
Identifying Cancer Type-Specific Transcriptional Programs through Network Analysis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Training the Cancer Cellnet Model
- Generation of metadata file (training sample table)
- 2.
- Pre-processing of training data
- 3.
- GRN construction, training, and validation:
- 4.
- Precision recall curves
2.2. Querying Normal Tissue Using Training Cancer Data
2.3. Gene Ontology Functional Annotation
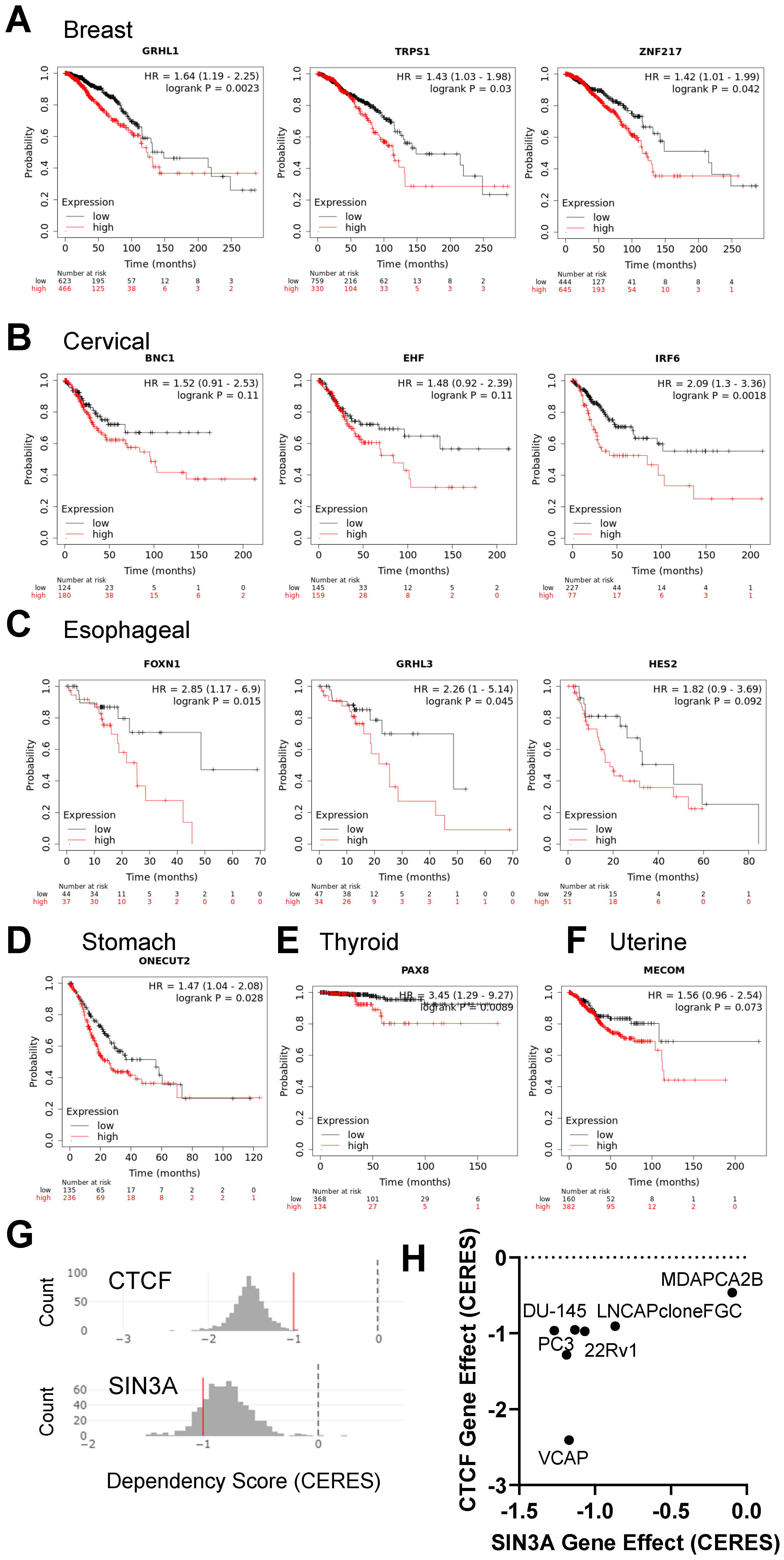
2.4. Kaplan–Meier Plots
2.5. Cancer Dependency Analysis Using the Depmap Database
3. Results
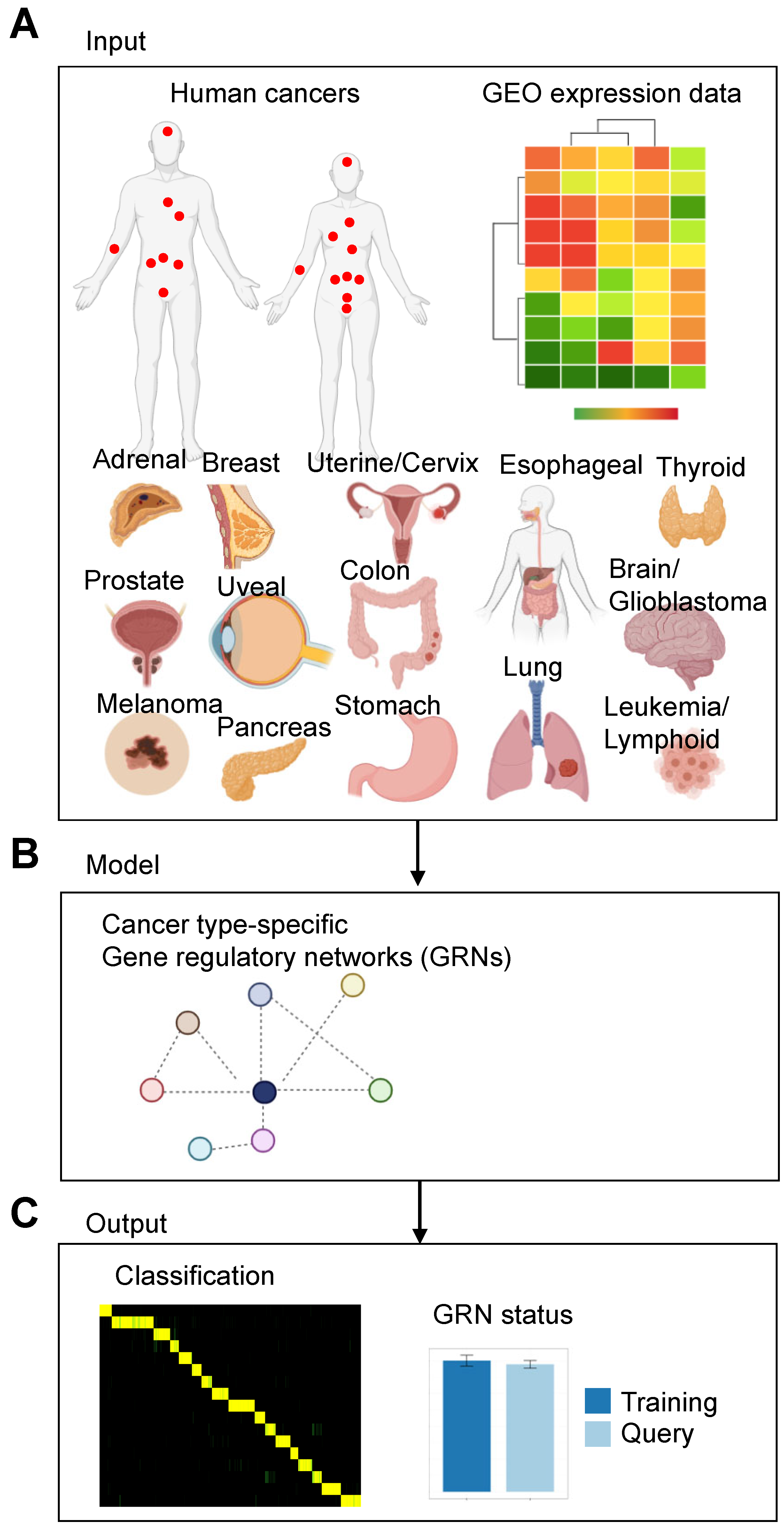
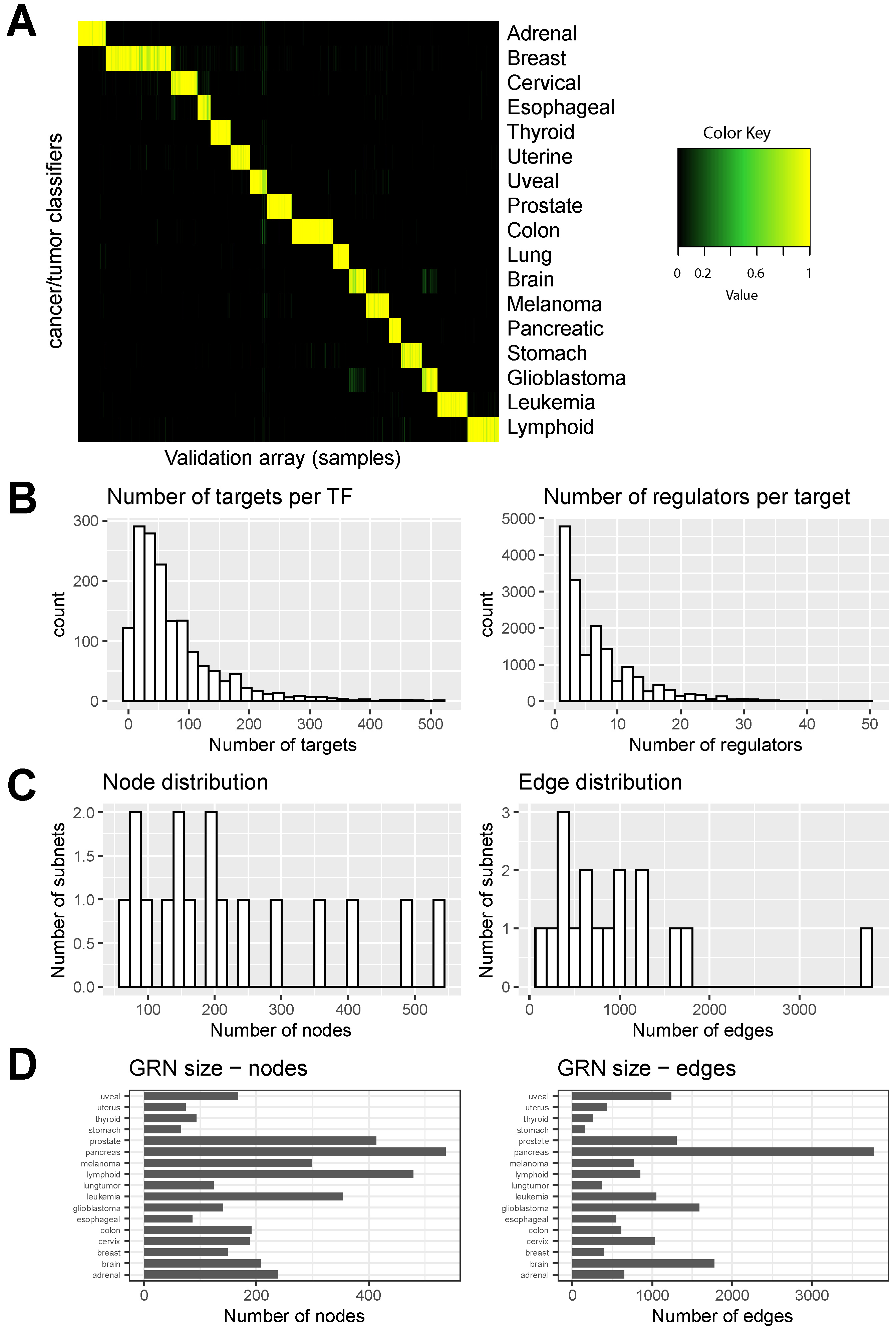
3.1. Application of a Network Biology Platform to Identify Cancer Type-Specific Gene Regulatory Networks
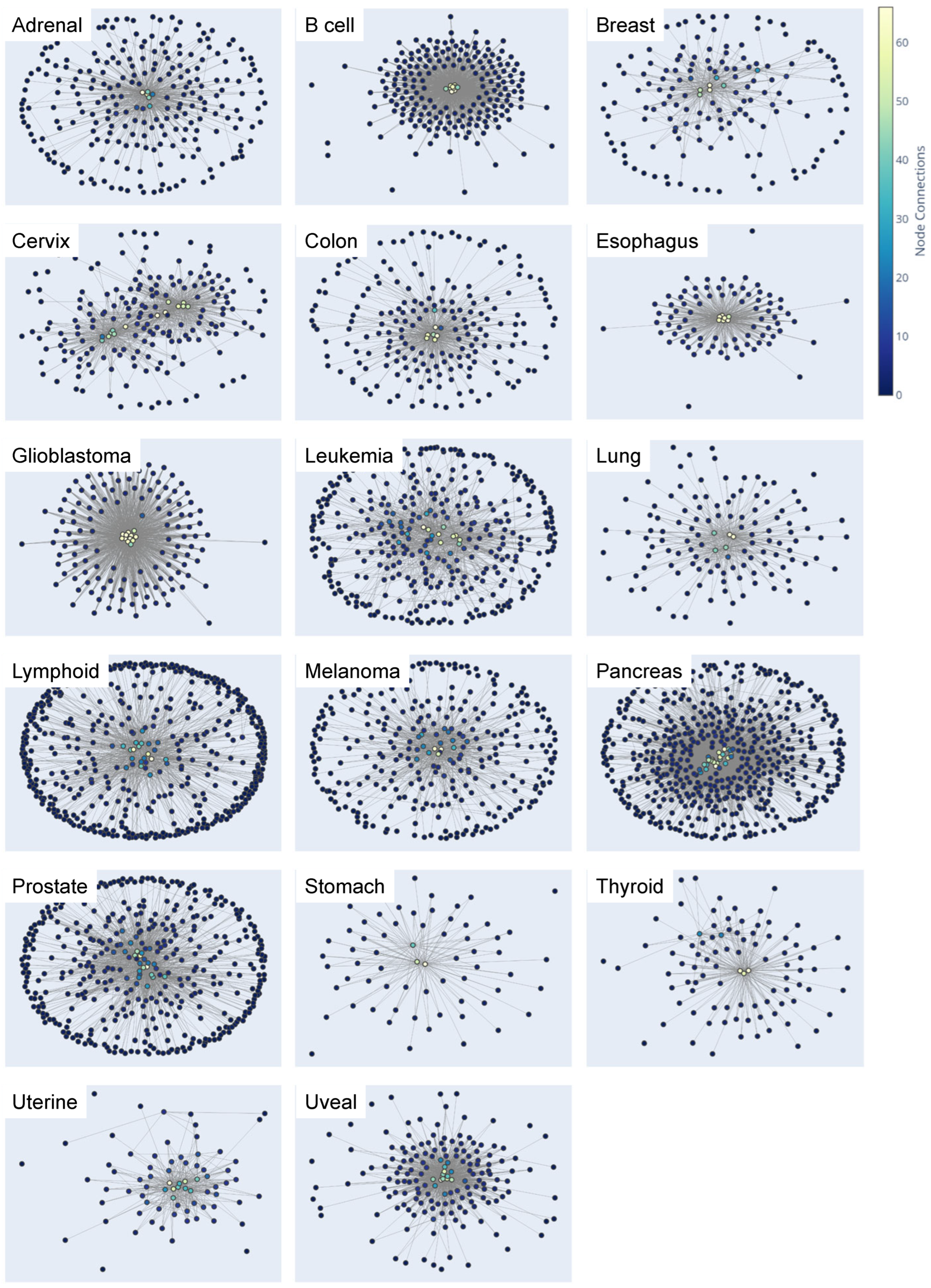
3.2. Exploration and Analysis of Network Influencing Genes
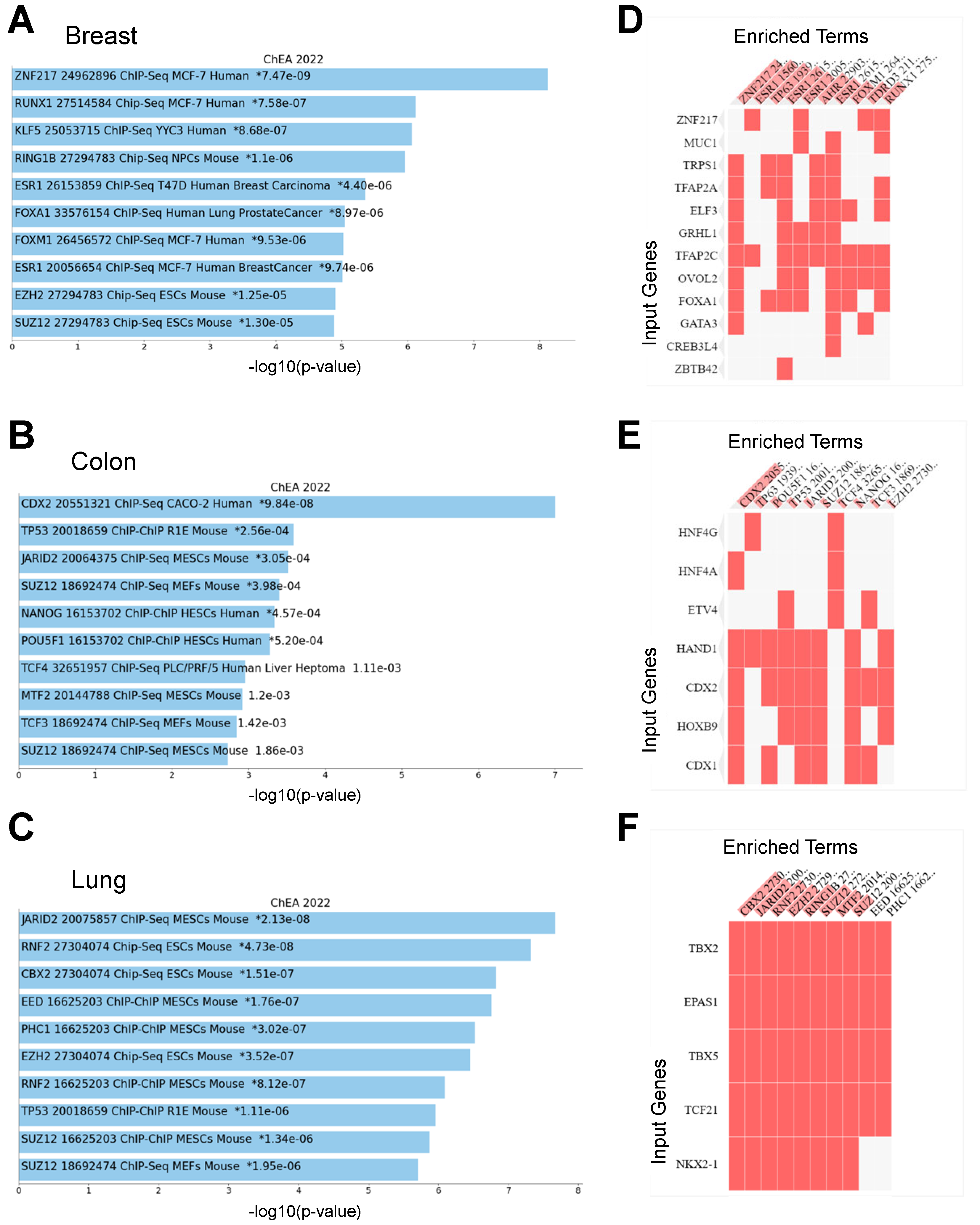
3.3. Functional Annotation of Network Influencing Genes
3.4. Implications of Elevated Gene Expression on Survival Rates of Cancer Patients
4. Discussion
4.1. Performance Evaluation and Insights Derived from the Classifier and Gene Regulatory Networks
4.2. Network Influencing Genes within Cancer Gene Regulatory Networks
4.3. Gene Regulatory Networks in Prostate Cancer
4.4. Gene Regulatory Network Performance Measures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilic, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef]
- Loh, Y.H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Sebastiano, V.; Wu, G.; Arauzo-Bravo, M.J.; Sasse, P.; Gentile, L.; Ko, K.; Ruau, D.; Ehrich, M.; van den Boom, D.; et al. Oct4-induced pluripotency in adult neural stem cells. Cell 2009, 136, 411–419. [Google Scholar] [CrossRef]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef]
- Huang, P.; He, Z.; Ji, S.; Sun, H.; Xiang, D.; Liu, C.; Hu, Y.; Wang, X.; Hui, L. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 2011, 475, 386–389. [Google Scholar] [CrossRef]
- Sekiya, S.; Suzuki, A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 2011, 475, 390–393. [Google Scholar] [CrossRef]
- Szabo, E.; Rampalli, S.; Risueno, R.M.; Schnerch, A.; Mitchell, R.; Fiebig-Comyn, A.; Levadoux-Martin, M.; Bhatia, M. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 2010, 468, 521–526. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Maherali, N.; Kulalert, W.; Hochedlinger, K. Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J. Cell Sci. 2009, 122, 3502–3510. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Ishii, H.; Nagai, K.; Hoshino, H.; Mimori, K.; Tanaka, F.; Nagano, H.; Sekimoto, M.; Doki, Y.; Mori, M. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Pruszak, J.; Varadarajan, M.; Blomen, V.A.; Gokhale, S.; Camargo, F.D.; Wernig, M.; Jaenisch, R.; Brummelkamp, T.R. Generation of iPSCs from cultured human malignant cells. Blood 2010, 115, 4039–4042. [Google Scholar] [CrossRef]
- Saito, S.; Lin, Y.C.; Nakamura, Y.; Eckner, R.; Wuputra, K.; Kuo, K.K.; Lin, C.S.; Yokoyama, K.K. Potential application of cell reprogramming techniques for cancer research. Cell Mol. Life Sci. 2019, 76, 45–65. [Google Scholar] [CrossRef]
- Borges, G.T.; Vencio, E.F.; Vencio, R.Z.; Vessella, R.L.; Ware, C.B.; Liu, A.Y. Reprogramming of prostate cancer cells—Technical challenges. Curr. Urol. Rep. 2015, 16, 468. [Google Scholar] [CrossRef]
- Iskender, B.; Izgi, K.; Canatan, H. Reprogramming bladder cancer cells for studying cancer initiation and progression. Tumour Biol. 2016, 37, 13237–13245. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Cufi, S.; Oliveras-Ferraros, C.; Cuyas, E.; Lopez-Bonet, E.; Lupu, R.; Alarcon, T.; Vellon, L.; Iglesias, J.M.; Leis, O.; et al. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle 2013, 12, 3109–3124. [Google Scholar] [CrossRef]
- Kim, J.; Hoffman, J.P.; Alpaugh, R.K.; Rhim, A.D.; Reichert, M.; Stanger, B.Z.; Furth, E.E.; Sepulveda, A.R.; Yuan, C.X.; Won, K.J.; et al. An iPSC line from human pancreatic ductal adenocarcinoma undergoes early to invasive stages of pancreatic cancer progression. Cell Rep. 2013, 3, 2088–2099. [Google Scholar] [CrossRef]
- Zhang, X.; Cruz, F.D.; Terry, M.; Remotti, F.; Matushansky, I. Terminal differentiation and loss of tumorigenicity of human cancers via pluripotency-based reprogramming. Oncogene 2013, 32, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M.; et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, H.; Nagano, H.; Haraguchi, N.; Nishikawa, S.; Tomokuni, A.; Kano, Y.; Fukusumi, T.; Saito, T.; Ozaki, M.; Sakai, D.; et al. Hypoxia and TP53 deficiency for induced pluripotent stem cell-like properties in gastrointestinal cancer. Int. J. Oncol. 2012, 40, 1423–1430. [Google Scholar] [CrossRef]
- Cahan, P.; Li, H.; Morris, S.A.; Lummertz da Rocha, E.; Daley, G.Q.; Collins, J.J. CellNet: Network biology applied to stem cell engineering. Cell 2014, 158, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Radley, A.H.; Schwab, R.M.; Tan, Y.; Kim, J.; Lo, E.K.W.; Cahan, P. Assessment of engineered cells using CellNet and RNA-seq. Nat. Protoc. 2017, 12, 1089–1102. [Google Scholar] [CrossRef]
- Morris, S.A.; Cahan, P.; Li, H.; Zhao, A.M.; San Roman, A.K.; Shivdasani, R.A.; Collins, J.J.; Daley, G.Q. Dissecting engineered cell types and enhancing cell fate conversion via CellNet. Cell 2014, 158, 889–902. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef]
- Nagy, A.; Munkacsy, G.; Gyorffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef]
- Lanczky, A.; Gyorffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- Margolin, A.A.; Wang, K.; Lim, W.K.; Kustagi, M.; Nemenman, I.; Califano, A. Reverse engineering cellular networks. Nat. Protoc. 2006, 1, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Margolin, A.A.; Califano, A. Theory and limitations of genetic network inference from microarray data. Ann. N. Y. Acad. Sci. 2007, 1115, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Rosvall, M.; Bergstrom, C.T. Maps of random walks on complex networks reveal community structure. Proc. Natl. Acad. Sci. USA 2008, 105, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Davidson, E.H.; Erwin, D.H. Gene regulatory networks and the evolution of animal body plans. Science 2006, 311, 796–800. [Google Scholar] [CrossRef]
- Mouse, E.C.; Stamatoyannopoulos, J.A.; Snyder, M.; Hardison, R.; Ren, B.; Gingeras, T.; Gilbert, D.M.; Groudine, M.; Bender, M.; Kaul, R.; et al. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol. 2012, 13, 418. [Google Scholar] [CrossRef]
- Xu, H.; Baroukh, C.; Dannenfelser, R.; Chen, E.Y.; Tan, C.M.; Kou, Y.; Kim, Y.E.; Lemischka, I.R.; Ma’ayan, A. ESCAPE: Database for integrating high-content published data collected from human and mouse embryonic stem cells. Database 2013, 2013, bat045. [Google Scholar] [CrossRef]
- Correa-Cerro, L.S.; Piao, Y.; Sharov, A.A.; Nishiyama, A.; Cadet, J.S.; Yu, H.; Sharova, L.V.; Xin, L.; Hoang, H.G.; Thomas, M.; et al. Generation of mouse ES cell lines engineered for the forced induction of transcription factors. Sci. Rep. 2011, 1, 167. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Jeanquartier, F.; Jean-Quartier, C.; Holzinger, A. Integrated web visualizations for protein-protein interaction databases. BMC Bioinform. 2015, 16, 195. [Google Scholar] [CrossRef]
- Li, R.; Campos, J.; Iida, J. A Gene Regulatory Program in Human Breast Cancer. Genetics 2015, 201, 1341–1348. [Google Scholar] [CrossRef]
- Bonhomme, C.; Duluc, I.; Martin, E.; Chawengsaksophak, K.; Chenard, M.P.; Kedinger, M.; Beck, F.; Freund, J.N.; Domon-Dell, C. The Cdx2 homeobox gene has a tumour suppressor function in the distal colon in addition to a homeotic role during gut development. Gut 2003, 52, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Hoflmayer, D.; Steinhoff, A.; Hube-Magg, C.; Kluth, M.; Simon, R.; Burandt, E.; Tsourlakis, M.C.; Minner, S.; Sauter, G.; Buscheck, F.; et al. Expression of CCCTC-binding factor (CTCF) is linked to poor prognosis in prostate cancer. Mol. Oncol. 2020, 14, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Taberlay, P.C.; Achinger-Kawecka, J.; Lun, A.T.; Buske, F.A.; Sabir, K.; Gould, C.M.; Zotenko, E.; Bert, S.A.; Giles, K.A.; Bauer, D.C.; et al. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016, 26, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Rhie, S.K.; Perez, A.A.; Lay, F.D.; Schreiner, S.; Shi, J.; Polin, J.; Farnham, P.J. A high-resolution 3D epigenomic map reveals insights into the creation of the prostate cancer transcriptome. Nat. Commun. 2019, 10, 4154. [Google Scholar] [CrossRef]
- Guo, Y.; Perez, A.A.; Hazelett, D.J.; Coetzee, G.A.; Rhie, S.K.; Farnham, P.J. CRISPR-mediated deletion of prostate cancer risk-associated CTCF loop anchors identifies repressive chromatin loops. Genome Biol. 2018, 19, 160. [Google Scholar] [CrossRef]
- Al Olama, A.A.; Kote-Jarai, Z.; Berndt, S.I.; Conti, D.V.; Schumacher, F.; Han, Y.; Benlloch, S.; Hazelett, D.J.; Wang, Z.; Saunders, E.; et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat. Genet. 2014, 46, 1103–1109. [Google Scholar] [CrossRef]
- Thomas, G.; Jacobs, K.B.; Yeager, M.; Kraft, P.; Wacholder, S.; Orr, N.; Yu, K.; Chatterjee, N.; Welch, R.; Hutchinson, A.; et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet. 2008, 40, 310–315. [Google Scholar] [CrossRef]
- Eeles, R.A.; Kote-Jarai, Z.; Al Olama, A.A.; Giles, G.G.; Guy, M.; Severi, G.; Muir, K.; Hopper, J.L.; Henderson, B.E.; Haiman, C.A.; et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat. Genet. 2009, 41, 1116–1121. [Google Scholar] [CrossRef]
- Berndt, S.I.; Wang, Z.; Yeager, M.; Alavanja, M.C.; Albanes, D.; Amundadottir, L.; Andriole, G.; Beane Freeman, L.; Campa, D.; Cancel-Tassin, G.; et al. Two susceptibility loci identified for prostate cancer aggressiveness. Nat. Commun. 2015, 6, 6889. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurup, J.T.; Kim, S.; Kidder, B.L. Identifying Cancer Type-Specific Transcriptional Programs through Network Analysis. Cancers 2023, 15, 4167. https://doi.org/10.3390/cancers15164167
Kurup JT, Kim S, Kidder BL. Identifying Cancer Type-Specific Transcriptional Programs through Network Analysis. Cancers. 2023; 15(16):4167. https://doi.org/10.3390/cancers15164167
Chicago/Turabian StyleKurup, Jiji T., Seongho Kim, and Benjamin L. Kidder. 2023. "Identifying Cancer Type-Specific Transcriptional Programs through Network Analysis" Cancers 15, no. 16: 4167. https://doi.org/10.3390/cancers15164167
APA StyleKurup, J. T., Kim, S., & Kidder, B. L. (2023). Identifying Cancer Type-Specific Transcriptional Programs through Network Analysis. Cancers, 15(16), 4167. https://doi.org/10.3390/cancers15164167