and Challenges. Nat. Rev. Clin. Oncol. 2023, 20, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural Innate and Adaptive Immunity to Cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic Viruses as Engineering Platforms for Combination Immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef]
- Shi, T.; Song, X.; Wang, Y.; Liu, F.; Wei, J. Combining Oncolytic Viruses with Cancer Immunotherapy: Establishing a New Generation of Cancer Treatment. Front. Immunol. 2020, 11, 683. [Google Scholar] [CrossRef]
- Ren, Y.; Miao, J.M.; Wang, Y.Y.; Fan, Z.; Kong, X.B.; Yang, L.; Cheng, G. Oncolytic Viruses Combined with Immune Checkpoint Therapy for Colorectal Cancer Is a Promising Treatment Option. Front. Immunol. 2022, 13, 961796. [Google Scholar] [CrossRef]
- Li, Q.; Tan, F.; Wang, Y.; Liu, X.; Kong, X.; Meng, J.; Yang, L.; Cen, S. The Gamble between Oncolytic Virus Therapy and IFN. Front. Immunol. 2022, 13, 971674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.; Chen, K.; Qian, L.; Wang, P. Oncolytic Virotherapy Reverses the Immunosuppressive Tumor Microenvironment and Its Potential in Combination with Immunotherapy. Cancer Cell Int. 2021, 21, 262. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Wellenstein, M.D.; De Visser, K.E. Neutrophils in Cancer: Neutral No More. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Hofman, L.; Lawler, S.E.; Lamfers, M.L.M. The Multifaceted Role of Macrophages in Oncolytic Virotherapy. Viruses 2021, 13, 1570. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.; Errington, F.; Ilett, E.; Morgan, R.; Scott, K.; Kottke, T.; Thompson, J.; Morrison, E.; Harrington, K.; Pandha, H.; et al. Tumor Infection by Oncolytic Reovirus Primes Adaptive Anti-Tumor Immunity. Clin. Cancer Res. 2008, 14, 7358–7366. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final Analyses of OPTiM: A Randomized Phase III Trial of Talimogene Laherparepvec versus Granulocyte-Macrophage Colony-Stimulating Factor in Unresectable Stage III-IV Melanoma. J. ImmunoTher. Cancer 2019, 7, 145. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral Oncolytic Herpes Virus G47Δ for Residual or Recurrent Glioblastoma: A Phase 2 Trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized Dose-Finding Clinical Trial of Oncolytic Immunotherapeutic Vaccinia JX-594 in Liver Cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef]
- Holloway, R.W.; Kendrick, J.E.; Stephens, A.; Kennard, J.; Burt, J.; LeBlanc, J.; Sellers, K.; Smith, J.; Coakley, S. Phase 1b Study of Oncolytic Vaccinia Virus GL-ONC1 in Recurrent Ovarian Cancer (ROC). J. Clin. Oncol. 2018, 36, 5577. [Google Scholar] [CrossRef]
- Streby, K.A.; Currier, M.A.; Triplet, M.; Ott, K.; Dishman, D.J.; Vaughan, M.R.; Ranalli, M.A.; Setty, B.; Skeens, M.A.; Whiteside, S.; et al. First-in-Human Intravenous Seprehvir in Young Cancer Patients: A Phase 1 Clinical Trial. Mol. Ther. 2019, 27, 1930–1938. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Alfred Yung, W.K.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Hartmann, L.C.; Cliby, W.A.; Long, H.J.; Prema, P.; Barrette, B.A.; Kaur, J.S.; Haluska, P.J., Jr.; Aderca, I.; Zollman, P.J.; et al. Phase I Trial of Intraperitoneal Administration of an Oncolytic Measles Virus Strain Engineered to Express Carcinoembryonic Antigen for Recurrent Ovarian Cancer. Cancer Res. 2010, 70, 875–882. [Google Scholar] [CrossRef]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimäki, A.; et al. Phase I Study with ONCOS-102 for the Treatment of Solid Tumors—An Evaluation of Clinical Response and Exploratory Analyses of Immune Markers. J. ImmunoTher. Cancer 2016, 4, 17. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Ross, M.I.; Agarwala, S.S.; Taylor, M.H.; Vetto, J.T.; Neves, R.I.; Daud, A.; Khong, H.T.; Ungerleider, R.S.; Tanaka, M. Efficacy and Genetic Analysis for a Phase II Multicenter Trial of HF10, a Replication-Competent HSV-1 Oncolytic Immunotherapy, and Ipilimumab Combination Treatment in Patients with Stage IIIb-IV Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2018, 36, 9541. [Google Scholar] [CrossRef]
- Cook, J.; Peng, K.W.; Witzig, T.E.; Broski, S.M.; Villasboas, J.C.; Paludo, J.; Patnaik, M.; Rajkumar, V.; Dispenzieri, A.; Leung, N.; et al. Clinical Activity of Single-Dose Systemic Oncolytic VSV Virotherapy in Patients with Relapsed Refractory T-Cell Lymphoma. Blood Adv. 2022, 6, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Gross, N.D.; Nemunaitis, J.J.; Andtbacka, R.H.I.; Argiris, A.; Ohr, J.; Vetto, J.T.; Senzer, N.N.; Bedell, C.; Ungerleider, R.S.; et al. Phase I Trial of Intratumoral Therapy Using HF10, an Oncolytic HSV-1, Demonstrates Safety in HSV+/HSV− Patients with Refractory and Superficial Cancers. J. Clin. Oncol. 2014, 32, 6082. [Google Scholar] [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A Phase 1 Trial of Oncolytic HSV-1, G207, given in Combination with Radiation for Recurrent GBM Demonstrates Safety and Radiographic Responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef]
- Cui, C.L.; Wang, X.; Lian, B.; Ji, Q.; Zhou, L.; Chi, Z.; Si, L.; Sheng, X.; Kong, Y.; Yu, J.; et al. OrienX010, an Oncolytic Virus, in Patients with Unresectable Stage IIIC-IV Melanoma: A Phase Ib Study. J. ImmunoTher. Cancer 2022, 10, e004307. [Google Scholar] [CrossRef]
- Shirakawa, Y.; Tazawa, H.; Tanabe, S.; Kanaya, N.; Noma, K.; Koujima, T.; Kashima, H.; Kato, T.; Kuroda, S.; Kikuchi, S.; et al. Phase I Dose-Escalation Study of Endoscopic Intratumoral Injection of OBP-301 (Telomelysin) with Radiotherapy in Oesophageal Cancer Patients Unfit for Standard Treatments. Eur. J. Cancer 2021, 153, 98–108. [Google Scholar] [CrossRef]
- Musher, B.L.; Smaglo, B.G.; Abidi, W.; Othman, M.; Patel, K.; Jawaid, S.; Jing, J.; Brisco, A.; Wenthe, J.; Eriksson, E.; et al. A Phase I/II Study of LOAd703, a TMZ-CD40L/4-1BBL-Armed Oncolytic Adenovirus, Combined with Nab-Paclitaxel and Gemcitabine in Advanced Pancreatic Cancer. J. Clin. Oncol. 2022, 40, 4138. [Google Scholar] [CrossRef]
- Annels, N.E.; Mansfield, D.; Arif, M.; Ballesteros-Merino, C.; Simpson, G.R.; Denyer, M.; Sandhu, S.S.; Melcher, A.A.; Harrington, K.J.; Davies, B.; et al. Phase I Trial of an ICAM-1-Targeted Immunotherapeutic-Coxsackievirus A21 (CVA21) as an Oncolytic Agent Against Non Muscle-Invasive Bladder Cancer. Clin. Cancer Res. 2019, 25, 5818–5831. [Google Scholar] [CrossRef]
- Moreno, V.; Barretina-Ginesta, M.P.; García-Donas, J.; Jayson, G.C.; Roxburgh, P.; Vázquez, R.M.; Michael, A.; Antón-Torres, A.; Brown, R.; Krige, D.; et al. Safety and Efficacy of the Tumor-Selective Adenovirus Enadenotucirev with or without Paclitaxel in Platinum-Resistant Ovarian Cancer: A Phase 1 Clinical Trial. J. ImmunoTher. Cancer 2021, 9, e003645. [Google Scholar] [CrossRef]
- Bazan-Peregrino, M.; Garcia-Carbonero, R.; Laquente, B.; Álvarez, R.; Mato-Berciano, A.; Gimenez-Alejandre, M.; Morgado, S.; Rodríguez-García, A.; Maliandi, M.V.; Riesco, M.C.; et al. VCN-01 Disrupts Pancreatic Cancer Stroma and Exerts Antitumor Effects. J. ImmunoTher. Cancer 2021, 9, e003254. [Google Scholar] [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Hirooka, Y.; Kasuya, H.; Ishikawa, T.; Kawashima, H.; Ohno, E.; Villalobos, I.B.; Naoe, Y.; Ichinose, T.; Koyama, N.; Tanaka, M.; et al. A Phase I Clinical Trial of EUS-Guided Intratumoral Injection of the Oncolytic Virus, HF10 for Unresectable Locally Advanced Pancreatic Cancer. BMC Cancer 2018, 18, 596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Huang, J.; Tang, J.; Hu, S.; Luo, S.; Luo, Z.; Zhou, F.; Tan, S.; Ying, J.; Chang, Q.; et al. Intratumoral OH2, an Oncolytic Herpes Simplex Virus 2, in Patients with Advanced Solid Tumors: A Multicenter, Phase I/II Clinical Trial. J. ImmunoTher. Cancer 2021, 9, e002224. [Google Scholar] [CrossRef] [PubMed]
- Mayo Clinic Phase I Trial of a Measles Virus Derivative Producing CEA (MV-CEA) in Patients with Recurrent Glioblastoma Multiforme (GBM). 2019. Available online: https://clinicaltrials.gov (accessed on 1 June 2023).
- Garcia-Carbonero, R.; Gil Martín, M.; Alvarez Gallego, R.; Macarulla Mercade, T.; Riesco Martinez, M.C.; Guillen-Ponce, C.; Vidal, N.; Real, F.X.; Moreno, R.; Maliandi, V.; et al. Systemic Administration of the Hyaluronidase-Expressing Oncolytic Adenovirus VCN-01 in Patients with Advanced or Metastatic Pancreatic Cancer: First-in-Human Clinical Trial. Ann. Oncol. 2019, 30, v271–v272. [Google Scholar] [CrossRef]
- Packiam, V.T.; Lamm, D.L.; Barocas, D.A.; Trainer, A.; Fand, B.; Davis, R.L.; Clark, W.; Kroeger, M.; Dumbadze, I.; Chamie, K.; et al. An Open Label, Single-Arm, Phase II Multicenter Study of the Safety and Efficacy of CG0070 Oncolytic Vector Regimen in Patients with BCG-Unresponsive Non–Muscle-Invasive Bladder Cancer: Interim Results. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 440–447. [Google Scholar] [CrossRef]
- Moehler, M.; Heo, J.; Lee, H.C.; Tak, W.Y.; Chao, Y.; Paik, S.W.; Yim, H.J.; Byun, K.S.; Baron, A.; Ungerechts, G.; et al. Vaccinia-Based Oncolytic Immunotherapy Pexastimogene Devacirepvec in Patients with Advanced Hepatocellular Carcinoma after Sorafenib Failure: A Randomized Multicenter Phase IIb Trial (TRAVERSE). OncoImmunology 2019, 8, 1615817. [Google Scholar] [CrossRef]
- Li, S.J.; Sun, Z.J. Fueling Immune Checkpoint Blockade with Oncolytic Viruses: Current Paradigms and Challenges Ahead. Cancer Lett. 2022, 550, 215937. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. Cancer Clin. Trials 2016, 39, 98–106. [Google Scholar] [CrossRef]
- De Silva, P.; Aiello, M.; Gu-Trantien, C.; Migliori, E.; Willard-Gallo, K.; Solinas, C. Targeting CTLA-4 in Cancer: Is It the Ideal Companion for PD-1 Blockade Immunotherapy Combinations? Int. J. Cancer 2021, 149, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Schalper, K.A.; Kaftan, E.; Herbst, R.S. Predictive Biomarkers for PD-1 Axis Therapies: The Hidden Treasure or a Call for Research. Clin. Cancer Res. 2016, 22, 2102–2104. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Sanmamed, M.F.; Hastings, K.; Politi, K.; Rimm, D.L.; Chen, L.; Melero, I.; Schalper, K.A.; Herbst, R.S. Immunotherapy in Non–Small Cell Lung Cancer: Facts and Hopes. Clin. Cancer Res. 2019, 25, 4592–4602. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Ungerechts, G.; Nettelbeck, D.M. Viro-Antibody Therapy: Engineering Oncolytic Viruses for Genetic Delivery of Diverse Antibody-Based Biotherapeutics. mAbs 2021, 13, 1982447. [Google Scholar] [CrossRef]
- Li, R.; Steinberg, G.; Uchio, E.; Lamm, D.; Paras, S.; Kamat, A.; Bivalacqua, T.; Packiam, V.; Chisamore, M.; McAdory, J.; et al. 666 Phase 2, Single Arm Study of CG0070 Combined with Pembrolizumab in Patients with Non-Muscle Invasive Bladder Cancer (NMIBC) Unresponsive to Bacillus Calmette-Guerin (BCG). J. ImmunoTher. Cancer 2022, 10. [Google Scholar]
- Nassiri, F.; Patil, V.; Yefet, L.S.; Singh, O.; Liu, J.; Dang, R.M.A.; Yamaguchi, T.N.; Daras, M.; Cloughesy, T.F.; Colman, H.; et al. Oncolytic DNX-2401 Virotherapy plus Pembrolizumab in Recurrent Glioblastoma: A Phase 1/2 Trial. Nat. Med. 2023, 29, 1370–1378. [Google Scholar] [CrossRef]
- Chesney, J.A.; Ribas, A.; Long, G.V.; Kirkwood, J.M.; Dummer, R.; Puzanov, I.; Hoeller, C.; Gajewski, T.F.; Gutzmer, R.; Rutkowski, P.; et al. Randomized, Double-Blind, Placebo-Controlled, Global Phase III Trial of Talimogene Laherparepvec Combined with Pembrolizumab for Advanced Melanoma. J. Clin. Oncol. 2023, 41, 528–540. [Google Scholar] [CrossRef]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination with Ipilimumab versus Ipilimumab Alone in Patients with Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Olszanski, A.J.; Nyakas, M.; Hornyak, T.J.; Wolchok, J.D.; Levitsky, V.; Kuryk, L.; Hansen, T.B.; Jäderberg, M. Pilot Study of ONCOS-102 and Pembrolizumab: Remodeling of the Tumor Microenvironment and Clinical Outcomes in Anti-PD-1-Resistant Advanced Melanoma. Clin. Cancer Res. 2023, 29, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.M.; Antonescu, C.R.; Bowler, T.; Munhoz, R.; Chi, P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Movva, S.; Dholakia, R.; et al. Objective Response Rate Among Patients with Locally Advanced or Metastatic Sarcoma Treated with Talimogene Laherparepvec in Combination With Pembrolizumab: A Phase 2 Clinical Trial. JAMA Oncol. 2020, 6, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.G.; Wang, D.; Harb, W.; Rosen, L.; Mahadevan, D.; Berlin, J.D.; Basciano, P.; Brown, R.; Arogundade, O.; Cox, C.; et al. SPICE, a Phase I Study of Enadenotucirev in Combination with Nivolumab in Tumours of Epithelial Origin: Analysis of the Metastatic Colorectal Cancer Patients in the Dose Escalation Phase. Ann. Oncol. 2019, 30, v231. [Google Scholar] [CrossRef]
- Lillie, T.; Stone, A.; Lockwood, S.; Brown, R.; Fox, A.; Bournazou, E.; Beadle, J. 329 Prolonged Overall Survival (OS) in Patients with Metastatic Colorectal Cancer (MCRC) in SPICE, a Phase I Study of Enadenotucirev in Combination with Nivolumab. J. ImmunoTher. Cancer 2020, 8, A202. [Google Scholar]
- Krige, D.; Fakih, M.; Rosen, L.; Wang, D.; Harb, W.; Babiker, H.; Berlin, J.; Di Genova, G.; Miles, D.; Mark, P.; et al. Combining Enadenotucirev and Nivolumab increased tumour immune cell infiltration/activation in patients with microsatellite- stable/instability-low metastatic colorectal cancer in a Phase 1 study. J. ImmunoTher. 2021, 9. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- Monge, C.; Xie, C.; Myojin, Y.; Coffman, K.; Hrones, D.M.; Wang, S.; Hernandez, J.M.; Wood, B.J.; Levy, E.B.; Juburi, I.; et al. Phase I/II Study of PexaVec in Combination with Immune Checkpoint Inhibition in Refractory Metastatic Colorectal Cancer. J. ImmunoTher. Cancer 2023, 11, e005640. [Google Scholar] [CrossRef]
- Fransen, M.F.; Van Der Sluis, T.C.; Ossendorp, F.; Arens, R.; Melief, C.J.M. Controlled Local Delivery of CTLA-4 Blocking Antibody Induces CD8 + T-Cell-Dependent Tumor Eradication and Decreases Risk of Toxic Side Effects. Clin. Cancer Res. 2013, 19, 5381–5389. [Google Scholar] [CrossRef]
- Han, X.; Li, H.; Zhou, D.; Chen, Z.; Gu, Z. Local and Targeted Delivery of Immune Checkpoint Blockade Therapeutics. Acc. Chem. Res. 2020, 53, 2521–2533. [Google Scholar] [CrossRef]
- Du, T.; Shi, G.; Li, Y.M.; Zhang, J.F.; Tian, H.W.; Wei, Y.Q.; Deng, H.; Yu, D.C. Tumor-Specific Oncolytic Adenoviruses Expressing Granulocyte Macrophage Colony-Stimulating Factor or Anti-CTLA4 Antibody for the Treatment of Cancers. Cancer Gene Ther. 2014, 21, 340–348. [Google Scholar] [CrossRef]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Löskog, A.; et al. Targeted Cancer Immunotherapy with Oncolytic Adenovirus Coding for a Fully Human Monoclonal Antibody Specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a New Fusion-Enhanced Oncolytic Immunotherapy Platform Based on Herpes Simplex Virus Type 1. J. ImmunoTher. Cancer 2019, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.R.; Vijayakumar, G.; Palese, P. A Recombinant Antibody-Expressing Influenza Virus Delays Tumor Growth in a Mouse Model. Cell Rep. 2018, 22, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Wang, X.; Xing, M.; Yang, X.; Wu, M.; Shi, H.; Zhu, C.; Wang, X.; Guo, Y.; Tang, S.; et al. Intratumoral Delivery of a Novel Oncolytic Adenovirus Encoding Human Antibody against PD-1 Elicits Enhanced Antitumor Efficacy. Mol. Ther. Oncolytics 2022, 25, 236–248. [Google Scholar] [CrossRef]
- Lin, C.; Ren, W.; Luo, Y.; Li, S.; Chang, Y.; Li, L.; Xiong, D.; Huang, X.; Xu, Z.; Yu, Z.; et al. Intratumoral Delivery of a PD-1-Blocking ScFv Encoded in Oncolytic HSV-1 Promotes Antitumor Immunity and Synergizes with TIGIT Blockade. Cancer Immunol. Res. 2020, 8, 632–648. [Google Scholar] [CrossRef]
- Ju, F.; Luo, Y.; Lin, C.; Jia, X.; Xu, Z.; Tian, R.; Lin, Y.; Zhao, M.; Chang, Y.; Huang, X.; et al. Oncolytic Virus Expressing PD-1 Inhibitors Activates a Collaborative Intratumoral Immune Response to Control Tumor and Synergizes with CTLA-4 or TIM-3 Blockade. J. ImmunoTher. Cancer 2022, 10, e004762. [Google Scholar] [CrossRef]
- Tian, C.; Liu, J.; Zhou, H.; Li, J.; Sun, C.; Zhu, W.; Yin, Y.; Li, X. Enhanced Anti-Tumor Response Elicited by a Novel Oncolytic HSV-1 Engineered with an Anti-PD-1 Antibody. Cancer Lett. 2021, 518, 49–58. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, X.; Feng, L.; Yang, Z.; Zhou, L.; Duan, X.; Cheng, S.; Zhang, W.; Liu, B.; Zhang, K. Enhanced Therapeutic Efficacy of a Novel Oncolytic Herpes Simplex Virus Type 2 Encoding an Antibody Against Programmed Cell Death 1. Mol. Ther. Oncolytics 2019, 15, 201–213. [Google Scholar] [CrossRef]
- Woo, Y.; Zhang, Z.; Yang, A.; Chaurasiya, S.; Park, A.K.; Lu, J.; Kim, S.I.; Warner, S.G.; Von Hoff, D.; Fong, Y. Novel Chimeric Immuno-Oncolytic Virus CF33-HNIS-AntiPDL1 for the Treatment of Pancreatic Cancer. J. Am. Coll. Surg. 2020, 230, 709–717. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Yang, A.; Zhang, Z.; Lu, J.; Valencia, H.; Kim, S.I.; Woo, Y.; Warner, S.G.; Olafsen, T.; Zhao, Y.; et al. A Comprehensive Preclinical Study Supporting Clinical Trial of Oncolytic Chimeric Poxvirus CF33-HNIS-Anti-PD-L1 to Treat Breast Cancer. Mol. Ther. Methods Clin. Dev. 2022, 24, 102–116. [Google Scholar] [CrossRef]
- Yang, A.; Zhang, Z.; Chaurasiya, S.; Park, A.K.; Kim, I.; Priceman, S.; Fong, Y.; Jung, A.; Lu, J. Development of the Oncolytic Virus, CF33, and Its Derivatives for Peritoneal-Directed Treatment of Gastric Cancer Peritoneal Metastases. J. ImmunoTher. Cancer 2023, 11, e006280. [Google Scholar] [CrossRef]
- Wu, C.; Wu, M.; Liang, M.; Xiong, S.; Dong, C. A Novel Oncolytic Virus Engineered with PD-L1 ScFv Effectively Inhibits Tumor Growth in a Mouse Model. Cell Mol. Immunol. 2019, 16, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Veinalde, R.; Pidelaserra-Martí, G.; Moulin, C.; Jeworowski, L.M.; Küther, L.; Buchholz, C.J.; Jäger, D.; Ungerechts, G.; Engeland, C.E. Oncolytic Measles Vaccines Encoding PD-1 and PD-L1 Checkpoint Blocking Antibodies to Increase Tumor-Specific T Cell Memory. Mol. Ther. Oncolytics 2022, 24, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.L.; Wang, L.P.; Dong, S.H.; Sun, F.; Cheng, J.X.; Yang, X.L.; Zhang, S.G.; Wang, X.L.; Wang, X.X.; Yang, P.H. A Recombinant Influenza Virus with a CTLA4-Specific ScFv Inhibits Tumor Growth in a Mouse Model. Cell Biol. Int. 2021, 45, 1202–1210. [Google Scholar] [CrossRef]
- Kleinpeter, P.; Fend, L.; Thioudellet, C.; Geist, M.; Sfrontato, N.; Koerper, V.; Fahrner, C.; Schmitt, D.; Gantzer, M.; Remy-Ziller, C.; et al. Vectorization in an Oncolytic Vaccinia Virus of an Antibody, a Fab and a ScFv against Programmed Cell Death -1 (PD-1) Allows Their Intratumoral Delivery and an Improved Tumor-Growth Inhibition. OncoImmunology 2016, 5, e1220467. [Google Scholar] [CrossRef]
- Vijayakumar, G.; McCroskery, S.; Palese, P. Engineering Newcastle Disease Virus as an Oncolytic Vector for Intratumoral Delivery of Immune Checkpoint Inhibitors and Immunocytokines. J. Virol. 2020, 94, 1128. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Scialò, F.; Passariello, M.; Leggiero, E.; D’Agostino, A.; Tripodi, L.; Gentile, L.; Bianco, A.; Castaldo, G.; Cerullo, V.; et al. Oncolytic Adenoviral Vector-Mediated Expression of an Anti-PD-L1-ScFv Improves Anti-Tumoral Efficacy in a Melanoma Mouse Model. Front. Oncol. 2022, 12, 902190. [Google Scholar] [CrossRef] [PubMed]
- Passaro, C.; Alayo, Q.; De Laura, I.; McNulty, J.; Grauwet, K.; Ito, H.; Bhaskaran, V.; Mineo, M.; Lawler, S.E.; Shah, K.; et al. Arming an Oncolytic Herpes Simplex Virus Type 1 with a Single-Chain Fragment Variable Antibody against PD-1 for Experimental Glioblastoma Therapy. Clin. Cancer Res. 2019, 25, 290–299. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, A.; Chaurasiya, S.; Park, A.K.; Lu, J.; Kim, S.I.; Warner, S.G.; Yuan, Y.C.; Liu, Z.; Han, H.; et al. CF33-HNIS-AntiPDL1 Virus Primes Pancreatic Ductal Adenocarcinoma for Enhanced Anti-PD-L1 Therapy. Cancer Gene Ther. 2022, 29, 722–733. [Google Scholar] [CrossRef]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 Checkpoint Blockade Enhances Oncolytic Measles Virus Therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.Y. An Engineered Oncolytic Virus Expressing PD-L1 Inhibitors Activates Tumor Neoantigen-Specific T Cell Responses. Nat. Commun. 2020, 11, 1395. [Google Scholar] [CrossRef]
- Vijayakumar, G.; Palese, P.; Goff, P.H. Oncolytic Newcastle Disease Virus Expressing a Checkpoint Inhibitor as a Radioenhancing Agent for Murine Melanoma. EBioMedicine 2019, 49, 96–105. [Google Scholar] [CrossRef]
- Bates, E.A.; Lovatt, C.; Plein, A.R.; Davies, J.A.; Siebzehnrubl, F.A.; Parker, A.L. Engineering Adenoviral Vectors with Improved GBM Selectivity. Viruses 2023, 15, 1086. [Google Scholar] [CrossRef] [PubMed]
- Fender, P.; Jeanson, L.; Ivanov, M.A.; Colin, P.; Mallet, J.; Dedieu, J.F.; Latta-Mahieu, M. Controlled Transgene Expression by E1-E4-Defective Adenovirus Vectors Harbouring a “Tet-on” Switch System. J. Gene Med. 2002, 4, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Verma, R.; Sznol, M.; Boddupalli, C.S.; Gettinger, S.N.; Kluger, H.; Callahan, M.; Wolchok, J.D.; Halaban, R.; Dhodapkar, M.V. Combination Therapy with Anti–CTLA-4 and Anti–PD-1 Leads to Distinct Immunologic Changes in Vivo. J. Immunol. 2015, 194, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in Cancer Immunotherapy. J. ImmunoTher. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Wei, M.; He, B.; Chen, A.; Wang, S.; Kong, L.; Zhang, Y.; Meng, G.; Xu, T.; Wu, J.; et al. Enhanced Antitumor Efficacy of a Novel Oncolytic Vaccinia Virus Encoding a Fully Monoclonal Antibody against T-Cell Immunoglobulin and ITIM Domain (TIGIT). EBioMedicine 2021, 64, 103240. [Google Scholar] [CrossRef]
- Zuo, S.; Wei, M.; Xu, T.; Kong, L.; He, B.; Wang, S.; Wang, S.; Wu, J.; Dong, J.; Wei, J. An Engineered Oncolytic Vaccinia Virus Encoding a Single-Chain Variable Fragment against TIGIT Induces Effective Antitumor Immunity and Synergizes with PD-1 or LAG-3 Blockade. J. ImmunoTher. Cancer 2021, 9, e002843. [Google Scholar] [CrossRef]
- Lee, J.B.; Ha, S.-J.; Kim, H.R. Clinical Insights Into Novel Immune Checkpoint Inhibitors. Front. Pharmacol. 2021, 12, 681320. [Google Scholar] [CrossRef]
- Popat, S.; Grohé, C.; Corral, J.; Reck, M.; Novello, S.; Gottfried, M.; Radonjic, D.; Kaiser, R. Anti-Angiogenic Agents in the Age of Resistance to Immune Checkpoint Inhibitors: Do They Have a Role in Non-Oncogene-Addicted Non-Small Cell Lung Cancer? Lung Cancer 2020, 144, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
| Virus | Diameter | Genome | Genome Size | Transgene Capacity |
|---|---|---|---|---|
| Adenovirus | 90–100 nm | dsDNA | 30–36 kb | ~2.5 kb |
| Herpes simplex virus | 200 nm | dsDNA | ~152 kb | ~30 kb |
| Vaccinia virus | 350 nm | dsDNA | ~192 kb | ~25 kb |
| Influenza A virus | 80–120 nm | ss(–)RNA | ~13.5 kb | ~2.4 kb |
| Newcastle disease virus | 100–500 nm | ss(–)RNA | ~15 kb | ~4.5 kb |
| Measles virus | 100–200 nm | ss(–)RNA | ~16 kb | ~6 kb |
| Vesicular stomatitis virus | 70–200 nm | ss(–)RNA | ~11.1 kb | ~4.5 kb |
| Coxsackie virus | 22–30 nm | ss(+)RNA | ~7.5 kb | <1 kb |
| Reovirus | 80 nm | dsRNA | 24 kb | ~1.5 kb |
| Virus | OV | Engineered Specificity | Transgene | Indication | Delivery | Key Findings | Ref. |
|---|---|---|---|---|---|---|---|
| Adenovirus | CG0070 | Ad5 with E1a under E2F-1 promoter | GM-CSF | NMIBC Phase II | IVS | Disease control: 47% 6-month CR; 29% 12-month CR | [49] |
| DNX-2401 | Ad5 with 24 bp E1a deletion; RGD integrin-binding motif | GBM Phase I | IT | Survival: 20% >3-year survival Disease control: 12% demonstrated >95% tumour reduction Immune response: increased tumour CD8+ and T-bet+ cells; decreased TIM-3+ cells | [31] | ||
| EnAd | Ad11p/3 chimera generated through directed evolution | Ovarian Phase I | IV | Survival: 64% PFS Disease control: 10% ORR; 35% achieved stable disease; 65% saw reduction in tumour burden; Immune responses: 83.3% demonstrated increased CD8+ TILs | [42] | ||
| LoAd-703 | Ad5 with 24 bp E1a deletion; Pseudo-typed Ad35 knob | TMZ-CD40L; 4-1BBL | PDAC Phase I/II | IT | Survival: OS 8.7 months Disease control: 44% ORR; 94% DCR Immune response: increased effector memory T cells; decreased Tregs and MDSCs | [40] | |
| ONCOS-102 | Ad5 with 24 bp E1a deletion; Pseudo-typed Ad3 knob | GM-CSF | Solid tumours Phase I | IT | Immune response: increase in TILs; increase in systemic tumour-specific CD8+ T cells; increased tumour PD-L1 expression | [33] | |
| Telomelysin | Ad5 with E1a under hTERT promoter | Oesophageal Phase I | IT | Disease control: 91.7% ORR; 83.3% Stage I and 60% Stage II/III CRR Immune response: increased tumour CD8+ T cells; increased tumour PD-L1 expression | [39] | ||
| VCN-01 | Ad5 with 24 bp E1a deletion; E2F1 promoter insertion; RGDK integrin-binding motif | Hyaluronidase | PDAC Phase I | IT | Disease control: injected tumours reduced in size or remained stable; reduction in tumour stiffness | [43] | |
| IV | Disease control: 40–45% ORR including 1 complete response Immune response: CD8+ T cell tumour infiltration and IDO upregulation in 64% of patients | [48] | |||||
| Coxsackie virus | CVA21 | NMIBC Phase I | IVS | Disease control: 1/15 demonstrated CR; viral protein detected in 86% of tumours with no viral protein seen in stroma Immune response: CR patient demonstrated increased immune infiltration; RNA-seq demonstrated increased intrinsic apoptotic cell death pathway and PD-L1, LAG-3 and IDO within the TME | [41] | ||
| Herpes simplex virus | T-VEC | HSV1 with ICP34.5 deletion; US11 deletion | GM-CSF | Melanoma Phase III | IT | Survival: median OS 23.3 months Disease control: 19% DRR; 31.5% ORR; 50% demonstrated CR of which 88.5% were estimated to survive at 5-years; median time to CR 8.6 months Approved for the local treatment of unresectable metastatic stage IIIB/C–IVM1a melanoma in Europe and US | [26] |
| G207 | HSV1 with ICP34.5 deletion; UL39 deletion; | GBM Phase I (+Rad) | IT | Survival: median OS 7.5 months Disease control: median PFS 2.5 months; 67% demonstrated stable or partial response at ≥ 1 time point | [37] | ||
| Paediatric glioma Phase I | IT | Survival: Median OS 12.2 months; 36% still alive at 18 months Disease control: 18% demonstrated stable disease at 12 months Immune response: increased CD4+ and CD8+ T cell tumour infiltration | [44] | ||||
| G47Δ | G207 with additional α47 deletion; US11 promoter deletion | GBM Phase II | IT | Survival: median OS 20.2 months; 84.2% survival at 12 months Disease control: median PFS 4.7 months; stable disease in 18 patients at 2 years Immune response: increased CD4+, CD8+ and decreased Foxp3+ TIL Conditional and time-limited approval for treatment of GBM in Japan | [27] | ||
| HF10 | HSV1 with UL43, UL49.5, UL55 & UL56 deletions; Latency-associated transcripts deletions; UL53 & UL54 overexpression | Pancreatic cancer Phase I | IT | Survival: median OS 15.5 months; 2 patients were alive at 3 year follow up Disease control: median PFS 6.3 months; 33.3% PR; 44.4% SD; 2 patients demonstrated surgical CR Immune response: increased CD4+, CD8+ TILs | [45] | ||
| Superficial solid tumours Phase II | IT | Disease control: 33.3% SD; 1 patient demonstrated pathological CR after 4 months; 30–61% reduction in tumour size in those demonstrating responses | [36] | ||||
| Seprehvir | HSV1 with ICP34.5 deletion | Paediatric solid tumours Phase I | IT | Survival: median OS 7 months Disease control: 80% demonstrated SD at 14 days; 43% SD at 28 days | [30] | ||
| OrienX010 | HSV1 with ICP34.5 deletion; US12 deletion | GM-CSF | Melanoma Phase I | IT | Survival: median OS 19.2 months Disease control: median PFS 2.9 months; 54.6% of injected tumours regressed, 25.8% of which regressed by ≥30%; 54.1% of non-injected regional tumours regressed, 32.8% of which regressed by ≥30%; 1 distant non-injected metastases regressed by 58% | [38] | |
| OH2 | HSV2 with ICP34.5 & ICP47 deletion; | GM-CSF | Solid tumours Phase I/II | IT | Disease control: 1 PR; 33% stable disease Immune response: 79% saw increased CD8+ TILs; 86% increased CD3+ TILs; 71.4% increased PD-L1+ cells | [46] | |
| Newcastle disease virus | PV701 | Solid tumours Phase I | IV | Disease control: 61% PFS at 4 months; 33% OR; 1 CR cervical cancer; 2 PRs colorectal; 1 PR melanoma | [8] | ||
| Measles virus | MV-CEA | Carcinoembryonic antigen | Ovarian cancer Phase I/II | IP | Survival: median OS 12.15 months Disease control: 67% SD; 36% demonstrated >30% tumour reduction | [9] | |
| GBM Phase I | IT | Survival: median OS 11.6 months Disease control: 59% 3-month PFS; 23% 6-month PFS | [47] | ||||
| MV-NIS | Sodium iodide symporter | Ovarian cancer Phase I | IP | Survival: median OS 26.2 months Disease control: 81% SD | [32] | ||
| Vaccinia virus | GL-ONC1 | Β-galactosidase; β-glucuronidase | Ovarian cancer Phase I | IP | Disease control: median PFS 11.6 months; 78% 6-month PFS; 63% ORR; 52% CR Immune response: increased CD4+ & CD8+ TILs | [29] | |
| JX-594 | TK1 deletion | GM-CSF | HCC Phase II | IT | Survival: median OS 9 months; ~35% alive at 2 years Disease control: 46% demonstrated tumour control at 8 weeks; average 32.2% decrease in tumour size Immune response: increased tumour specific CD8+ TILs | [28] | |
| HCC Refractory to Sorafenib treatment Phase IIb | IT | Survival: no significant increase in survival compared to BSC Disease control: 13% DCR compared to 18% DCR with BSC Immune response: OV treated patients demonstrated a significant increase in vaccinia-specific T cells; 21.7% OV treated patients demonstrated tumour associated antigen-specific T cells | [50] | ||||
| Vesicular stomatitis virus | VSV- IFNβ-NIS | IFN-β; sodium iodide symporter | TCL Phase I | IV | Disease control: 1 6-month PR; 1 20-month CR; 71.4% reduction in ≥1 tumour | [35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lovatt, C.; Parker, A.L. Oncolytic Viruses and Immune Checkpoint Inhibitors: The “Hot” New Power Couple. Cancers 2023, 15, 4178. https://doi.org/10.3390/cancers15164178
Lovatt C, Parker AL. Oncolytic Viruses and Immune Checkpoint Inhibitors: The “Hot” New Power Couple. Cancers. 2023; 15(16):4178. https://doi.org/10.3390/cancers15164178
Chicago/Turabian StyleLovatt, Charlotte, and Alan L. Parker. 2023. "Oncolytic Viruses and Immune Checkpoint Inhibitors: The “Hot” New Power Couple" Cancers 15, no. 16: 4178. https://doi.org/10.3390/cancers15164178
APA StyleLovatt, C., & Parker, A. L. (2023). Oncolytic Viruses and Immune Checkpoint Inhibitors: The “Hot” New Power Couple. Cancers, 15(16), 4178. https://doi.org/10.3390/cancers15164178