

Development of a Patient-Derived 3D Immuno-Oncology Platform to Potentiate Immunotherapy Responses in Ascites-Derived Circulating Tumor Cells

 , , , , , , ,
, , , , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Digital Spatial Profiling of the Tumor-Immune Microenvironment
2.2. Tumor Cell Culture Assays
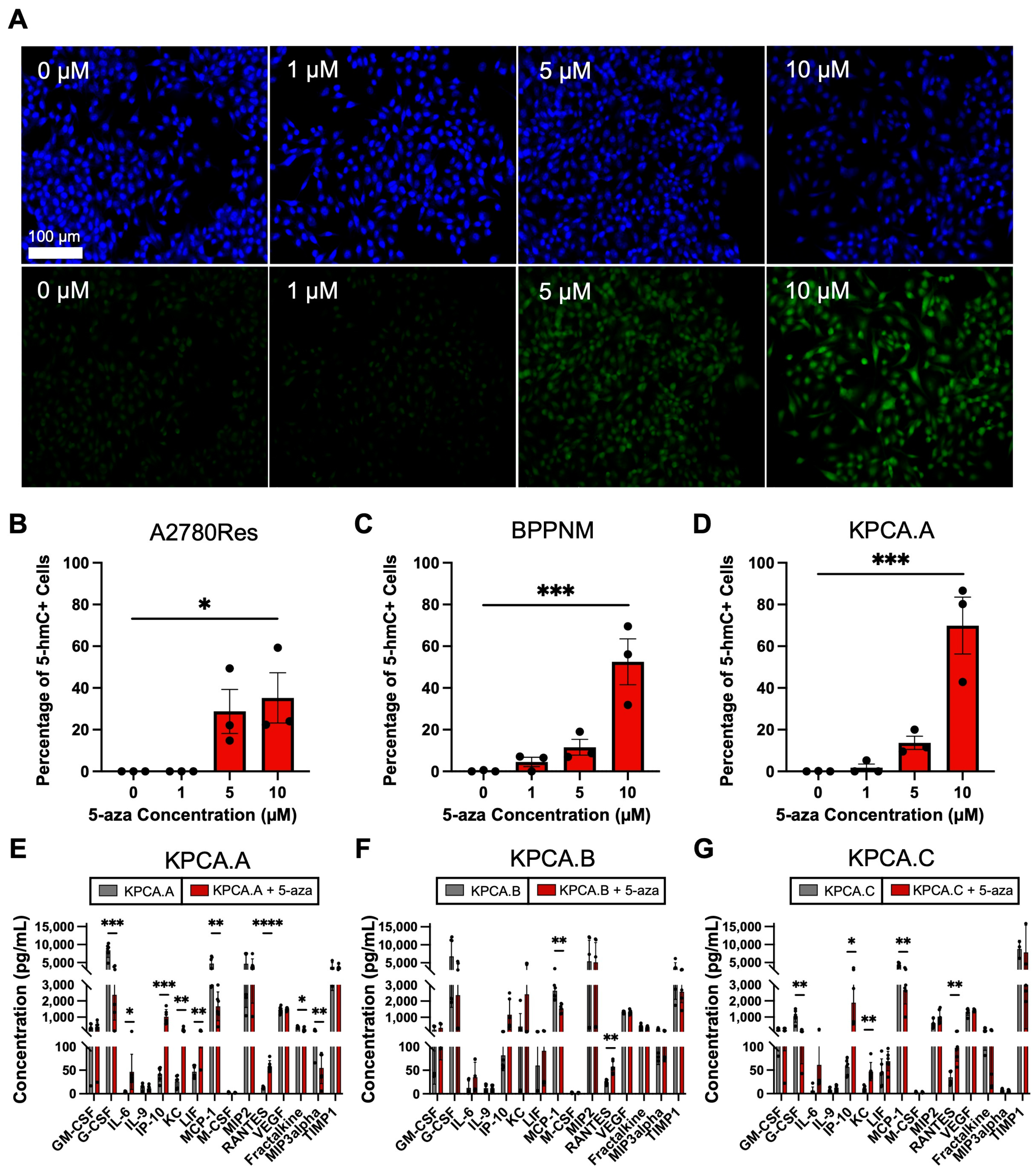
2.3. Assessment of Global Methylation (5-hmC) Levels in HGSOC
2.4. Tumor-Derived Secreted Cytokine/Chemokine Profiling in Response to DNMTIs
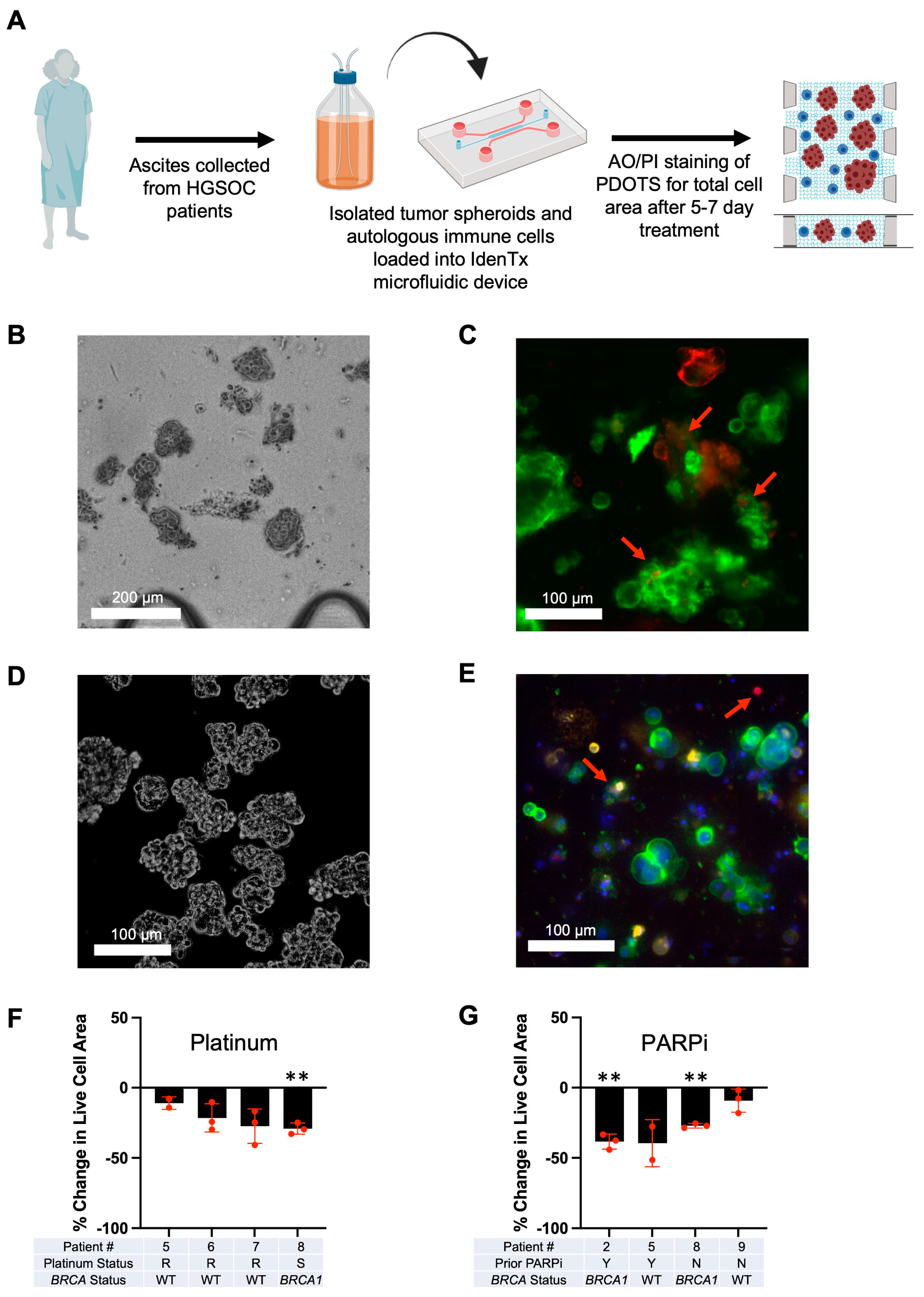
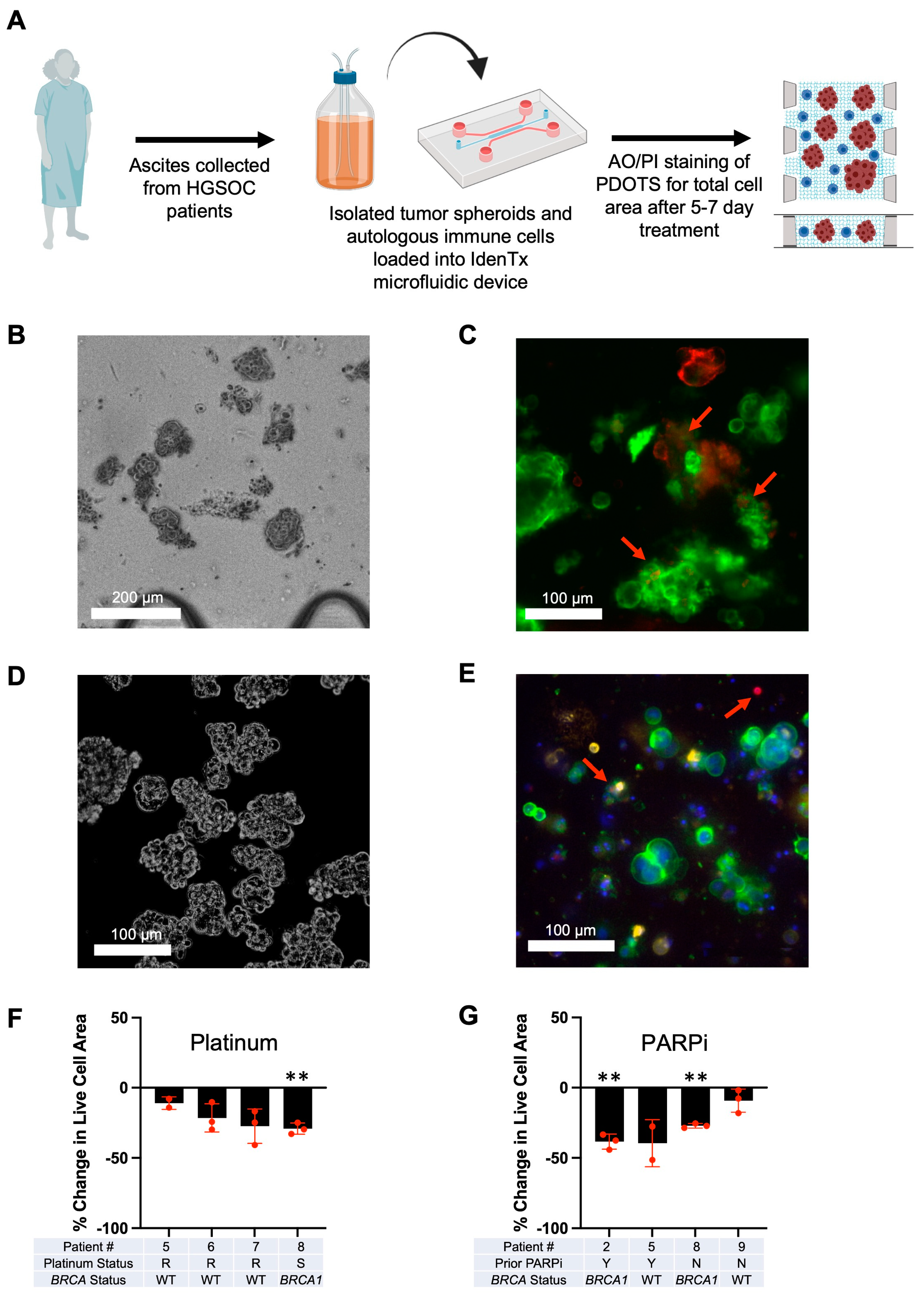
2.5. Optimization of Patient-Derived Organotypic HGSOC Spheroids (PDOTS) in 3D Microfluidic Devices
2.6. Immunofluorescent Imaging of PDOTS
2.7. Identification of Early Markers of Immune Activation in CD4+ and CD8+ T Cells
2.8. Flow Cytometry of Immune Cells
2.9. Assessment of Therapeutic Efficacy in Syngeneic HGSOC Models
2.10. Statistical Analysis
3. Results
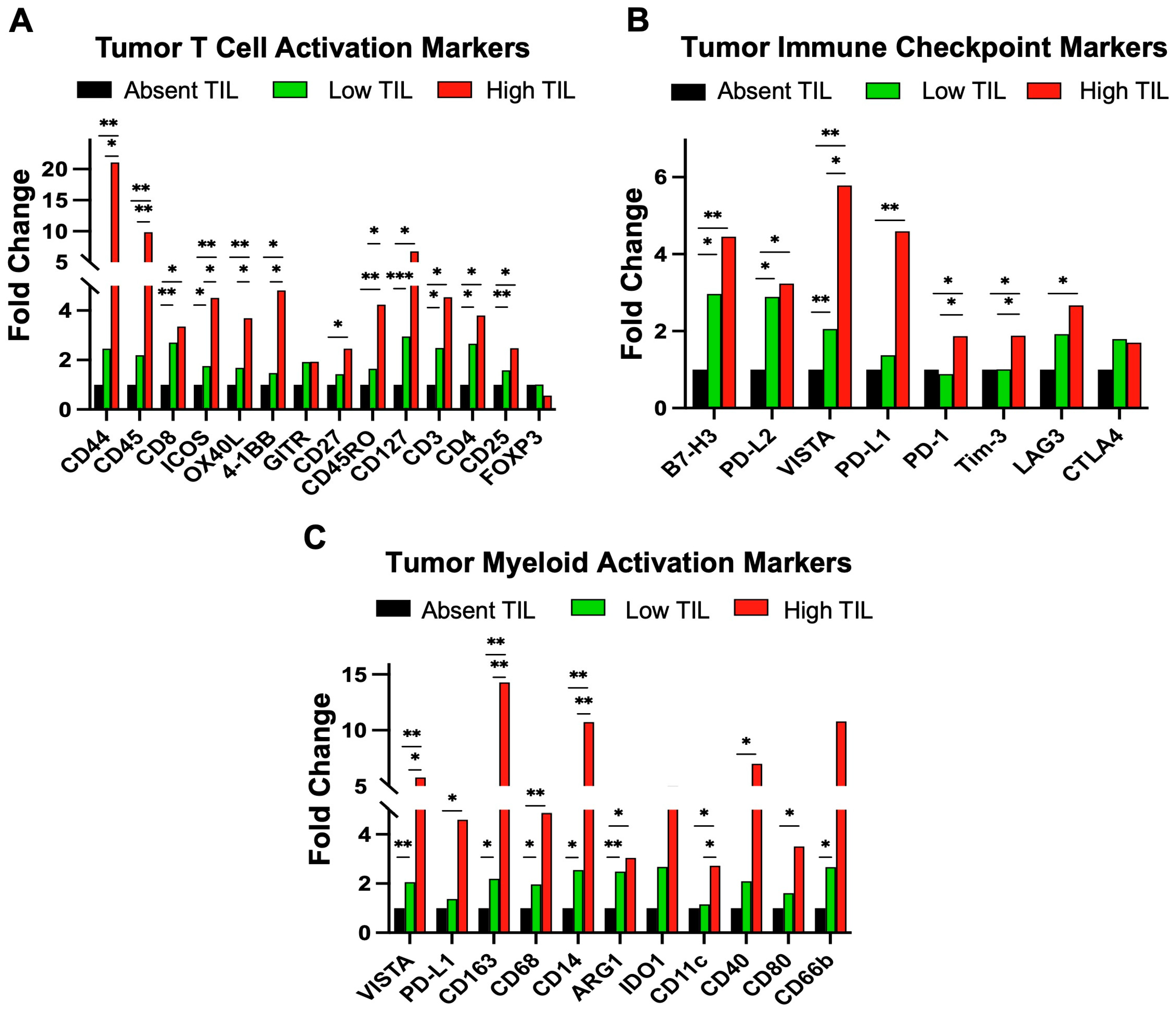
3.1. Digital Spatial Proteomic Profiling of Patient TILs in HGSOC
3.2. Epigenetic Priming Reverses Loss of 5-hmC and Mediates Upregulation of Key Tumor Cytokines
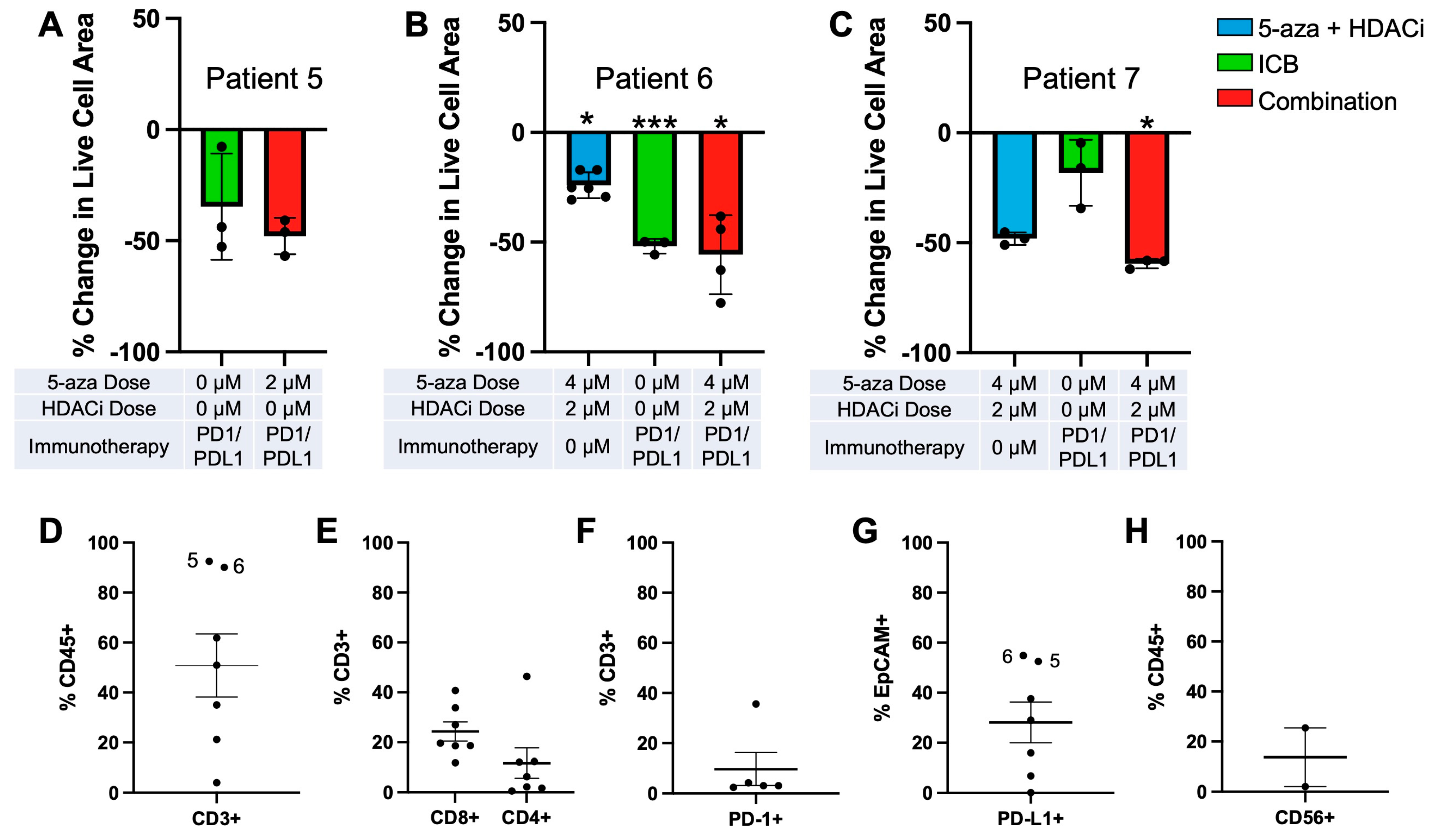
3.3. Optimization of Patient-Derived 3D Ex Vivo Platforms for Rapid Immunotherapeutic Testing of HGSOC Tumors and Ascites
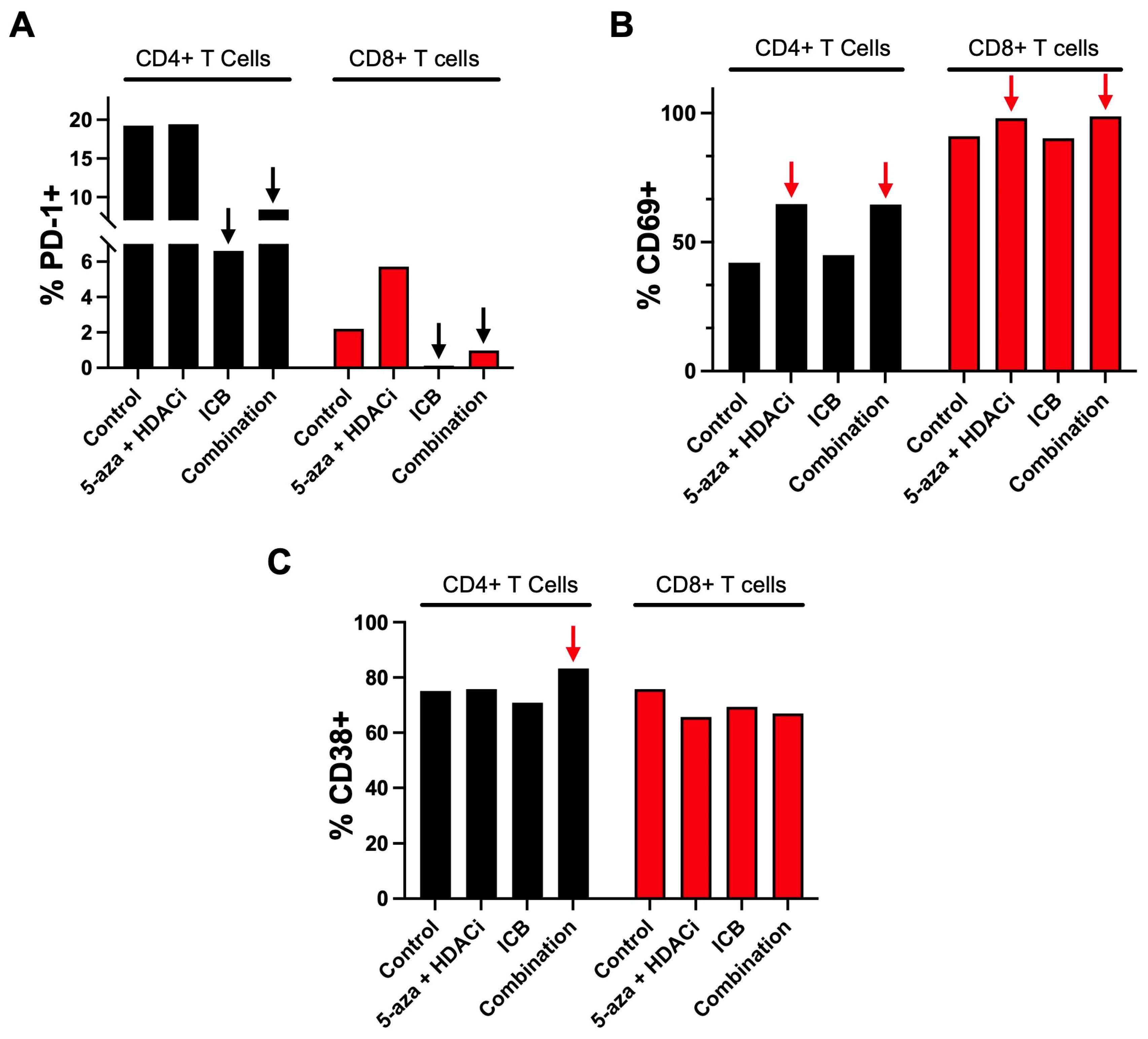
3.4. Epigenetic Priming Potentiates ICB in PDOTS and Upregulates Early Activation Immune T Cell Markers
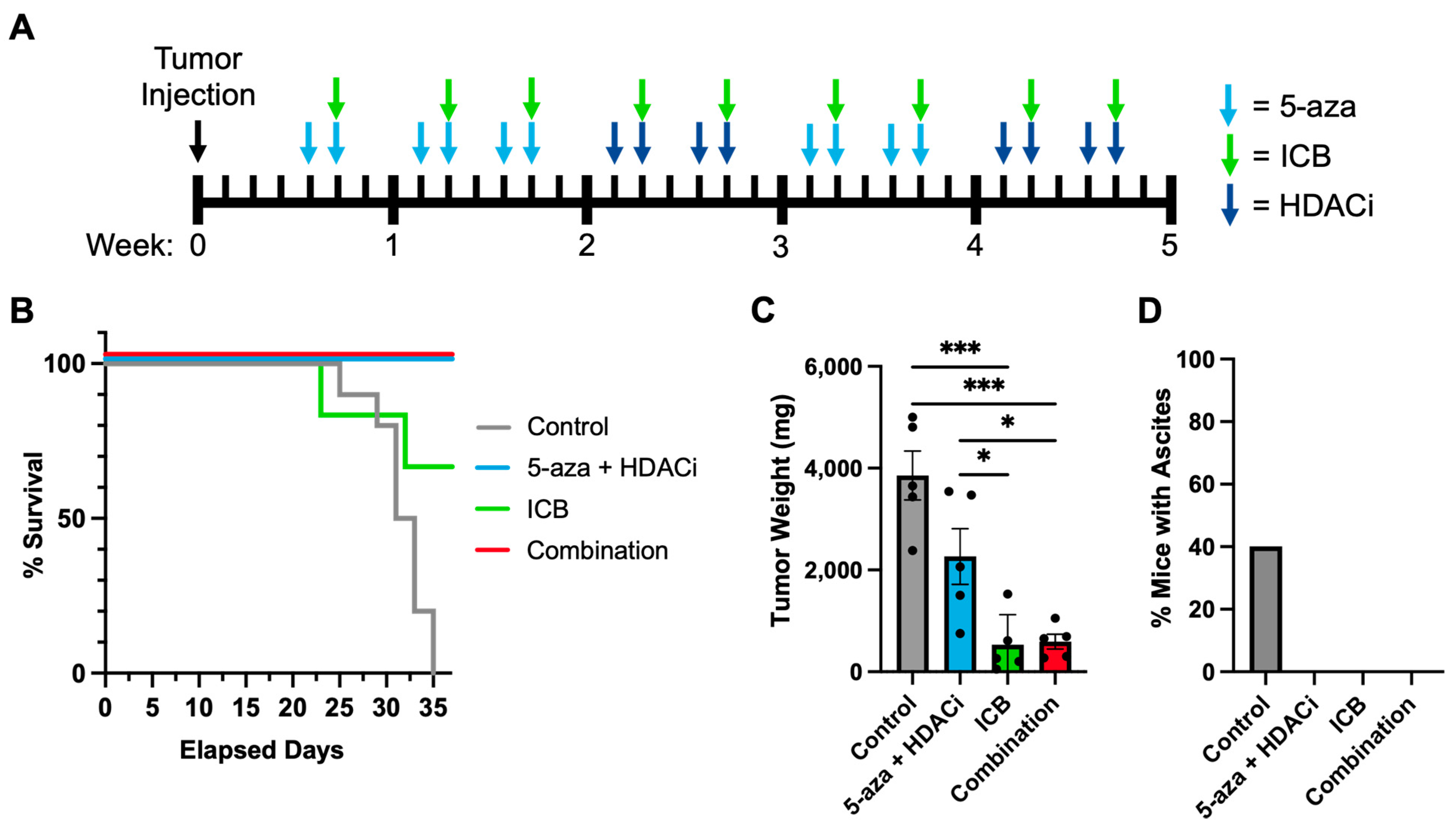
3.5. Epigenetic Priming Increases Overall Survival by Potentiating ICB in Syngeneic HGSOC Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, L.-Y.; Han, C.-S.; Li, P.-L.; Zhang, X.-C. 5-Hydroxymethylcytosine Expression Is Associated with Poor Survival in Cervical Squamous Cell Carcinoma. Jpn. J. Clin. Oncol. 2016, 46, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Houseman, E.A.; King, J.E.; von Herrmann, K.M.; Fadul, C.E.; Christensen, B.C. 5-Hydroxymethylcytosine Localizes to Enhancer Elements and Is Associated with Survival in Glioblastoma Patients. Nat. Commun. 2016, 7, 13177. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wu, K.; Ji, M.; Jin, W.; He, N.; Shi, B.; Hou, P. Decreased 5-Hydroxymethylcytosine (5-HmC) Is an Independent Poor Prognostic Factor in Gastric Cancer Patients. J. Biomed. Nanotechnol. 2013, 9, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J. Origin and Molecular Pathogenesis of Ovarian High-Grade Serous Carcinoma. Ann. Oncol. 2013, 24, x16–x21. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.R. Ovarian Cancer Update: Lessons From Morphology, Molecules, and Mice. Arch. Pathol. Lab. Med. 2009, 133, 1775–1781. [Google Scholar] [CrossRef]
- Cho, K.R.; Shih, I.-M. Ovarian Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 287–313. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–Genome Characterization of Chemoresistant Ovarian Cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Min, A.; Im, S.-A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti–PD-1 Antibody, Nivolumab, in Patients with Platinum-Resistant Ovarian Cancer. JCO 2015, 33, 4015–4022. [Google Scholar] [CrossRef] [PubMed]
- Palaia, I.; Tomao, F.; Sassu, C.M.; Musacchio, L.; Benedetti Panici, P. Immunotherapy For Ovarian Cancer: Recent Advances and Combination Therapeutic Approaches. OTT 2020, 13, 6109–6129. [Google Scholar] [CrossRef] [PubMed]
- Chardin, L.; Leary, A. Immunotherapy in Ovarian Cancer: Thinking Beyond PD-1/PD-L1. Front. Oncol. 2021, 11, 795547. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Odunsi, K.; Coukos, G. Immune Therapy Opportunities in Ovarian Cancer. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e228–e240. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 Pathway Blockade for Cancer Therapy: Mechanisms, Response Biomarkers, and Combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive Biomarkers for Checkpoint Inhibitor-Based Immunotherapy. The Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Wang, L.; Amoozgar, Z.; Huang, J.; Saleh, M.H.; Xing, D.; Orsulic, S.; Goldberg, M.S. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer Immunol. Res. 2015, 3, 1030–1041. [Google Scholar] [CrossRef]
- Wittenberger, T.; Sleigh, S.; Reisel, D.; Zikan, M.; Wahl, B.; Alunni-Fabbroni, M.; Jones, A.; Evans, I.; Koch, J.; Paprotka, T.; et al. DNA Methylation Markers for Early Detection of Women’s Cancer: Promise and Challenges. Epigenomics 2014, 6, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Wu, H.; Zhang, H.; Lian, C.G.; Lu, Q. DNA Methylation/Hydroxymethylation in Melanoma. Oncotarget 2017, 8, 78163–78173. [Google Scholar] [CrossRef]
- Goode, E.L.; Block, M.S.; Kalli, K.R.; Vierkant, R.A.; Chen, W.; Fogarty, Z.C.; Gentry-Maharaj, A.; Toloczko, A.; Hein, A.; Bouligny, A.L.; et al. Dose-Response Association of CD8 + Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol. 2017, 3, e173290. [Google Scholar] [CrossRef] [PubMed]
- Vilain, R.E.; Menzies, A.M.; Wilmott, J.S.; Kakavand, H.; Madore, J.; Guminski, A.; Liniker, E.; Kong, B.Y.; Cooper, A.J.; Howle, J.R.; et al. Dynamic Changes in PD-L1 Expression and Immune Infiltrates Early During Treatment Predict Response to PD-1 Blockade in Melanoma. Clin. Cancer Res. 2017, 23, 5024–5033. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.J.; Shklovskaya, E.; Hersey, P. Epigenetic Modulation in Cancer Immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 48–56. [Google Scholar] [CrossRef]
- Matei, D.; Fang, F.; Shen, C.; Schilder, J.; Arnold, A.; Zeng, Y.; Berry, W.A.; Huang, T.; Nephew, K.P. Epigenetic Resensitization to Platinum in Ovarian Cancer. Cancer Res. 2012, 72, 2197–2205. [Google Scholar] [CrossRef]
- Wang, Y.; Cardenas, H.; Fang, F.; Condello, S.; Taverna, P.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. Epigenetic Targeting of Ovarian Cancer Stem Cells. Cancer Res. 2014, 74, 4922–4936. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.; Rao, S. Epigenetics and Immunotherapy: The Current State of Play. Mol. Immunol. 2017, 87, 227–239. [Google Scholar] [CrossRef]
- Peixoto, P.; Renaude, E.; Boyer-Guittaut, M.; Hervouet, E. Epigenetics, a Key Player of Immunotherapy Resistance. CDR 2018, 1, 219–229. [Google Scholar] [CrossRef]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of Metastatic Mouse Cancers Resistant to Immune Checkpoint Blockade by Suppression of Myeloid-Derived Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef]
- Oda, K.; Hamanishi, J.; Matsuo, K.; Hasegawa, K. Genomics to Immunotherapy of Ovarian Clear Cell Carcinoma: Unique Opportunities for Management. Gynecol. Oncol. 2018, 151, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.H.; Wang, L.; Goldberg, M.S. Improving Cancer Immunotherapy with DNA Methyltransferase Inhibitors. Cancer Immunol. Immunother. 2016, 65, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8 + Tumor-Infiltrating Lymphocytes and a High CD8 + /Regulatory T Cell Ratio Are Associated with Favorable Prognosis in Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch Repair Deficiency/Microsatellite Instability-High as a Predictor for Anti-PD-1/PD-L1 Immunotherapy Efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M.; Galaine, J.; Guenat, D.; Mougey, V.; Perrard, J.; et al. PD-L1 Expression Is Regulated by Both DNA Methylation and NF-KB during EMT Signaling in Non-Small Cell Lung Carcinoma. OncoImmunology 2018, 7, e1423170. [Google Scholar] [CrossRef]
- Emran, A.A.; Chatterjee, A.; Rodger, E.J.; Tiffen, J.C.; Gallagher, S.J.; Eccles, M.R.; Hersey, P. Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends Immunol. 2019, 40, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ren, S.; Chen, Y.; Liu, A.; Wu, Q.; Jiang, T.; Lv, P.; Song, D.; Hu, F.; Lan, J.; et al. PD-L1 Methylation Restricts PD-L1/PD-1 Interactions to Control Cancer Immune Surveillance. Sci. Adv. 2023, 9, eade4186. [Google Scholar] [CrossRef]
- Tucker, D.W.; Getchell, C.R.; McCarthy, E.T.; Ohman, A.W.; Sasamoto, N.; Xu, S.; Ko, J.Y.; Gupta, M.; Shafrir, A.; Medina, J.E.; et al. Epigenetic Reprogramming Strategies to Reverse Global Loss of 5-Hydroxymethylcytosine, a Prognostic Factor for Poor Survival in High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2018, 24, 1389–1401. [Google Scholar] [CrossRef]
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.-M.; Wang, T.-L.; et al. Epigenetic Therapy Activates Type I Interferon Signaling in Murine Ovarian Cancer to Reduce Immunosuppression and Tumor Burden. Proc. Natl. Acad. Sci. USA 2017, 114, E10981–E10990. [Google Scholar] [CrossRef]
- Iyer, S.; Zhang, S.; Yucel, S.; Horn, H.; Smith, S.G.; Reinhardt, F.; Hoefsmit, E.; Assatova, B.; Casado, J.; Meinsohn, M.-C.; et al. Genetically Defined Syngeneic Mouse Models of Ovarian Cancer as Tools for the Discovery of Combination Immunotherapy. Cancer Discov. 2021, 11, 384–407. [Google Scholar] [CrossRef] [PubMed]
- Campisi, M.; Shelton, S.E.; Chen, M.; Kamm, R.D.; Barbie, D.A.; Knelson, E.H. Engineered Microphysiological Systems for Testing Effectiveness of Cell-Based Cancer Immunotherapies. Cancers 2022, 14, 3561. [Google Scholar] [CrossRef] [PubMed]
- Aref, A.R.; Campisi, M.; Ivanova, E.; Portell, A.; Larios, D.; Piel, B.P.; Mathur, N.; Zhou, C.; Coakley, R.V.; Bartels, A.; et al. 3D Microfluidic Ex Vivo Culture of Organotypic Tumor Spheroids to Model Immune Checkpoint Blockade. Lab Chip 2018, 18, 3129–3143. [Google Scholar] [CrossRef]
- Knelson, E.H.; Ivanova, E.V.; Tarannum, M.; Campisi, M.; Lizotte, P.H.; Booker, M.A.; Ozgenc, I.; Noureddine, M.; Meisenheimer, B.; Chen, M.; et al. Activation of Tumor-Cell STING Primes NK-Cell Therapy. Cancer Immunol. Res. 2022, 10, 947–961. [Google Scholar] [CrossRef]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex Digital Spatial Profiling of Proteins and RNA in Fixed Tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; He, X.; Wang, Q. Immune Checkpoint Blockades in Gynecological Cancers: A Review of Clinical Trials. Acta Obstet. Gynecol. Scand. 2022, 101, 941–951. [Google Scholar] [CrossRef]
- Leary, A.; Tan, D.; Ledermann, J. Immune Checkpoint Inhibitors in Ovarian Cancer: Where Do We Stand? Ther. Adv. Med. Oncol. 2021, 13, 175883592110398. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef]
- Castellanos, J.R.; Purvis, I.J.; Labak, C.M.; Guda, M.R.; Tsung, A.J.; Velpula, K.K.; Asuthkar, S. B7-H3 Role in the Immune Landscape of Cancer. Am. J. Clin. Exp. Immunol. 2017, 6, 66–75. [Google Scholar]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T Cell Exhaustion: From Pathophysiological Basics to Tumor Immunotherapy. Cell Commun. Signal. 2017, 15, 1. [Google Scholar] [CrossRef]
- Horn, L.A.; Long, T.M.; Atkinson, R.; Clements, V.; Ostrand-Rosenberg, S. Soluble CD80 Protein Delays Tumor Growth and Promotes Tumor-Infiltrating Lymphocytes. Cancer Immunol. Res. 2018, 6, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Amobi-McCloud, A.; Muthuswamy, R.; Battaglia, S.; Yu, H.; Liu, T.; Wang, J.; Putluri, V.; Singh, P.K.; Qian, F.; Huang, R.-Y.; et al. IDO1 Expression in Ovarian Cancer Induces PD-1 in T Cells via Aryl Hydrocarbon Receptor Activation. Front. Immunol. 2021, 12, 678999. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 Pathway in Cancer: From Bench to Bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Saxena, S.; Singh, R.K. Neutrophils in the Tumor Microenvironment. In Tumor Microenvironment; Birbrair, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; Volume 1224, pp. 1–20. ISBN 978-3-030-35722-1. [Google Scholar]
- Templeton, A.J.; McNamara, M.G.; Šeruga, B.; Vera-Badillo, F.E.; Aneja, P.; Ocaña, A.; Leibowitz-Amit, R.; Sonpavde, G.; Knox, J.J.; Tran, B.; et al. Prognostic Role of Neutrophil-to-Lymphocyte Ratio in Solid Tumors: A Systematic Review and Meta-Analysis. JNCI J. Natl. Cancer Inst. 2014, 106, dju124. [Google Scholar] [CrossRef] [PubMed]
- Kwiecień, I.; Rutkowska, E.; Raniszewska, A.; Rzepecki, P.; Domagała-Kulawik, J. Modulation of the Immune Response by Heterogeneous Monocytes and Dendritic Cells in Lung Cancer. WJCO 2021, 12, 966–982. [Google Scholar] [CrossRef]
- Osugi, Y.; Vuckovic, S.; Hart, D.N.J. Myeloid Blood CD11c+ Dendritic Cells and Monocyte-Derived Dendritic Cells Differ in Their Ability to Stimulate T Lymphocytes. Blood 2002, 100, 2858–2866. [Google Scholar] [CrossRef]
- Kim, C.W.; Kim, K.-D.; Lee, H.K. The Role of Dendritic Cells in Tumor Microenvironments and Their Uses as Therapeutic Targets. BMB Rep. 2021, 54, 31–43. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Son, D.-S.; Parl, A.K.; Montgomery Rice, V.; Khabele, D. Keratinocyte Chemoattractant (KC)/Human Growth-Regulated Oncogene (GRO) Chemokines and pro-Inflammatory Chemokine Networks in Mouse and Human Ovarian Epithelial Cancer Cells. Cancer Biol. Ther. 2007, 6, 1308–1318. [Google Scholar] [CrossRef]
- Lunardi, S.; Lim, S.Y.; Muschel, R.J.; Brunner, T.B. IP-10/CXCL10 Attracts Regulatory T Cells: Implication for Pancreatic Cancer. OncoImmunology 2015, 4, e1027473. [Google Scholar] [CrossRef]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765. [Google Scholar] [CrossRef]
- Crawford, A.; Angelosanto, J.M.; Nadwodny, K.L.; Blackburn, S.D.; Wherry, E.J. A Role for the Chemokine RANTES in Regulating CD8 T Cell Responses during Chronic Viral Infection. PLoS Pathog. 2011, 7, e1002098. [Google Scholar] [CrossRef]
- Kumar, J.; Fraser, F.W.; Riley, C.; Ahmed, N.; McCulloch, D.R.; Ward, A.C. Granulocyte Colony-Stimulating Factor Receptor Signalling via Janus Kinase 2/Signal Transducer and Activator of Transcription 3 in Ovarian Cancer. Br. J. Cancer 2014, 110, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Alvero, A.B.; Montagna, M.K.; Craveiro, V.; Liu, L.; Mor, G. Distinct Subpopulations of Epithelial Ovarian Cancer Cells Can Differentially Induce Macrophages and T Regulatory Cells Toward a Pro-Tumor Phenotype: IMMUNE REGULATION BY OVARIAN CANCER CELLS. Am. J. Reprod. Immunol. 2012, 67, 256–265. [Google Scholar] [CrossRef]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, Regulation, and Involvement in Disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yang, F.; Zhang, F.; Guo, D.; Li, L.; Wang, X.; Liang, T.; Wang, J.; Cai, Z.; Jin, H. CD69 Enhances Immunosuppressive Function of Regulatory T-Cells and Attenuates Colitis by Prompting IL-10 Production. Cell Death Dis. 2018, 9, 905. [Google Scholar] [CrossRef]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From Activation Marker to Metabolic Gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Montes, C.; Santos-Argumedo, L. CD38 Is Expressed Selectively during the Activation of a Subset of Mature T Cells with Reduced Proliferation but Improved Potential to Produce Cytokines. J. Leukoc. Biol. 2004, 77, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xiong, C.; Hsu, Y.-C.; Wang, X.; Chen, L. Human Leukocyte Antigen (HLA) and Cancer Immunotherapy: HLA-Dependent and -Independent Adoptive Immunotherapies. Ann. Blood 2020, 5, 14. [Google Scholar] [CrossRef]
- Ford, C.E.; Werner, B.; Hacker, N.F.; Warton, K. The Untapped Potential of Ascites in Ovarian Cancer Research and Treatment. Br. J. Cancer 2020, 123, 9–16. [Google Scholar] [CrossRef]
- Rickard, B.P.; Conrad, C.; Sorrin, A.J.; Ruhi, M.K.; Reader, J.C.; Huang, S.A.; Franco, W.; Scarcelli, G.; Polacheck, W.J.; Roque, D.M.; et al. Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response. Cancers 2021, 13, 4318. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerton, T.J.; Green, A.; Campisi, M.; Chen, M.; Gjeci, I.; Mahadevan, N.; Lee, C.A.A.; Mishra, R.; Vo, H.V.; Haratani, K.; et al. Development of a Patient-Derived 3D Immuno-Oncology Platform to Potentiate Immunotherapy Responses in Ascites-Derived Circulating Tumor Cells. Cancers 2023, 15, 4128. https://doi.org/10.3390/cancers15164128
Gerton TJ, Green A, Campisi M, Chen M, Gjeci I, Mahadevan N, Lee CAA, Mishra R, Vo HV, Haratani K, et al. Development of a Patient-Derived 3D Immuno-Oncology Platform to Potentiate Immunotherapy Responses in Ascites-Derived Circulating Tumor Cells. Cancers. 2023; 15(16):4128. https://doi.org/10.3390/cancers15164128
Chicago/Turabian StyleGerton, Thomas J., Allen Green, Marco Campisi, Minyue Chen, Iliana Gjeci, Navin Mahadevan, Catherine A. A. Lee, Ranjan Mishra, Ha V. Vo, Koji Haratani, and et al. 2023. "Development of a Patient-Derived 3D Immuno-Oncology Platform to Potentiate Immunotherapy Responses in Ascites-Derived Circulating Tumor Cells" Cancers 15, no. 16: 4128. https://doi.org/10.3390/cancers15164128
APA StyleGerton, T. J., Green, A., Campisi, M., Chen, M., Gjeci, I., Mahadevan, N., Lee, C. A. A., Mishra, R., Vo, H. V., Haratani, K., Li, Z.-H., Hasselblatt, K. T., Testino, B., Connor, T., Lian, C. G., Elias, K. M., Lizotte, P., Ivanova, E. V., Barbie, D. A., & Dinulescu, D. M. (2023). Development of a Patient-Derived 3D Immuno-Oncology Platform to Potentiate Immunotherapy Responses in Ascites-Derived Circulating Tumor Cells. Cancers, 15(16), 4128. https://doi.org/10.3390/cancers15164128