




L-Pampo™, a Novel TLR2/3 Agonist, Acts as a Potent Cancer Vaccine Adjuvant by Activating Draining Lymph Node Dendritic Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cancer Cell Lines
2.3. Generation and Activation of Mouse Bone-Marrow-Derived Dendritic Cells (BMDCs)
2.4. Adjuvant Formulation and Immunization
2.5. Isolation of Lymph Node Cells from Inguinal Lymph Nodes
2.6. Flow Cytometry
2.7. T-Cell Proliferation Assay
2.8. ELISA Assay
2.9. Enzyme-Linked Immunospot (ELISpot) Assay
2.10. Tumor Models and Treatment
2.11. Statistical Analysis
3. Results
3.1. L-Pampo™ Promotes BMDC Maturation and T-Cell Proliferation
3.2. L-Pampo™ Promotes the Recruitment and Activation of DCs in Draining Lymph Nodes
3.3. L-Pampo™ Increases DC Subsets in Draining Lymph Nodes
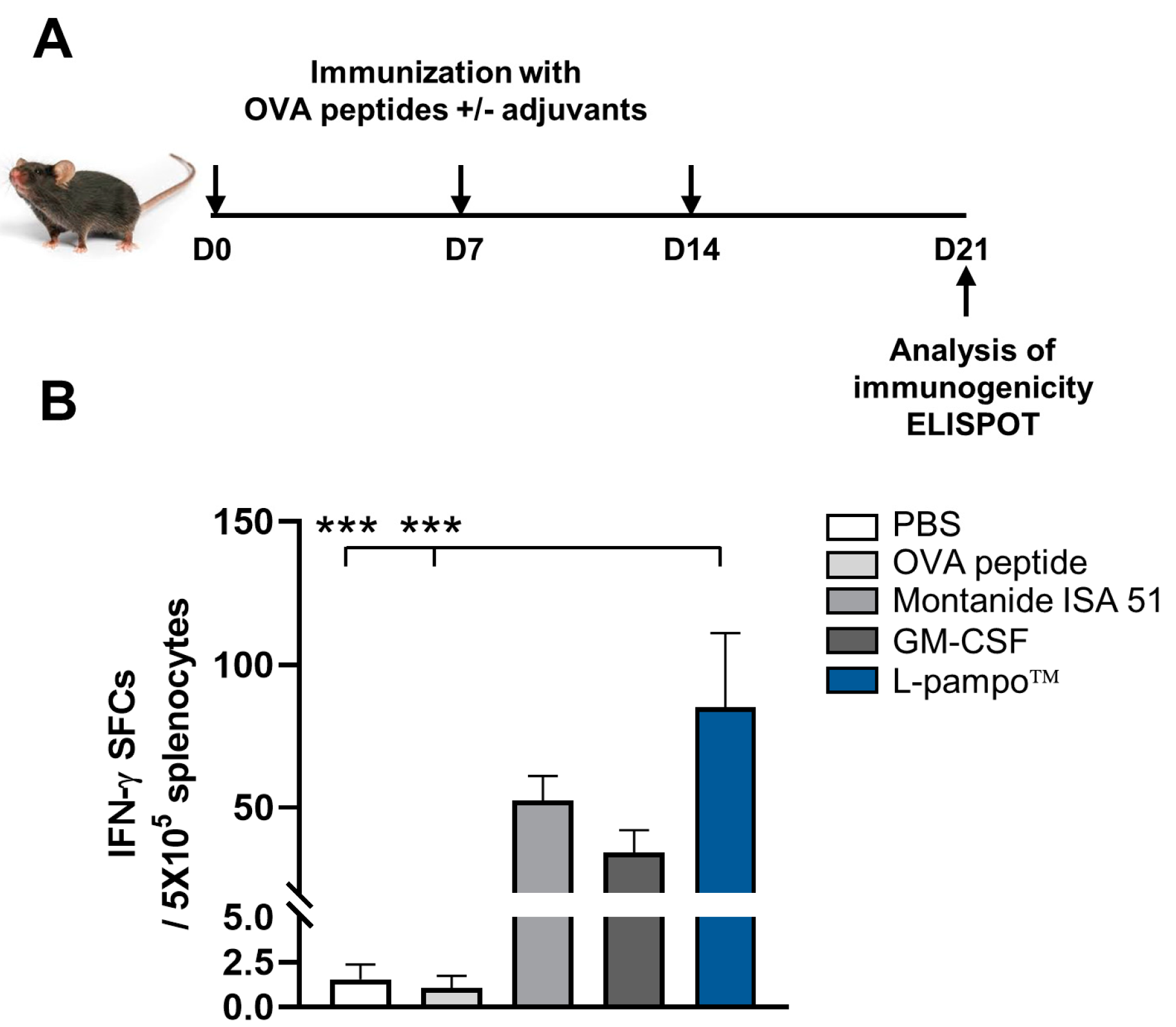
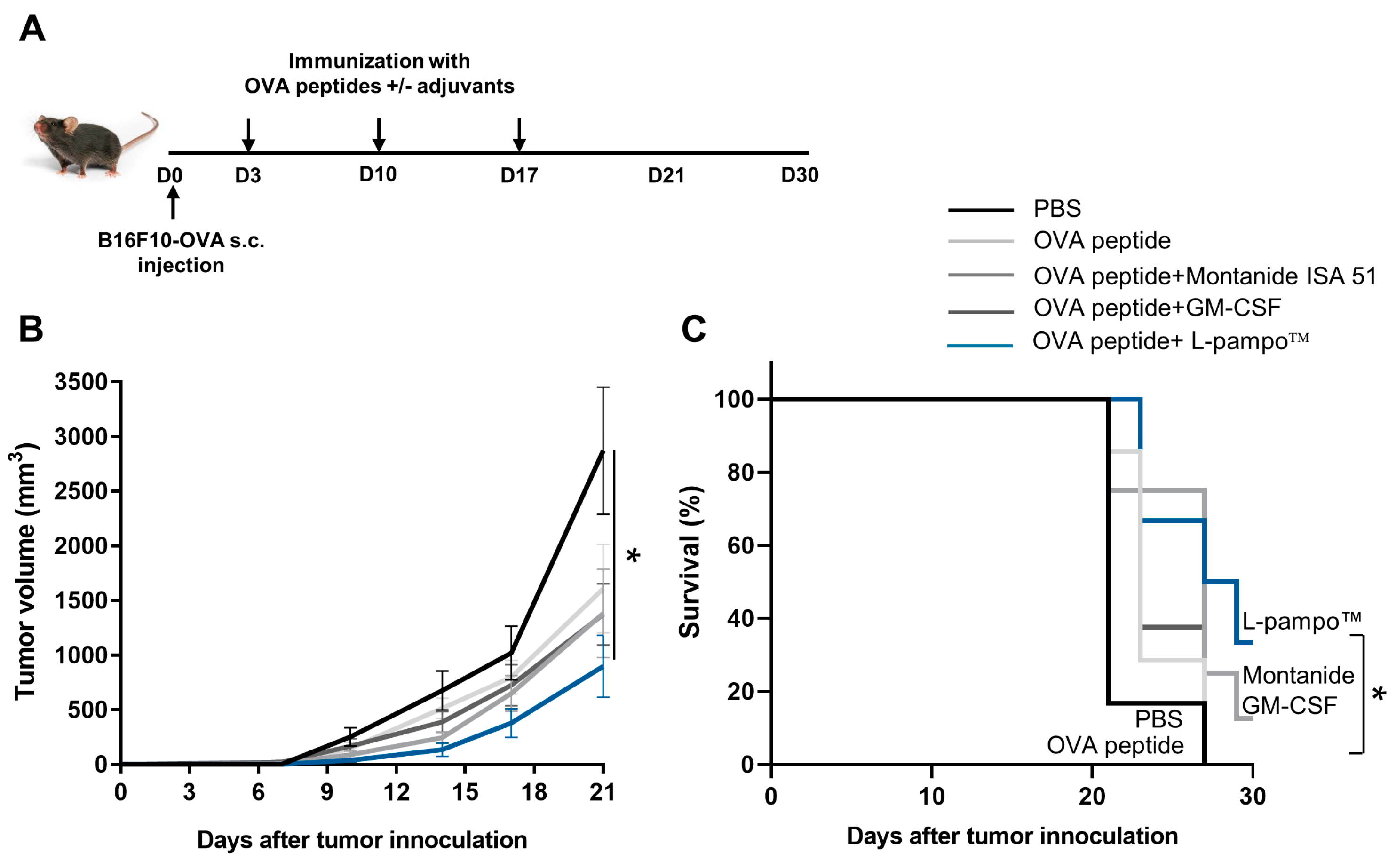
3.4. L-Pampo™ Increases OVA Peptide-Specific T-Cell Responses and Inhibits Tumor Growth
3.5. Combination of OVA Admixed with L-Pampo™ with Immune Checkpoint Inhibitors Enhances Antigen-Specific T-Cell Response and Anti-Tumor Efficacy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kirkwood, J.M.; Butterfield, L.H.; Tarhini, A.A.; Zarour, H.; Kalinski, P.; Ferrone, S. Immunotherapy of cancer in 2012. CA A Cancer J. Clin. 2012, 62, 309–335. [Google Scholar] [CrossRef] [PubMed]
- Melief, C.J.M.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Abd-Aziz, N.; Poh, C.L. Development of Peptide-Based Vaccines for Cancer. J. Oncol. 2022, 2022, 9749363. [Google Scholar] [CrossRef] [PubMed]
- Paston, S.J.; Brentville, V.A.; Symonds, P.; Durrant, L.G. Cancer Vaccines, Adjuvants, and Delivery Systems. Front. Immunol. 2021, 12, 627932. [Google Scholar] [CrossRef]
- van Doorn, E.; Liu, H.; Huckriede, A.; Hak, E. Safety and tolerability evaluation of the use of Montanide ISA (TM) 51 as vaccine adjuvant: A systematic review. Hum. Vaccines Immunother. 2016, 12, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Khani, A.T.; Ortiz, A.S.; Swaminathan, S. GM-CSF: A Double-Edged Sword in Cancer Immunotherapy. Front. Immunol. 2022, 13, 901277. [Google Scholar] [CrossRef]
- MacLeod, M.K.L.; McKee, A.S.; David, A.; Wang, J.R.; Mason, R.; Kappler, J.W.; Marrack, P. Vaccine adjuvants aluminum and monophosphoryl lipid A provide distinct signals to generate protective cytotoxic memory CD8 T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 7914–7919. [Google Scholar] [CrossRef]
- Gan, J.Y.; Du, G.S.; He, C.T.; Jiang, M.; Mou, X.Y.; Xue, J.; Sun, X. Tumor cell membrane enveloped aluminum phosphate nanoparticles for enhanced cancer vaccination. J. Control. Release 2020, 326, 297–309. [Google Scholar] [CrossRef]
- Luchner, M.; Reinke, S.; Milicic, A. TLR Agonists as Vaccine Adjuvants Targeting Cancer and Infectious Diseases. Pharmaceutics 2021, 13, 142. [Google Scholar] [CrossRef]
- Lee, B.R.; Jeong, S.K.; Ahn, B.C.; Lee, B.J.; Shin, S.J.; Yum, J.S.; Ha, S.J. Combination of TLR1/2 and TLR3 ligands enhances CD4(+) T cell longevity and antibody responses by modulating type I IFN production. Sci. Rep. 2016, 6, 32526. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.K.; Heo, Y.K.; Jeong, J.H.; Ham, S.J.; Yum, J.S.; Ahn, B.C.; Song, C.S.; Chun, E.Y. COVID-19 Subunit Vaccine with a Combination of TLR1/2 and TLR3 Agonists Induces Robust and Protective Immunity. Vaccines 2021, 9, 957. [Google Scholar] [CrossRef]
- Lee, W.S.; Kim, D.S.; Kim, J.H.; Heo, Y.; Yang, H.; Go, E.J.; Kim, J.H.; Lee, S.J.; Ahn, B.C.; Yum, J.S.; et al. Intratumoral immunotherapy using a TLR2/3 agonist, L-pampo, induces robust antitumor immune responses and enhances immune checkpoint blockade. J. Immunother. Cancer 2022, 10, e004799. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Salvi, V.; Soriani, A.; Laffranchi, M.; Sozio, F.; Bosisio, D.; Sozzani, S. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell. Mol. Immunol. 2023, 20, 432–447. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.Y.; Li, Z.T.; Jaklenec, A.; Hu, Q.Y. Vaccine delivery systems toward lymph nodes. Adv. Drug Deliv. Rev. 2021, 179, 113914. [Google Scholar] [CrossRef]
- Clancy-Thompson, E.; King, L.K.; Nunnley, L.D.; Mullins, I.M.; Slingluff, C.L., Jr.; Mullins, D.W. Peptide vaccination in montanide adjuvant induces and GM-CSF increases CXCR3 and cutaneous lymphocyte antigen expression by tumor antigen–specific CD8 T cells. Cancer Immunol. Res. 2013, 1, 332–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8(+) T cell responses through CD8 alpha(+) dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef]
- Manh, T.P.V.; Bertho, N.; Hosmalin, A.; Schwartz-Comil, I.; Dalod, M. Investigating evolutionary conservation of dendritic cell subset identity and functions. Front. Immunol. 2015, 6, 260. [Google Scholar] [CrossRef] [Green Version]
- Cancel, J.C.; Crozat, K.; Dalod, M.; Mattiuz, R. Are Conventional Type 1 Dendritic Cells Critical for Protective Antitumor Immunity and How? Front. Immunol. 2019, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Franzin, R.; Netti, G.S.; Spadaccino, F.; Porta, C.; Gesualdo, L.; Stallone, G.; Castellano, G.; Ranieri, E. The Use of Immune Checkpoint Inhibitors in Oncology and the Occurrence of AKI: Where Do We Stand? Front. Immunol. 2020, 11, 574271. [Google Scholar] [CrossRef]
- Marei, H.E.; Hasan, A.; Pozzoli, G.; Cenciarelli, C. Cancer immunotherapy with immune checkpoint inhibitors (ICIs): Potential, mechanisms of resistance, and strategies for reinvigorating T cell responsiveness when resistance is acquired. Cancer Cell Int. 2023, 23, 64. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.G.; Sang, Y.B.; Lee, J.H.; Chon, H.J. Combining Cancer Vaccines with Immunotherapy: Establishing a New Immunological Approach. Int. J. Mol. Sci. 2021, 22, 8035. [Google Scholar] [CrossRef] [PubMed]
- Hailemichael, Y.; Dai, Z.M.; Jaffarzad, N.; Ye, Y.; Medina, M.A.; Huang, X.F.; Dorta-Estremera, S.M.; Greeley, N.R.; Nitti, G.; Peng, W.Y.; et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat. Med. 2013, 19, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Salerno, E.P.; Shea, S.M.; Olson, W.C.; Petroni, G.R.; Smolkin, M.E.; McSkimming, C.; Chianese-Bullock, K.A.; Slingluff, C.L. Activation, dysfunction and retention of T cells in vaccine sites after injection of incomplete Freund’s adjuvant, with or without peptide. Cancer Immunol. Immunother. 2013, 62, 1149–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunsvig, P.F.; Guren, T.K.; Nyakas, M.; Steinfeldt-Reisse, C.H.; Rasch, W.; Kyte, J.A.; Juul, H.V.; Aamdal, S.; Gaudernack, G.; Inderberg, E.M. Long-Term Outcomes of a Phase I Study with UV1, a Second Generation Telomerase Based Vaccine, in Patients With Advanced Non-Small Cell Lung Cancer. Front. Immunol. 2020, 11, 572172. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.L.; Zhang, S.S.; Han, N.; Jiang, J.H.; Xu, Y.Y.; Ma, D.Y.; Lu, L.T.; Guo, X.J.; Qiu, M.; Huang, Q.X.; et al. A Neoantigen-Based Peptide Vaccine for Patients With Advanced Pancreatic Cancer Refractory to Standard Treatment. Front. Immunol. 2021, 12, 691605. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Ardavanis, A.; Litton, J.K.; Shumway, N.M.; Hale, D.F.; Murray, J.L.; Perez, S.A.; Ponniah, S.; Baxevanis, C.N.; Papamichail, M. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide GP2 vaccine in breast cancer patients to prevent recurrence. Oncotarget 2016, 7, 66192. [Google Scholar] [CrossRef] [Green Version]
- Gouttefangeas, C.; Rammensee, H.G. Personalized cancer vaccines: Adjuvants are important, too. Cancer Immunol. Immunother. 2018, 67, 1911–1918. [Google Scholar] [CrossRef]
- Gaudreau, S.; Guindi, C.; Menard, M.; Benabdallah, A.; Dupuis, G.; Amrani, A. GM-CSF induces bone marrow precursors of NOD mice to skew into tolerogenic dendritic cells that protect against diabetes. Cell. Immunol. 2010, 265, 31–36. [Google Scholar] [CrossRef]
- Zhao, W.D.; Zhao, G.; Wang, B. Revisiting GM-CSF as an adjuvant for therapeutic vaccines. Cell. Mol. Immunol. 2018, 15, 187–189. [Google Scholar] [CrossRef] [Green Version]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E.B. Cancer Vaccines: Adjuvant Potency, Importance of Age, Lifestyle, and Treatments. Front. Immunol. 2021, 11, 615240. [Google Scholar] [CrossRef] [PubMed]
- Marincola, F.M.; Jaffee, E.M.; Hicklin, D.J.; Ferrone, S. Escape of human solid tumors from T–cell recognition: Molecular mechanisms and functional significance. Adv. Immunol. 1999, 74, 181–273. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.L.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, Y.; Ko, E.; Park, S.; Park, S.-O.; Ahn, B.-C.; Yum, J.-S.; Chun, E. L-Pampo™, a Novel TLR2/3 Agonist, Acts as a Potent Cancer Vaccine Adjuvant by Activating Draining Lymph Node Dendritic Cells. Cancers 2023, 15, 3978. https://doi.org/10.3390/cancers15153978
Heo Y, Ko E, Park S, Park S-O, Ahn B-C, Yum J-S, Chun E. L-Pampo™, a Novel TLR2/3 Agonist, Acts as a Potent Cancer Vaccine Adjuvant by Activating Draining Lymph Node Dendritic Cells. Cancers. 2023; 15(15):3978. https://doi.org/10.3390/cancers15153978
Chicago/Turabian StyleHeo, Yoonki, Eunbyeol Ko, Sejung Park, Si-On Park, Byung-Cheol Ahn, Jung-Sun Yum, and Eunyoung Chun. 2023. "L-Pampo™, a Novel TLR2/3 Agonist, Acts as a Potent Cancer Vaccine Adjuvant by Activating Draining Lymph Node Dendritic Cells" Cancers 15, no. 15: 3978. https://doi.org/10.3390/cancers15153978
APA StyleHeo, Y., Ko, E., Park, S., Park, S.-O., Ahn, B.-C., Yum, J.-S., & Chun, E. (2023). L-Pampo™, a Novel TLR2/3 Agonist, Acts as a Potent Cancer Vaccine Adjuvant by Activating Draining Lymph Node Dendritic Cells. Cancers, 15(15), 3978. https://doi.org/10.3390/cancers15153978