
Herpes Simplex Virus, Human Papillomavirus, and Cervical Cancer: Overview, Relationship, and Treatment Implications
 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
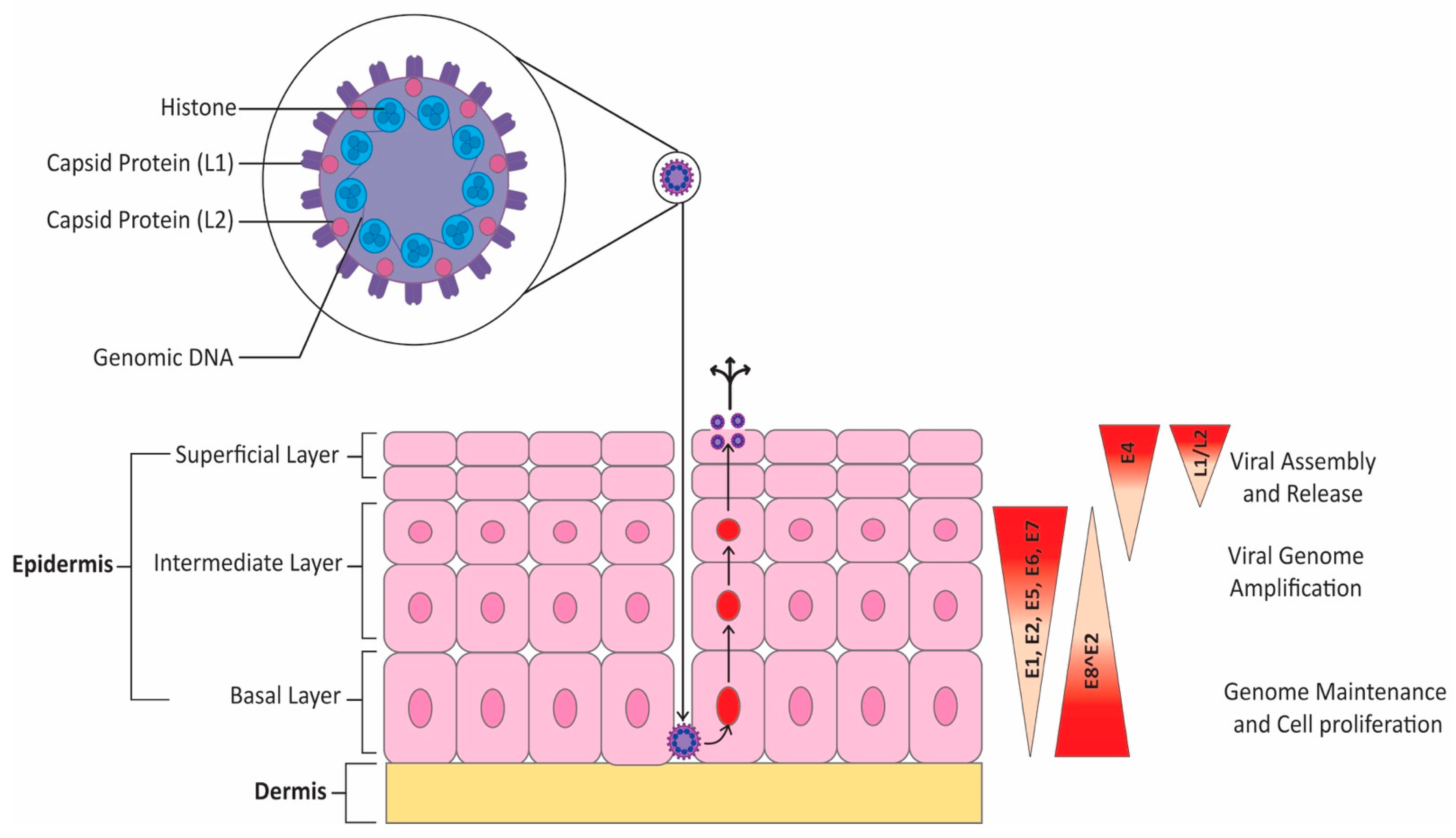
2. Overview of HPV Infection and Oncogenicity
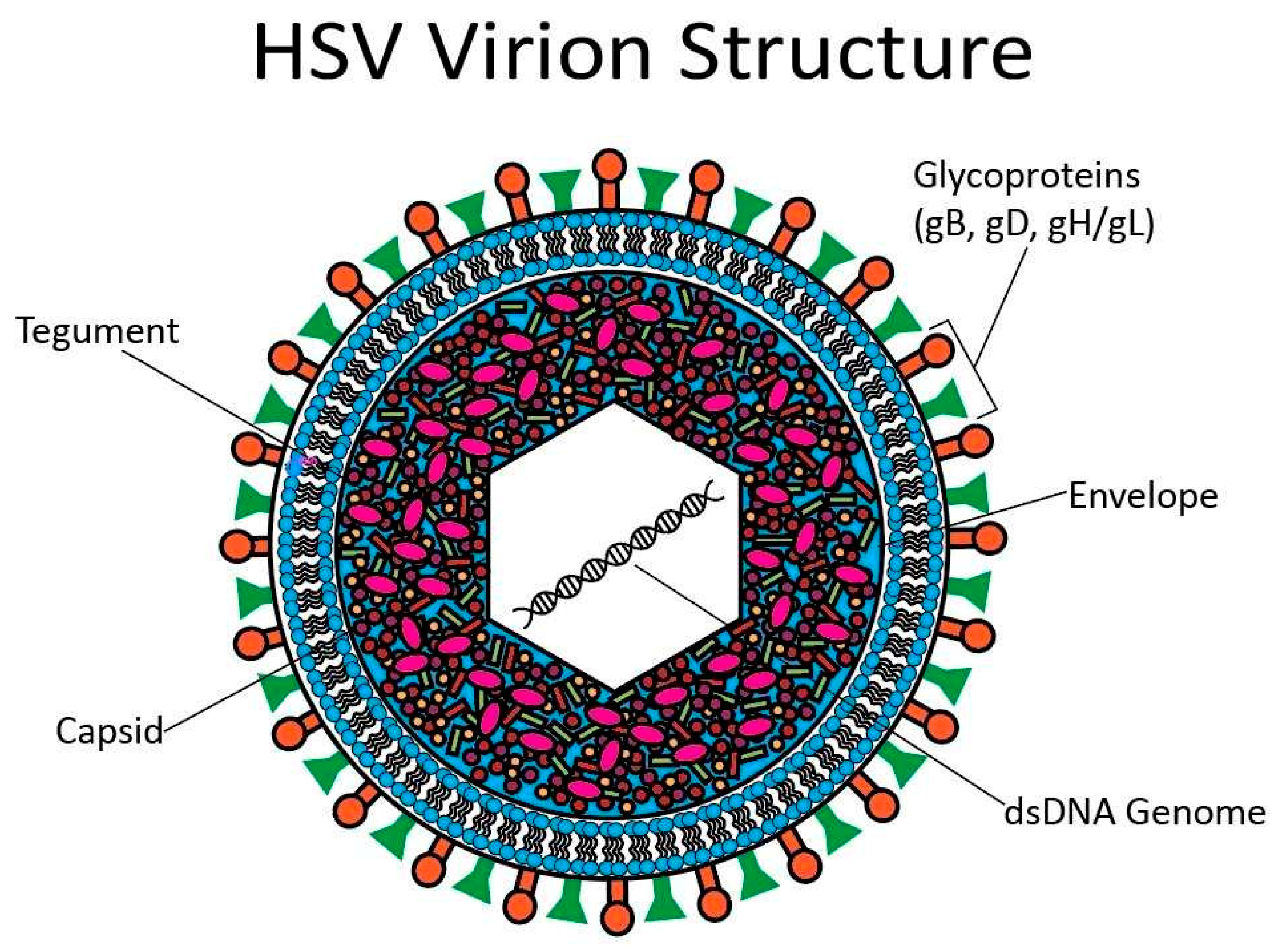
3. Overview of HSV Infection
4. Relationship between HPV and Cervical Cancer
5. Relationship between HSV and Malignancy
Relationship between HSV and Cervical Cancer
6. Targeting HPV and HSV for the Prevention of Cervical Cancer
6.1. Prophylaxis and Early Detection of Oncogenic HPV
6.2. Prophylaxis and Treatment of HSV Infection
7. Recent Advances in Cervical Cancer Therapeutics
7.1. Systemic Therapies
7.2. Other Therapeutic Advances
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.R.; Srivastava, S.; Shumayla, S.; Kurian, K.; Rehman, A.; Garg, R.; Rathi, S.K.; Mehra, S. Women’s Knowledge on Cervical Cancer Risk Factors and Symptoms: A Cross Sectional Study from Urban India. Asian Pac. J. Cancer Prev. 2022, 23, 1083–1090. [Google Scholar] [CrossRef]
- Vu, M.; Yu, J.; Awolude, O.A.; Chuang, L. Cervical cancer worldwide. Curr. Probl. Cancer 2018, 42, 457–465. [Google Scholar] [CrossRef]
- Bhatla, N.; Berek, J.S.; Cuello Fredes, M.; Denny, L.A.; Grenman, S.; Karunaratne, K.; Kehoe, S.T.; Konishi, I.; Olawaiye, A.B.; Prat, J.; et al. Revised FIGO staging for carcinoma of the cervix uteri. Int. J. Gynecol. Obstet. 2019, 145, 129–135. [Google Scholar] [CrossRef]
- Okunade, K.S. Human papillomavirus and cervical cancer. J. Obstet. Gynaecol. 2020, 40, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.A.; James, D.; Marzan, A.; Armaos, M. Cervical Cancer: An Overview of Pathophysiology and Management. Semin. Oncol. Nurs. 2019, 35, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Szymonowicz, K.A.; Chen, J. Biological and clinical aspects of HPV-related cancers. Cancer Biol. Med. 2020, 17, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.A.; Zhou, Y.; Jin, T. Epidemiology and Burden of Human Papillomavirus and Related Diseases, Molecular Pathogenesis, and Vaccine Evaluation. Front. Public Health 2020, 8, 552028. [Google Scholar] [CrossRef]
- Luo, C.; Yu, S.; Zhang, J.; Wu, X.; Dou, Z.; Li, Z.; Yang, E.; Zhang, L. Hepatitis B or C viral infection and the risk of cervical cancer. Infect. Agents Cancer 2022, 17, 54. [Google Scholar] [CrossRef]
- Feng, X.; Lu, H.; Wei, Y.; Guan, M.; Wang, J.; Liu, C.; Shen, T.; Chen, Q.; Rao, Q. Prognostic impact of hepatitis B virus infection in patients with primary cervical cancer. Cancer Med. 2021, 10, 8310–8319. [Google Scholar] [CrossRef]
- Fokom Domgue, J.; Cunningham, S.A.; Yu, R.K.; Shete, S. Prevalence and determinants of cervical cancer screening with a combination of cytology and human papillomavirus testing. Ann. Epidemiol. 2019, 36, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Siddique, A.; Khan, A.A.; Wang, Q.; Malik, A.; Jan, A.T.; Rudayni, H.A.; Chaudhary, A.A.; Khan, S. Chlamydia Trachomatis Infection: Their potential implication in the Etiology of Cervical Cancer. J. Cancer 2021, 12, 4891–4900. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Kahesa, C.; Mwaiselage, J.; West, J.T.; Wood, C.; Angeletti, P.C. How the Cervical Microbiota Contributes to Cervical Cancer Risk in Sub-Saharan Africa. Front. Cell. Infect. Microbiol. 2020, 10, 23. [Google Scholar] [CrossRef]
- Ahmadi, M.; Rasi, H.; Mostafazadeh, M.; Hajazimian, S.; Maroufi, N.F.; Nahaei, M.R.; Rahaee, S.; Isazadeh, A. Analysis of cervical lesions for presence of HSV-2 and HPV-16 and HPV-18 in Iranian patients by PCR. Horm. Mol. Biol. Clin. Investig. 2017, 31, 20170019. [Google Scholar] [CrossRef]
- DiPaolo, J.A.; Woodworth, C.D.; Popescu, N.C.; Koval, D.L.; Lopez, J.V.; Doniger, J. HSV-2-induced tumorigenicity in HPV16-immortalized human genital keratinocytes. Virology 1990, 177, 777–779. [Google Scholar] [CrossRef]
- Howley, P.M.; Schiller, J.T.; Lowy, D.R. Papillomaviruses. In Fields Virology; Howley, P.M., Knipe, D.M., Eds.; Wolters Kluwer, Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1662–1703. [Google Scholar]
- Petca, A.; Borislavschi, A.; Zvanca, M.E.; Petca, R.C.; Sandru, F.; Dumitrascu, M.C. Non-sexual HPV transmission and role of vaccination for a better future (Review). Exp. Ther. Med. 2020, 20, 186. [Google Scholar] [CrossRef] [PubMed]
- Balbi, G.; Schiattarella, A.; Fasulo, D.; Cafiero, A.; Mastrogiacomo, A.; Musone, R.; Carucci, A.; Cobellis, L. Vertical transmission of Human papillomavirus: Experience from a center of south Italy. Minerva Obstet. Gynecol. 2022, 75, 45–54. [Google Scholar] [CrossRef]
- Houlihan, C.F.; Baisley, K.; Bravo, I.G.; Pavon, M.A.; Changalucha, J.; Kapiga, S.; De Sanjose, S.; Ross, D.A.; Hayes, R.J.; Watson-Jones, D. Human papillomavirus DNA detected in fingertip, oral and bathroom samples from unvaccinated adolescent girls in Tanzania. Sex. Transm. Infect. 2019, 95, 374–379. [Google Scholar] [CrossRef]
- Hu, X.; Zhou, Q.; Yu, J.; Wang, J.; Tu, Q.; Zhu, X. Prevalence of HPV infections in surgical smoke exposed gynecologists. Int. Arch. Occup. Environ. Health 2021, 94, 107–115. [Google Scholar] [CrossRef]
- Brown, J.C.; Newcomb, W.W. Herpesvirus capsid assembly: Insights from structural analysis. Curr. Opin. Virol. 2011, 1, 142–149. [Google Scholar] [CrossRef]
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1802–1822. [Google Scholar]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef]
- Mathew, J., Jr.; Sapra, A. Herpes Simplex Type 2; StatPearls: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554427/ (accessed on 7 March 2023).
- Saleh, D.; Yarrarapu, S.N.S.; Sharma, S. Herpes Simplex Type 1; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Zhu, S.; Viejo-Borbolla, A. Pathogenesis and virulence of herpes simplex virus. Virulence 2021, 12, 2670–2702. [Google Scholar] [CrossRef]
- Hoyt, B.; Bhawan, J. Histological spectrum of cutaneous herpes infections. Am. J. Dermatopathol. 2014, 36, 609–619. [Google Scholar] [CrossRef]
- Krishnan, R.; Stuart, P.M. Developments in Vaccination for Herpes Simplex Virus. Front. Microbiol. 2021, 12, 798927. [Google Scholar] [CrossRef]
- McLendon, L.; Puckett, J.; Green, C.; James, J.; Head, K.J.; Yun Lee, H.; Young Pierce, J.; Beasley, M.; Daniel, C.L. Factors associated with HPV vaccination initiation among United States college students. Hum. Vaccines Immunother. 2021, 17, 1033–1043. [Google Scholar] [CrossRef]
- Quinlan, J.D. Human Papillomavirus: Screening, Testing, and Prevention. Am. Fam. Physician 2021, 104, 152–159. [Google Scholar] [PubMed]
- Burley, M.; Roberts, S.; Parish, J.L. Epigenetic regulation of human papillomavirus transcription in the productive virus life cycle. Semin. Immunopathol. 2020, 42, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fu, Y.; You, B.; Li, Y.; Yao, Y.; Wang, X.; Cheng, X. Clinical characteristics of single human papillomavirus 53 infection: A retrospective study of 419 cases. BMC Infect. Dis. 2021, 21, 1158. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Sekine, M.; Hanley, S.J.B.; Kudo, R.; Hara, M.; Adachi, S.; Ueda, Y.; Miyagi, E.; Enomoto, T. Risk factors for HPV infection and high-grade cervical disease in sexually active Japanese women. Sci. Rep. 2021, 11, 2898. [Google Scholar] [CrossRef] [PubMed]
- Pimple, S.; Mishra, G. Cancer cervix: Epidemiology and disease burden. Cytojournal 2022, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- So, K.A.; Kim, M.J.; Lee, K.H.; Lee, I.H.; Kim, M.K.; Lee, Y.K.; Hwang, C.S.; Jeong, M.S.; Kee, M.K.; Kang, C.; et al. The Impact of High-Risk HPV Genotypes Other Than HPV 16/18 on the Natural Course of Abnormal Cervical Cytology: A Korean HPV Cohort Study. Cancer Res. Treat. 2016, 48, 1313–1320. [Google Scholar] [CrossRef]
- Liu, F.; Chang, L.; Bai, T.; Liu, X.; Hu, J. Association of human papillomavirus genotype distribution and cervical cytology: A cross-sectional study. Epidemiol. Infect. 2021, 149, e95. [Google Scholar] [CrossRef]
- Aksoy, P.; Gottschalk, E.Y.; Meneses, P.I. HPV entry into cells. Mutat. Res. Mol. Mech. Mutagen. 2017, 772, 13–22. [Google Scholar] [CrossRef]
- Ryndock, E.J.; Meyers, C. A risk for non-sexual transmission of human papillomavirus? Expert Rev. Anti-Infect. Ther. 2014, 12, 1165–1170. [Google Scholar] [CrossRef]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Tuong, Z.K.; Frazer, I.H. Papillomavirus Immune Evasion Strategies Target the Infected Cell and the Local Immune System. Front. Oncol. 2019, 9, 682. [Google Scholar] [CrossRef] [PubMed]
- Bordignon, V.; Di Domenico, E.G.; Trento, E.; D’Agosto, G.; Cavallo, I.; Pontone, M.; Pimpinelli, F.; Mariani, L.; Ensoli, F. How Human Papillomavirus Replication and Immune Evasion Strategies Take Advantage of the Host DNA Damage Repair Machinery. Viruses 2017, 9, 390. [Google Scholar] [CrossRef] [PubMed]
- Plummer, M.; Schiffman, M.; Castle, P.E.; Maucort-Boulch, D.; Wheeler, C.M.; Group, A. A 2-year prospective study of human papillomavirus persistence among women with a cytological diagnosis of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesion. J. Infect. Dis. 2007, 195, 1582–1589. [Google Scholar] [CrossRef]
- Gravitt, P.E.; Winer, R.L. Natural History of HPV Infection across the Lifespan: Role of Viral Latency. Viruses 2017, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Vink, M.A.; Bogaards, J.A.; van Kemenade, F.J.; de Melker, H.E.; Meijer, C.J.; Berkhof, J. Clinical progression of high-grade cervical intraepithelial neoplasia: Estimating the time to preclinical cervical cancer from doubly censored national registry data. Am. J. Epidemiol. 2013, 178, 1161–1169. [Google Scholar] [CrossRef]
- New, C.; Lee, Z.Y.; Tan, K.S.; Wong, A.H.; Wang, Y.; Tran, T. Tetraspanins: Host Factors in Viral Infections. Int. J. Mol. Sci. 2021, 22, 11609. [Google Scholar] [CrossRef]
- Kumar, A.; Jacob, T.; Abban, C.Y.; Meneses, P.I. Intermediate heparan sulfate binding during HPV-16 infection in HaCaTs. Am. J. Ther. 2014, 21, 331–342. [Google Scholar] [CrossRef]
- Campos, S.K. Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2. Viruses 2017, 9, 370. [Google Scholar] [CrossRef]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Guion, L.G.M.; Keiffer, T.R.; Sapp, M. Human Papillomavirus Major Capsid Protein L1 Remains Associated with the Incoming Viral Genome throughout the Entry Process. J. Virol. 2017, 91, e00537-17. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009, 5, e1000524. [Google Scholar] [CrossRef]
- Scheffer, K.D.; Berditchevski, F.; Florin, L. The tetraspanin CD151 in papillomavirus infection. Viruses 2014, 6, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Fast, L.A.; Mikulicic, S.; Fritzen, A.; Schwickert, J.; Boukhallouk, F.; Hochdorfer, D.; Sinzger, C.; Suarez, H.; Monk, P.N.; Yanez-Mo, M.; et al. Inhibition of Tetraspanin Functions Impairs Human Papillomavirus and Cytomegalovirus Infections. Int. J. Mol. Sci. 2018, 19, 3007. [Google Scholar] [CrossRef]
- Kumar, A.; Hossain, R.A.; Yost, S.A.; Bu, W.; Wang, Y.; Dearborn, A.D.; Grakoui, A.; Cohen, J.I.; Marcotrigiano, J. Structural insights into hepatitis C virus receptor binding and entry. Nature 2021, 598, 521–525. [Google Scholar] [CrossRef]
- Finke, J.; Hitschler, L.; Boller, K.; Florin, L.; Lang, T. HPV caught in the tetraspanin web? Med. Microbiol. Immunol. 2020, 209, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Sterkand, R.T.; Ozbun, M.A. Interaction of human papillomavirus type 16 particles with heparan sulfate and syndecan-1 molecules in the keratinocyte extracellular matrix plays an active role in infection. J. Gen. Virol. 2015, 96, 2232–2241. [Google Scholar] [CrossRef] [PubMed]
- Woodham, A.W.; Da Silva, D.M.; Skeate, J.G.; Raff, A.B.; Ambroso, M.R.; Brand, H.E.; Isas, J.M.; Langen, R.; Kast, W.M. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS ONE 2012, 7, e43519. [Google Scholar] [CrossRef]
- Mikulicic, S.; Finke, J.; Boukhallouk, F.; Wustenhagen, E.; Sons, D.; Homsi, Y.; Reiss, K.; Lang, T.; Florin, L. ADAM17-dependent signaling is required for oncogenic human papillomavirus entry platform assembly. eLife 2019, 8, e22153. [Google Scholar] [CrossRef]
- Ozbun, M.A.; Campos, S.K. The long and winding road: Human papillomavirus entry and subcellular trafficking. Curr. Opin. Virol. 2021, 50, 76–86. [Google Scholar] [CrossRef]
- Calton, C.M.; Bronnimann, M.P.; Manson, A.R.; Li, S.; Chapman, J.A.; Suarez-Berumen, M.; Williamson, T.R.; Molugu, S.K.; Bernal, R.A.; Campos, S.K. Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathog. 2017, 13, e1006200. [Google Scholar] [CrossRef] [PubMed]
- Albert, E.; Laimins, L. Regulation of the Human Papillomavirus Life Cycle by DNA Damage Repair Pathways and Epigenetic Factors. Viruses 2020, 12, 744. [Google Scholar] [CrossRef] [PubMed]
- Mac, M.; Moody, C.A. Epigenetic Regulation of the Human Papillomavirus Life Cycle. Pathogens 2020, 9, 483. [Google Scholar] [CrossRef]
- McBride, A.A. Mechanisms and strategies of papillomavirus replication. Biol. Chem. 2017, 398, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Spink, K.M.; Laimins, L.A. Induction of the human papillomavirus type 31 late promoter requires differentiation but not DNA amplification. J. Virol. 2005, 79, 4918–4926. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Silla, T.; Ustav, E.; Ustav, M. Papillomavirus DNA replication—From initiation to genomic instability. Virology 2009, 384, 360–368. [Google Scholar] [CrossRef]
- Orav, M.; Gagnon, D.; Archambault, J. Interaction of the Human Papillomavirus E1 Helicase with UAF1-USP1 Promotes Unidirectional Theta Replication of Viral Genomes. mBio 2019, 10, e00152-19. [Google Scholar] [CrossRef]
- Orav, M.; Geimanen, J.; Sepp, E.M.; Henno, L.; Ustav, E.; Ustav, M. Initial amplification of the HPV18 genome proceeds via two distinct replication mechanisms. Sci. Rep. 2015, 5, 15952. [Google Scholar] [CrossRef] [PubMed]
- Sverdrup, F.; Khan, S.A. Replication of human papillomavirus (HPV) DNAs supported by the HPV type 18 E1 and E2 proteins. J. Virol. 1994, 68, 505–509. [Google Scholar] [CrossRef]
- Bristol, M.L.; Wang, X.; Smith, N.W.; Son, M.P.; Evans, M.R.; Morgan, I.M. DNA Damage Reduces the Quality, but Not the Quantity of Human Papillomavirus 16 E1 and E2 DNA Replication. Viruses 2016, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Hughes, F.J.; Romanos, M.A. E1 protein of human papillomavirus is a DNA helicase/ATPase. Nucleic Acids Res. 1993, 21, 5817–5823. [Google Scholar] [CrossRef] [PubMed]
- Piirsoo, A.; Kala, M.; Sankovski, E.; Ustav, M.; Piirsoo, M. Uncovering the Role of the E1 Protein in Different Stages of Human Papillomavirus 18 Genome Replication. J. Virol. 2020, 94, e00674-20. [Google Scholar] [CrossRef] [PubMed]
- Castro-Munoz, L.J.; Manzo-Merino, J.; Munoz-Bello, J.O.; Olmedo-Nieva, L.; Cedro-Tanda, A.; Alfaro-Ruiz, L.A.; Hidalgo-Miranda, A.; Madrid-Marina, V.; Lizano, M. The Human Papillomavirus (HPV) E1 protein regulates the expression of cellular genes involved in immune response. Sci. Rep. 2019, 9, 13620. [Google Scholar] [CrossRef]
- Jose, L.; Androphy, E.J.; DeSmet, M. Phosphorylation of the Human Papillomavirus E2 Protein at Tyrosine 138 Regulates Episomal Replication. J. Virol. 2020, 94, e0048-20. [Google Scholar] [CrossRef]
- Sanders, C.M.; Stenlund, A. Recruitment and loading of the E1 initiator protein: An ATP-dependent process catalysed by a transcription factor. EMBO J. 1998, 17, 7044–7055. [Google Scholar] [CrossRef]
- Yigitliler, A.; Renner, J.; Simon, C.; Schneider, M.; Stubenrauch, F.; Iftner, T. BRD4S interacts with viral E2 protein to limit human papillomavirus late transcription. J. Virol. 2021, 95, e02032-20. [Google Scholar] [CrossRef]
- Villota, C.; Varas-Godoy, M.; Jeldes, E.; Campos, A.; Villegas, J.; Borgna, V.; Burzio, L.O.; Burzio, V.A. HPV-18 E2 protein downregulates antisense noncoding mitochondrial RNA-2, delaying replicative senescence of human keratinocytes. Aging 2018, 11, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Liblekas, L.; Piirsoo, A.; Laanemets, A.; Tombak, E.M.; Laanevali, A.; Ustav, E.; Ustav, M.; Piirsoo, M. Analysis of the Replication Mechanisms of the Human Papillomavirus Genomes. Front. Microbiol. 2021, 12, 738125. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Foo, C.; Coleman, N.; Medcalf, L.; Hartley, O.; Prospero, T.; Napthine, S.; Sterling, J.; Winter, G.; Griffin, H. Characterization of events during the late stages of HPV16 infection in vivo using high-affinity synthetic Fabs to E4. Virology 1997, 238, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Keratinocyte Differentiation-Dependent Human Papillomavirus Gene Regulation. Viruses 2017, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Dreer, M.; van de Poel, S.; Stubenrauch, F. Control of viral replication and transcription by the papillomavirus E8^E2 protein. Virus Res. 2017, 231, 96–102. [Google Scholar] [CrossRef]
- Straub, E.; Dreer, M.; Fertey, J.; Iftner, T.; Stubenrauch, F. The viral E8^E2C repressor limits productive replication of human papillomavirus 16. J. Virol. 2014, 88, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Dreer, M.; Fertey, J.; van de Poel, S.; Straub, E.; Madlung, J.; Macek, B.; Iftner, T.; Stubenrauch, F. Interaction of NCOR/SMRT Repressor Complexes with Papillomavirus E8^E2C Proteins Inhibits Viral Replication. PLoS Pathog. 2016, 12, e1005556. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2019, 10, 3116. [Google Scholar] [CrossRef]
- Ilahi, N.E.; Bhatti, A. Impact of HPV E5 on viral life cycle via EGFR signaling. Microb. Pathog. 2020, 139, 103923. [Google Scholar] [CrossRef]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. S1), 2–23. [Google Scholar] [CrossRef]
- Aschner, C.B.; Herold, B.C. Alphaherpesvirus Vaccines. Curr. Issues Mol. Biol. 2021, 41, 469–508. [Google Scholar] [CrossRef]
- Smith, J.S.; Robinson, N.J. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: A global review. J. Infect. Dis. 2002, 186 (Suppl. S1), S3–S28. [Google Scholar] [CrossRef]
- Ramchandani, M.; Kong, M.; Tronstein, E.; Selke, S.; Mikhaylova, A.; Magaret, A.; Huang, M.L.; Johnston, C.; Corey, L.; Wald, A. Herpes Simplex Virus Type 1 Shedding in Tears and Nasal and Oral Mucosa of Healthy Adults. Sex. Transm. Dis. 2016, 43, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Jaishankar, D.; Shukla, D. Genital Herpes: Insights into Sexually Transmitted Infectious Disease. Microb. Cell 2016, 3, 438–450. [Google Scholar] [CrossRef]
- Corey, L.; Huang, M.L.; Selke, S.; Wald, A. Differentiation of herpes simplex virus types 1 and 2 in clinical samples by a real-time taqman PCR assay. J. Med. Virol. 2005, 76, 350–355. [Google Scholar] [CrossRef]
- Cole, S. Herpes Simplex Virus: Epidemiology, Diagnosis, and Treatment. Nurs. Clin. N. Am. 2020, 55, 337–345. [Google Scholar] [CrossRef]
- Whitley, R.; Baines, J. Clinical management of herpes simplex virus infections: Past, present, and future. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Goodyear, H.M.; Breuer, J. Herpes Simplex Virus Infections. In Harper’s Textbook of Pediatric Dermatology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2019. [Google Scholar] [CrossRef]
- Cohen, J.I. Herpesvirus latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef]
- Nicoll, M.P.; Hann, W.; Shivkumar, M.; Harman, L.E.; Connor, V.; Coleman, H.M.; Proenca, J.T.; Efstathiou, S. The HSV-1 Latency-Associated Transcript Functions to Repress Latent Phase Lytic Gene Expression and Suppress Virus Reactivation from Latently Infected Neurons. PLoS Pathog. 2016, 12, e1005539. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Sgarbanti, R.; Civitelli, L.; Ripoli, C.; Piacentini, R.; Garaci, E.; Grassi, C.; Palamara, A.T. Infectious agents and neurodegeneration. Mol. Neurobiol. 2012, 46, 614–638. [Google Scholar] [CrossRef]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell. Infect. Microbiol. 2020, 10, 617578. [Google Scholar] [CrossRef] [PubMed]
- Musarrat, F.; Chouljenko, V.; Kousoulas, K.G. Cellular and Viral Determinants of HSV-1 Entry and Intracellular Transport towards Nucleus of Infected Cells. J. Virol. 2021, 95, e02434-20. [Google Scholar] [CrossRef] [PubMed]
- Ojala, P.M.; Sodeik, B.; Ebersold, M.W.; Kutay, U.; Helenius, A. Herpes simplex virus type 1 entry into host cells: Reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell. Biol. 2000, 20, 4922–4931. [Google Scholar] [CrossRef]
- Jovasevic, V.; Liang, L.; Roizman, B. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 2008, 82, 3311–3319. [Google Scholar] [CrossRef] [PubMed]
- Packard, J.E.; Dembowski, J.A. HSV-1 DNA Replication-Coordinated Regulation by Viral and Cellular Factors. Viruses 2021, 13, 2015. [Google Scholar] [CrossRef]
- Lang, F.C.; Li, X.; Vladmirova, O.; Li, Z.R.; Chen, G.J.; Xiao, Y.; Li, L.H.; Lu, D.F.; Han, H.B.; Zhou, J.M. Selective recruitment of host factors by HSV-1 replication centers. Zool. Res. 2015, 36, 142–151. [Google Scholar]
- Lee, J.H.; Shim, J.; Kim, S.J. Stunning symmetries involved in the self-assembly of the HSV-1 capsid. J. Korean Phys. Soc. 2021, 78, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Crump, C. Virus Assembly and Egress of HSV. Adv. Exp. Med. Biol. 2018, 1045, 23–44. [Google Scholar] [CrossRef]
- Kwong, A.D.; Kruper, J.A.; Frenkel, N. Herpes simplex virus virion host shutoff function. J. Virol. 1988, 62, 912–921. [Google Scholar] [CrossRef]
- Taddeo, B.; Roizman, B. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J. Virol. 2006, 80, 9341–9345. [Google Scholar] [CrossRef]
- He, T.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; Zhang, S.; et al. Host shutoff activity of VHS and SOX-like proteins: Role in viral survival and immune evasion. Virol. J. 2020, 17, 68. [Google Scholar] [CrossRef]
- zur Hausen, H. Condylomata acuminata and human genital cancer. Cancer Res. 1976, 36, 794. [Google Scholar] [PubMed]
- Kocjan, B.J.; Bzhalava, D.; Forslund, O.; Dillner, J.; Poljak, M. Molecular methods for identification and characterization of novel papillomaviruses. Clin. Microbiol. Infect. 2015, 21, 808–816. [Google Scholar] [CrossRef]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J.; International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, M.; Cao, D.; Wei, X.; Wang, L.; Li, Y.; Yang, T.; Zhao, J.; Pei, M.; Jia, H.; et al. Assessment of the effectiveness of HPV16/18 infection referred for colposcopy in cervical cancer screening in Northwest of China. J. Med. Virol. 2018, 90, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.G.; Bensumaidea, S.H.; Alshammari, F.D.; Alenazi, F.S.H.; Almutlaq, B.A.; Alturkstani, M.Z.; Aladani, I.A. Prevalence of Human Papillomavirus subtypes 16 and 18 among Yemeni Patients with Cervical Cancer. Asian Pac. J. Cancer Prev. 2017, 18, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Partricia, S.; Mathan, G. Overview of high-risk HPV’s 16 and 18 infected cervical cancer: Pathogenesis to prevention. Biomed. Pharmacother. 2015, 70, 103–110. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.L.; Coleman, D.T.; Kelly, K.C.; Carroll, J.L.; Woodby, B.; Songock, W.K.; Cardelli, J.A.; Bodily, J.M. Human papillomavirus type 16 E5-mediated upregulation of Met in human keratinocytes. Virology 2018, 519, 1–11. [Google Scholar] [CrossRef]
- Hochmann, J.; Parietti, F.; Martinez, J.; Lopez, A.C.; Carreno, M.; Quijano, C.; Boccardo, E.; Sichero, L.; Moller, M.N.; Mirazo, S.; et al. Human papillomavirus type 18 E5 oncoprotein cooperates with E6 and E7 in promoting cell viability and invasion and in modulating the cellular redox state. Memórias Do Inst. Oswaldo Cruz 2020, 115, e190405. [Google Scholar] [CrossRef]
- Sudarshan, S.R.; Schlegel, R.; Liu, X. Two conserved amino acids differentiate the biology of high-risk and low-risk HPV E5 proteins. J. Med. Virol. 2022, 94, 4565–4575. [Google Scholar] [CrossRef]
- Li, S.; Hong, X.; Wei, Z.; Xie, M.; Li, W.; Liu, G.; Guo, H.; Yang, J.; Wei, W.; Zhang, S. Ubiquitination of the HPV Oncoprotein E6 Is Critical for E6/E6AP-Mediated p53 Degradation. Front. Microbiol. 2019, 10, 2483. [Google Scholar] [CrossRef] [PubMed]
- Hadami, K.; Saby, C.; Dakka, N.; Collin, G.; Attaleb, M.; Khyatti, M.; Filali-Maltouf, A.; Morjani, H.; El Mzibri, M. Degradation of p53 by HPV16-E6 variants isolated from cervical cancer specimens of Moroccan women. Gene 2021, 791, 145709. [Google Scholar] [CrossRef]
- Artaza-Irigaray, C.; Molina-Pineda, A.; Aguilar-Lemarroy, A.; Ortiz-Lazareno, P.; Limon-Toledo, L.P.; Pereira-Suarez, A.L.; Rojo-Contreras, W.; Jave-Suarez, L.F. E6/E7 and E6(*) From HPV16 and HPV18 Upregulate IL-6 Expression Independently of p53 in Keratinocytes. Front. Immunol. 2019, 10, 1676. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Wasson, C.W.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 activation by E6 is essential for the differentiation-dependent HPV18 life cycle. PLoS Pathog. 2018, 14, e1006975. [Google Scholar] [CrossRef]
- Fan, Q.; Huang, T.; Sun, X.; Wang, Y.W.; Wang, J.; Liu, Y.; Ni, T.; Gu, S.L.; Li, Y.H.; Wang, Y.D. HPV-16/18 E6-induced APOBEC3B expression associates with proliferation of cervical cancer cells and hypomethylation of Cyclin D1. Mol. Carcinog. 2021, 60, 313–330. [Google Scholar] [CrossRef]
- Montalto, F.I.; De Amicis, F. Cyclin D1 in Cancer: A Molecular Connection for Cell Cycle Control, Adhesion and Invasion in Tumor and Stroma. Cells 2020, 9, 2648. [Google Scholar] [CrossRef]
- Morgan, E.L.; Scarth, J.A.; Patterson, M.R.; Wasson, C.W.; Hemingway, G.C.; Barba-Moreno, D.; Macdonald, A. E6-mediated activation of JNK drives EGFR signalling to promote proliferation and viral oncoprotein expression in cervical cancer. Cell Death Differ. 2021, 28, 1669–1687. [Google Scholar] [CrossRef]
- Li, D.; Liu, S.H.; Liu, Q.Y.; Zou, Q.Q.; Lv, L.; Liu, G.L.; Wu, Y. Analysis of the Role and Regulatory Mechanism of hsa-miR-504 in Cervical Cancer Based on The Cancer Genome Atlas Database. Cancer Biother. Radiopharm. 2021, 36, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lai, Y.; Sun, Y.; Xu, B.; Qiang, X.; Zhou, X.; Wang, T. HPV16 E6 regulates the proliferation, invasion, and apoptosis of cervical cancer cells by downregulating miR-504. Transl. Cancer Res. 2020, 9, 7588–7595. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.T.; Lin, J.F.; Li, T.; Li, J.J.; Xu, R.H.; Ju, H.Q. LncRNA-mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun. 2021, 41, 109–120. [Google Scholar] [CrossRef]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Y.; Chen, M.; Cui, J. Identification of Novel Long Non-coding and Circular RNAs in Human Papillomavirus-Mediated Cervical Cancer. Front. Microbiol. 2017, 8, 1720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Zhang, J.; Mao, L. E6 hijacks KDM5C/lnc_000231/miR-497-5p/CCNE1 axis to promote cervical cancer progression. J. Cell. Mol. Med. 2020, 24, 11422–11433. [Google Scholar] [CrossRef] [PubMed]
- Yim, E.K.; Park, J.S. The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer Res. Treat. 2005, 37, 319–324. [Google Scholar] [CrossRef]
- Fischer, M.; Uxa, S.; Stanko, C.; Magin, T.M.; Engeland, K. Human papilloma virus E7 oncoprotein abrogates the p53-p21-DREAM pathway. Sci. Rep. 2017, 7, 2603. [Google Scholar] [CrossRef]
- Lin, Z.; Zhao, Y.; Li, Q.; Ci, X.; Ye, X.; Chen, G.; Tu, Q.; Feng, W.; Jiang, P.; Zhu, S.; et al. Sustained expression of HPV16 E7 oncoprotein promotes p-AKT(Ser473)/p-Src(Tyr527) signaling to drive precancerous lesions to invasive cervical cancer. Carcinogenesis 2022, 43, 479–493. [Google Scholar] [CrossRef]
- Tian, S.; Zhang, L.; Li, Y.; Cao, D.; Quan, S.; Guo, Y.; Yang, X.; Yang, T. Human Papillomavirus E7 Oncoprotein Promotes Proliferation and Migration through the Transcription Factor E2F1 in Cervical Cancer Cells. Anti-Cancer Agents Med. Chem. 2021, 21, 1689–1696. [Google Scholar] [CrossRef]
- Zuberi, Z.; Mremi, A.; Chilongola, J.O.; Semango, G.; Sauli, E. Expression analysis of p16 and TOP2A protein biomarkers in cervical cancer lesions and their correlation with clinico-histopathological characteristics in a referral hospital, Tanzania. PLoS ONE 2021, 16, e0259096. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, H.; Yi, S.; Gu, L.; Zhou, M. Mutual regulation of MDM4 and TOP2A in cancer cell proliferation. Mol. Oncol. 2019, 13, 1047–1058. [Google Scholar] [CrossRef]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132, jcs223826. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, W.; Huang, W.; Hua, Z.; Li, S. LncRNA MALAT1 was regulated by HPV16 E7 independently of pRB in cervical cancer cells. J. Cancer 2021, 12, 6344–6355. [Google Scholar] [CrossRef]
- Shen, F.; Zheng, H.; Zhou, L.; Li, W.; Xu, X. Overexpression of MALAT1 contributes to cervical cancer progression by acting as a sponge of miR-429. J. Cell. Physiol. 2019, 234, 11219–11226. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Song, L.; Zeng, S.; Zhang, L. MALAT1-miR-124-RBG2 axis is involved in growth and invasion of HR-HPV-positive cervical cancer cells. Tumor Biol. 2016, 37, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiang, F.; Liu, X.; Ma, X.; Cai, X.; Yang, Y.; Shen, X.; Yuan, C.; Xiang, Y.; Xiao, H. HPV E7 affects the function of cervical cancer cells via the TAL1/lncEBIC/KLHDC7B axis. Oncol. Rep. 2021, 45, 51. [Google Scholar] [CrossRef]
- Rapp, F. Herpesviruses and cancer. Connect. Med. 1980, 44, 131–133. [Google Scholar]
- Cesarman, E.; Chadburn, A.; Rubinstein, P.G. KSHV/HHV8-mediated hematologic diseases. Blood 2022, 139, 1013–1025. [Google Scholar] [CrossRef]
- Alibek, K.; Baiken, Y.; Kakpenova, A.; Mussabekova, A.; Zhussupbekova, S.; Akan, M.; Sultankulov, B. Implication of human herpesviruses in oncogenesis through immune evasion and supression. Infect. Agents Cancer 2014, 9, 3. [Google Scholar] [CrossRef]
- Sausen, D.G.; Basith, A.; Muqeemuddin, S. EBV and Lymphomagenesis. Cancers 2023, 15, 2133. [Google Scholar] [CrossRef]
- Shechter, O.; Sausen, D.G.; Gallo, E.S.; Dahari, H.; Borenstein, R. Epstein-Barr Virus (EBV) Epithelial Associated Malignancies: Exploring Pathologies and Current Treatments. Int. J. Mol. Sci. 2022, 23, 14389. [Google Scholar] [CrossRef]
- Javier, R.T.; Butel, J.S. The history of tumor virology. Cancer Res. 2008, 68, 7693–7706. [Google Scholar] [CrossRef]
- Galloway, D.A.; McDougall, J.K. The oncogenic potential of herpes simplex viruses: Evidence for a ‘hit-and-run’ mechanism. Nature 1983, 302, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Maitland, N.J.; Kinross, J.H.; Busuttil, A.; Ludgate, S.M.; Smart, G.E.; Jones, K.W. The detection of DNA tumour virus-specific RNA sequences in abnormal human cervical biopsies by in situ hybridization. J. Gen. Virol. 1981, 55, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, N.; Roizman, B.; Cassai, E.; Nahmias, A. A DNA fragment of Herpes simplex 2 and its transcription in human cervical cancer tissue. Proc. Natl. Acad. Sci. USA 1972, 69, 3784–3789. [Google Scholar] [CrossRef]
- Dhanwada, K.R.; Garrett, L.; Smith, P.; Thompson, K.D.; Doster, A.; Jones, C. Characterization of human keratinocytes transformed by high risk human papillomavirus types 16 or 18 and herpes simplex virus type 2. J. Gen. Virol. 1993, 74 Pt 6, 955–963. [Google Scholar] [CrossRef]
- Michutova, M.; Mrazova, V.; Kudelova, M.; Smolinska, M.; Supolikova, M.; Vrbova, M.; Golais, F. Herpes simplex viruses type 1 and 2 photoinactivated in the presence of methylene blue transform human and mouse cells in vitro. Acta Virol. 2017, 61, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Sheng, X.; Justice, J.L.; Lum, K.K.; Metzger, P.J.; Cook, K.C.; Kostas, J.C.; Cristea, I.M. Intercellular communication within the virus microenvironment affects the susceptibility of cells to secondary viral infections. Sci. Adv. 2023, 9, eadg3433. [Google Scholar] [CrossRef] [PubMed]
- Skinner, G.R. Transformation of primary hamster embryo fibroblasts by type 2 simplex virus: Evidence for a “hit and run” mechanism. Br. J. Exp. Pathol. 1976, 57, 361–376. [Google Scholar]
- Guidry, J.T.; Scott, R.S. The interaction between human papillomavirus and other viruses. Virus Res. 2017, 231, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Golais, F.; Mrazova, V. Human alpha and beta herpesviruses and cancer: Passengers or foes? Folia Microbiol. 2020, 65, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Luo, J.H.; Hunter, J.C.; Ordonez, J.V.; Aurelian, L. The transmembrane domain of the large subunit of HSV-2 ribonucleotide reductase (ICP10) is required for protein kinase activity and transformation-related signaling pathways that result in ras activation. Virology 1994, 200, 598–612. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.; Pereira, E.F.; Gober, M.; Yarowsky, P.J.; Aurelian, L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J. Virol. 2002, 76, 1435–1449. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.; Pereira, E.F.; Aurelian, L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) functions as a dominant regulator of apoptosis in hippocampal neurons involving activation of the ERK survival pathway and upregulation of the antiapoptotic protein Bag-1. J. Virol. 2003, 77, 1292–1305. [Google Scholar] [CrossRef]
- McDougall, J.K.; Galloway, D.A.; Fenoglio, C.M. Cervical carcinoma: Detection of herpes simplex virus RNA in cells undergoing neoplastic change. Int. J. Cancer 1980, 25, 1–8. [Google Scholar] [CrossRef]
- Li, S.; Wen, X. Seropositivity to herpes simplex virus type 2, but not type 1 is associated with cervical cancer: NHANES (1999–2014). BMC Cancer 2017, 17, 726. [Google Scholar] [CrossRef] [PubMed]
- Bahena-Roman, M.; Sanchez-Aleman, M.A.; Contreras-Ochoa, C.O.; Lagunas-Martinez, A.; Olamendi-Portugal, M.; Lopez-Estrada, G.; Delgado-Romero, K.; Guzman-Olea, E.; Madrid-Marina, V.; Torres-Poveda, K. Prevalence of active infection by herpes simplex virus type 2 in patients with high-risk human papillomavirus infection: A cross-sectional study. J. Med. Virol. 2020, 92, 1246–1252. [Google Scholar] [CrossRef]
- Bi, H.; Zhang, D.; Xiao, B. Association between human papillomavirus infection and common sexually transmitted infections, and the clinical significance of different Mycoplasma subtypes. Front. Cell. Infect. Microbiol. 2023, 13, 1145215. [Google Scholar] [CrossRef]
- Jary, A.; Teguete, I.; Sidibe, Y.; Kodio, A.; Dolo, O.; Burrel, S.; Boutolleau, D.; Beauvais-Remigereau, L.; Sayon, S.; Kampo, M.; et al. Prevalence of cervical HPV infection, sexually transmitted infections and associated antimicrobial resistance in women attending cervical cancer screening in Mali. Int. J. Infect. Dis. 2021, 108, 610–616. [Google Scholar] [CrossRef]
- de Abreu, A.L.; Malaguti, N.; Souza, R.P.; Uchimura, N.S.; Ferreira, E.C.; Pereira, M.W.; Carvalho, M.D.; Pelloso, S.M.; Bonini, M.G.; Gimenes, F.; et al. Association of human papillomavirus, Neisseria gonorrhoeae and Chlamydia trachomatis co-infections on the risk of high-grade squamous intraepithelial cervical lesion. Am. J. Cancer Res. 2016, 6, 1371–1383. [Google Scholar] [PubMed]
- Pisani, S.; Imperi, M.; Seganti, L.; Superti, F.; Tinari, A.; Bucci, M.; Degener, A.M. Effect of HSV-2 infection on the expression of HPV 16 genes in CaSki cells. Int. J. Immunopathol. Pharmacol. 2004, 17, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Paba, P.; Bonifacio, D.; Di Bonito, L.; Ombres, D.; Favalli, C.; Syrjanen, K.; Ciotti, M. Co-expression of HSV2 and Chlamydia trachomatis in HPV-positive cervical cancer and cervical intraepithelial neoplasia lesions is associated with aberrations in key intracellular pathways. Intervirology 2008, 51, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Kimoto, T.; Okuno, Y.; Minekawa, Y. Effect of herpes simplex virus on the DNA of human papillomavirus 18. J. Med. Virol. 1997, 53, 4–12. [Google Scholar] [CrossRef]
- Koanga, M.M.L.; Ngono, N.A.; Djiakam, N.G.; Wankam, M.; Brulet, E.C.; Amvam, Z.P.H. Association of Cervical Inflammation and Cervical Abnormalities in Women Infected with Herpes Simplex Virus Type 2. Int. J. Trop. Med. Public Health 2014, 1, 1. [Google Scholar]
- Okoye, J.O.; Ngokere, A.A.; Erinle, C.; Mbamalu, C. Co-existence of Herpes simplex virus type 2 and two other oncoviruses is associated with cervical lesions in women living with HIV in South-Western Nigeria. Afr. Health Sci. 2020, 20, 1015–1023. [Google Scholar] [CrossRef]
- Taku, O.; Brink, A.; Meiring, T.L.; Phohlo, K.; Businge, C.B.; Mbulawa, Z.Z.A.; Williamson, A.L. Detection of sexually transmitted pathogens and co-infection with human papillomavirus in women residing in rural Eastern Cape, South Africa. PeerJ 2021, 9, e10793. [Google Scholar] [CrossRef] [PubMed]
- McClymont, E.; Tan, D.H.; Bondy, S.; Albert, A.; Coutlee, F.; Lee, M.; Walmsley, S.; Ogilvie, G.; Money, D. HSV-2 infection and HPV incidence, persistence, and precancerous lesions in a cohort of HPV-vaccinated women living with HIV. Int. J. STD AIDS 2023, 34, 402–407. [Google Scholar] [CrossRef]
- Daiane de Peder, L.; Mesquita da Silva, C.; Nascimento, B.L.; Malizan, J.A.; Madeira, H.S.; Horvath, J.D.; Silva, E.S.; Vieira Teixeira, J.J. Prevalence of Sexually Transmitted Infections and Risk Factors Among Young People in a Public Health Center in Brazil: A Cross-Sectional Study. J. Pediatr. Adolesc. Gynecol. 2020, 33, 354–362. [Google Scholar] [CrossRef]
- Levy, S.B.; Gunta, J.; Edemekong, P. Screening for Sexually Transmitted Diseases. Prim. Care 2019, 46, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Gan, Y.; Dong, X.; Lu, Z. Herpes simplex virus type 2 and the risk of cervical cancer: A meta-analysis of observational studies. Arch. Gynecol. Obstet. 2014, 290, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.; Koskela, P.; Jellum, E.; Bloigu, A.; Anttila, T.; Hallmans, G.; Luukkaala, T.; Thoresen, S.; Youngman, L.; Dillner, J.; et al. Herpes simplex virus and risk of cervical cancer: A longitudinal, nested case-control study in the nordic countries. Am. J. Epidemiol. 2002, 156, 687–692. [Google Scholar] [CrossRef]
- Joharinia, N.; Faghihinejad, S.; Seyedi, K.; Farhadi, A.; Hosseini, S.Y.; Safaei, A.; Bahrampour, H.; Sarvari, J. Co-existing of HSV1/2 or EBV Infection with the Presence of High-Risk HPV DNA in Cervical Lesions in the Southwest of Iran. Asian Pac. J. Cancer Prev. 2020, 21, 1459–1464. [Google Scholar] [CrossRef]
- Lei, J.; Ploner, A.; Elfstrom, K.M.; Wang, J.; Roth, A.; Fang, F.; Sundstrom, K.; Dillner, J.; Sparen, P. HPV Vaccination and the Risk of Invasive Cervical Cancer. N. Engl. J. Med. 2020, 383, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.A.; Pimple, S.A.; Shastri, S.S. An overview of prevention and early detection of cervical cancers. Indian J. Med. Paediatr. Oncol. 2011, 32, 125–132. [Google Scholar] [CrossRef]
- Basu, P.; Malvi, S.G.; Joshi, S.; Bhatla, N.; Muwonge, R.; Lucas, E.; Verma, Y.; Esmy, P.O.; Poli, U.R.R.; Shah, A.; et al. Vaccine efficacy against persistent human papillomavirus (HPV) 16/18 infection at 10 years after one, two, and three doses of quadrivalent HPV vaccine in girls in India: A multicentre, prospective, cohort study. Lancet Oncol. 2021, 22, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Mac Eochagain, C.; Power, R.; Parker, I.; Brennan, D. HPV vaccination among seropositive, DNA negative cohorts: A systematic review & meta-analysis. J. Gynecol. Oncol. 2022, 33, e24. [Google Scholar] [CrossRef] [PubMed]
- Dilley, S.; Miller, K.M.; Huh, W.K. Human papillomavirus vaccination: Ongoing challenges and future directions. Gynecol. Oncol. 2020, 156, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, R.; Dhanani, S. The Development of Human Papillomavirus (HPV) Vaccines and Current Barriers to Implementation. Immunol. Investig. 2021, 50, 821–832. [Google Scholar] [CrossRef]
- de Oliveira, C.M.; Fregnani, J.; Villa, L.L. HPV Vaccine: Updates and Highlights. Acta Cytol. 2019, 63, 159–168. [Google Scholar] [CrossRef]
- Kirnbauer, R.; Booy, F.; Cheng, N.; Lowy, D.R.; Schiller, J.T. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc. Natl. Acad. Sci. USA 1992, 89, 12180–12184. [Google Scholar] [CrossRef]
- Force, U.S.P.S.T.; Curry, S.J.; Krist, A.H.; Owens, D.K.; Barry, M.J.; Caughey, A.B.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W., Jr.; Kemper, A.R.; et al. Screening for Cervical Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 320, 674–686. [Google Scholar] [CrossRef]
- Devarapalli, P.; Labani, S.; Nagarjuna, N.; Panchal, P.; Asthana, S. Barriers affecting uptake of cervical cancer screening in low and middle income countries: A systematic review. Indian J. Cancer 2018, 55, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Kirubarajan, A.; Leung, S.; Li, X.; Yau, M.; Sobel, M. Barriers and facilitators for cervical cancer screening among adolescents and young people: A systematic review. BMC Women’s Health 2021, 21, 122. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.M.; Castillo, E.; Duarte, L.F.; Arriagada, J.; Corrales, N.; Farias, M.A.; Henriquez, A.; Agurto-Munoz, C.; Gonzalez, P.A. Current Antivirals and Novel Botanical Molecules Interfering with Herpes Simplex Virus Infection. Front. Microbiol. 2020, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Borenstein, R.; Hanson, B.A.; Markosyan, R.M.; Gallo, E.S.; Narasipura, S.D.; Bhutta, M.; Shechter, O.; Lurain, N.S.; Cohen, F.S.; Al-Harthi, L.; et al. Ginkgolic acid inhibits fusion of enveloped viruses. Sci. Rep. 2020, 10, 4746. [Google Scholar] [CrossRef] [PubMed]
- Prichard, M.N.; Kern, E.R.; Hartline, C.B.; Lanier, E.R.; Quenelle, D.C. CMX001 potentiates the efficacy of acyclovir in herpes simplex virus infections. Antimicrob. Agents Chemother. 2011, 55, 4728–4734. [Google Scholar] [CrossRef]
- Lee, Y.J.; Neofytos, D.; Kim, S.J.; Cheteyan, L.; Huang, Y.T.; Papadopoulos, E.B.; Jakubowski, A.A.; Papanicolaou, G.A. Efficacy of brincidofovir as prophylaxis against HSV and VZV in hematopoietic cell transplant recipients. Transpl. Infect. Dis. 2018, 20, e12977. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, K.; Chono, K.; Suzuki, H. Antiviral efficacy of the helicase-primase inhibitor amenamevir in murine models of severe herpesvirus infection. Biochem. Pharmacol. 2018, 158, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Dropulic, L.K.; Oestreich, M.C.; Pietz, H.L.; Laing, K.J.; Hunsberger, S.; Lumbard, K.; Garabedian, D.; Turk, S.P.; Chen, A.; Hornung, R.L.; et al. A Randomized, Double-Blinded, Placebo-Controlled, Phase 1 Study of a Replication-Defective Herpes Simplex Virus (HSV) Type 2 Vaccine, HSV529, in Adults with or without HSV Infection. J. Infect. Dis. 2019, 220, 990–1000. [Google Scholar] [CrossRef]
- Egan, K.P.; Hook, L.M.; Naughton, A.; Pardi, N.; Awasthi, S.; Cohen, G.H.; Weissman, D.; Friedman, H.M. An HSV-2 nucleoside-modified mRNA genital herpes vaccine containing glycoproteins gC, gD, and gE protects mice against HSV-1 genital lesions and latent infection. PLoS Pathog. 2020, 16, e1008795. [Google Scholar] [CrossRef]
- Marrazzo, J.M.; Rabe, L.; Kelly, C.; Richardson, B.; Deal, C.; Schwartz, J.L.; Chirenje, Z.M.; Piper, J.; Morrow, R.A.; Hendrix, C.W.; et al. Tenofovir Gel for Prevention of Herpes Simplex Virus Type 2 Acquisition: Findings From the VOICE Trial. J. Infect. Dis. 2019, 219, 1940–1947. [Google Scholar] [CrossRef]
- Hill, E.K. Updates in Cervical Cancer Treatment. Clin. Obstet. Gynecol. 2020, 63, 3–11. [Google Scholar] [CrossRef]
- Mauricio, D.; Zeybek, B.; Tymon-Rosario, J.; Harold, J.; Santin, A.D. Immunotherapy in Cervical Cancer. Curr. Oncol. Rep. 2021, 23, 61. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Rotman, J.; den Otter, L.A.S.; Bleeker, M.C.G.; Samuels, S.S.; Heeren, A.M.; Roemer, M.G.M.; Kenter, G.G.; Zijlmans, H.; van Trommel, N.E.; de Gruijl, T.D.; et al. PD-L1 and PD-L2 Expression in Cervical Cancer: Regulation and Biomarker Potential. Front. Immunol. 2020, 11, 596825. [Google Scholar] [CrossRef] [PubMed]
- Kwok, G.; Yau, T.C.; Chiu, J.W.; Tse, E.; Kwong, Y.L. Pembrolizumab (Keytruda). Hum. Vaccines Immunother. 2016, 12, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Borcoman, E.; Le Tourneau, C. Keynote-158 study, FDA granted accelerated approval of pembrolizumab for the treatment of patients with advanced PD-L1-positive cervical cancer. Ann. Transl. Med. 2020, 8, 1611. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Ros, W.; Delord, J.P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef]
- Colombo, N.; Dubot, C.; Lorusso, D.; Caceres, M.V.; Hasegawa, K.; Shapira-Frommer, R.; Tewari, K.S.; Salman, P.; Hoyos Usta, E.; Yanez, E.; et al. Pembrolizumab for Persistent, Recurrent, or Metastatic Cervical Cancer. N. Engl. J. Med. 2021, 385, 1856–1867. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, H.; Chen, B. Nivolumab as Programmed Death-1 (PD-1) Inhibitor for Targeted Immunotherapy in Tumor. J. Cancer 2017, 8, 410–416. [Google Scholar] [CrossRef]
- Naumann, R.W.; Hollebecque, A.; Meyer, T.; Devlin, M.J.; Oaknin, A.; Kerger, J.; Lopez-Picazo, J.M.; Machiels, J.P.; Delord, J.P.; Evans, T.R.J.; et al. Safety and Efficacy of Nivolumab Monotherapy in Recurrent or Metastatic Cervical, Vaginal, or Vulvar Carcinoma: Results From the Phase I/II CheckMate 358 Trial. J. Clin. Oncol. 2019, 37, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Deng, W.; Frumovitz, M.; Buza, N.; Bellone, S.; Huh, W.; Khleif, S.; Lankes, H.A.; Ratner, E.S.; O’Cearbhaill, R.E.; et al. Phase II evaluation of nivolumab in the treatment of persistent or recurrent cervical cancer (NCT02257528/NRG-GY002). Gynecol. Oncol. 2020, 157, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69 (Suppl. S3), 4–10. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.; Shen, J.; Wang, Y.; Li, J.; Liu, Z.; He, M.; Cao, X.; Ling, J.; Huang, J.; Zheng, M.; et al. Camrelizumab Plus Apatinib in Patients with Advanced Cervical Cancer (CLAP): A Multicenter, Open-Label, Single-Arm, Phase II Trial. J. Clin. Oncol. 2020, 38, 4095–4106. [Google Scholar] [CrossRef] [PubMed]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Graziani, G.; Tentori, L.; Navarra, P. Ipilimumab: A novel immunostimulatory monoclonal antibody for the treatment of cancer. Pharmacol. Res. 2012, 65, 9–22. [Google Scholar] [CrossRef]
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab: First global approval. Drugs 2011, 71, 1093–1104. [Google Scholar] [CrossRef]
- Mayadev, J.S.; Enserro, D.; Lin, Y.G.; Da Silva, D.M.; Lankes, H.A.; Aghajanian, C.; Ghamande, S.; Moore, K.N.; Kennedy, V.A.; Fracasso, P.M.; et al. Sequential Ipilimumab After Chemoradiotherapy in Curative-Intent Treatment of Patients with Node-Positive Cervical Cancer. JAMA Oncol. 2020, 6, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Naumann, R.; Oaknin, A.; Meyer, T.; Lopez-Picazo, J.; Lao, C.; Bang, Y.-J.; Boni, V.; Sharfman, W.; Park, J.; Devriese, L.; et al. LBA62—Efficacy and safety of nivolumab (Nivo) + ipilimumab (Ipi) in patients (pts) with recurrent/metastatic (R/M) cervical cancer: Results from CheckMate 358. Ann. Oncol. 2019, 30, v898–v899. [Google Scholar] [CrossRef]
- Kasthuri, R.S.; Taubman, M.B.; Mackman, N. Role of tissue factor in cancer. J. Clin. Oncol. 2009, 27, 4834–4838. [Google Scholar] [CrossRef]
- de Bono, J.S.; Concin, N.; Hong, D.S.; Thistlethwaite, F.C.; Machiels, J.P.; Arkenau, H.T.; Plummer, R.; Jones, R.H.; Nielsen, D.; Windfeld, K.; et al. Tisotumab vedotin in patients with advanced or metastatic solid tumours (InnovaTV 201): A first-in-human, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 383–393. [Google Scholar] [CrossRef]
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; Gonzalez-Martin, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Tewari, K.S.; Sill, M.W.; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; Michael, H.E.; et al. Bevacizumab for advanced cervical cancer: Final overall survival and adverse event analysis of a randomised, controlled, open-label, phase 3 trial (Gynecologic Oncology Group 240). Lancet 2017, 390, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Kramer, I.; Lipp, H.P. Bevacizumab, a humanized anti-angiogenic monoclonal antibody for the treatment of colorectal cancer. J. Clin. Pharm. Ther. 2007, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Zhu, L.; Chen, L. Progress in research on paclitaxel and tumor immunotherapy. Cell. Mol. Biol. Lett. 2019, 24, 40. [Google Scholar] [CrossRef]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Cervical Cancer (Version 1.2023). Available online: https://www.nccn.org/professionals/physician_gls/pdf/cervical.pdf (accessed on 29 June 2023).
- Potter, R.; Tanderup, K.; Schmid, M.P.; Jurgenliemk-Schulz, I.; Haie-Meder, C.; Fokdal, L.U.; Sturdza, A.E.; Hoskin, P.; Mahantshetty, U.; Segedin, B.; et al. MRI-guided adaptive brachytherapy in locally advanced cervical cancer (EMBRACE-I): A multicentre prospective cohort study. Lancet Oncol. 2021, 22, 538–547. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, J.; Yang, S.; Chen, X.; Gu, C.; Liang, T.; Li, L.; Liu, D.; Cao, Y. Therapeutic effects and prognostic factors of (125)I brachytherapy for pelvic recurrence after early cervical cancer surgery. Sci. Rep. 2021, 11, 11356. [Google Scholar] [CrossRef]
- Huang, H.; Feng, Y.L.; Wan, T.; Zhang, Y.N.; Cao, X.P.; Huang, Y.W.; Xiong, Y.; Huang, X.; Zheng, M.; Li, Y.F.; et al. Effectiveness of Sequential Chemoradiation vs Concurrent Chemoradiation or Radiation Alone in Adjuvant Treatment After Hysterectomy for Cervical Cancer: The STARS Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 361–369. [Google Scholar] [CrossRef]
- Mileshkin, L.R.; Moore, K.N.; Barnes, E.H.; Gebski, V.; Narayan, K.; King, M.T.; Bradshaw, N.; Lee, Y.C.; Diamante, K.; Fyles, A.W.; et al. Adjuvant chemotherapy following chemoradiotherapy as primary treatment for locally advanced cervical cancer versus chemoradiotherapy alone (OUTBACK): An international, open-label, randomised, phase 3 trial. Lancet Oncol. 2023, 24, 468–482. [Google Scholar] [CrossRef]
- Santos Apolonio, J.; Lima de Souza Goncalves, V.; Cordeiro Santos, M.L.; Silva Luz, M.; Silva Souza, J.V.; Rocha Pinheiro, S.L.; de Souza, W.R.; Sande Loureiro, M.; de Melo, F.F. Oncolytic virus therapy in cancer: A current review. World J. Virol. 2021, 10, 229–255. [Google Scholar] [CrossRef]
- Kagabu, M.; Yoshino, N.; Saito, T.; Miura, Y.; Takeshita, R.; Murakami, K.; Kawamura, H.; Baba, T.; Sugiyama, T. The efficacy of a third-generation oncolytic herpes simplex viral therapy for an HPV-related uterine cervical cancer model. Int. J. Clin. Oncol. 2021, 26, 591–597. [Google Scholar] [CrossRef]
- Ni, J.; Feng, H.; Xu, X.; Liu, T.; Ye, T.; Chen, K.; Li, G. Oncolytic Vaccinia Virus Harboring Aphrocallistes vastus Lectin Inhibits the Growth of Cervical Cancer Cells Hela S3. Mar. Drugs 2021, 19, 532. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Saga, Y.; Uchibori, R.; Tsukahara, T.; Urabe, M.; Kume, A.; Fujiwara, H.; Suzuki, M.; Ozawa, K.; Mizukami, H. Eradication of cervical cancer in vivo by an AAV vector that encodes shRNA targeting human papillomavirus type 16 E6/E7. Int. J. Oncol. 2018, 52, 687–696. [Google Scholar] [CrossRef]
- Trimble, C.L.; Morrow, M.P.; Kraynyak, K.A.; Shen, X.; Dallas, M.; Yan, J.; Edwards, L.; Parker, R.L.; Denny, L.; Giffear, M.; et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 2015, 386, 2078–2088. [Google Scholar] [CrossRef]
- Bhuyan, P.K.; Dallas, M.; Kraynyak, K.; Herring, T.; Morrow, M.; Boyer, J.; Duff, S.; Kim, J.; Weiner, D.B. Durability of response to VGX-3100 treatment of HPV16/18 positive cervical HSIL. Hum. Vaccines Immunother. 2021, 17, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Komdeur, F.L.; Singh, A.; van de Wall, S.; Meulenberg, J.J.M.; Boerma, A.; Hoogeboom, B.N.; Paijens, S.T.; Oyarce, C.; de Bruyn, M.; Schuuring, E.; et al. First-in-Human Phase I Clinical Trial of an SFV-Based RNA Replicon Cancer Vaccine against HPV-Induced Cancers. Mol. Ther. 2021, 29, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.W.; Hur, S.Y.; Woo, J.W.; Kim, Y.M.; Lim, M.C.; Park, S.Y.; Seo, S.S.; No, J.H.; Kim, B.G.; Lee, J.K.; et al. Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: Interim results of a single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1653–1660. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Relationship Exists | Relationship Uncertain | Findings | Reference |
|---|---|---|---|---|
| McDougall et al. | X | Presence of HSV-2 RNA in cells undergoing pre-malignant changes; no evidence of HSV-2 RNA in squamous cell cancer cells | [159] | |
| Li et al. | X | HSV-2 associated with the occurrence of cervical cancer; coinfection with both HPV and HSV-2 had a higher relative risk than either infection with HSV-2 or HPV alone | [160] | |
| Bahena-Román et al. | X | Patients seropositive for HSV-2 had a higher risk of high-risk HPV; patients with high-risk HPV had a higher rate of active HSV-2 infection | [161] | |
| de Abreu et al. | X | Increased risk of developing ≥ ASC-US cytology (initial cellular transformation) but not other cervical abnormalities such as HSIL. The authors concluded that HSV-2 may be involved in initial cellular transformation but not progression | [164] | |
| DiPaolo et al. | X | Bg/II N region of HSV-2 converted immortalized genital epithelial cells into tumorigenic squamous cells | [15] | |
| Pisani et al. | X | HSV-2 infection increased HPV gene expression of E1, E2 and E6 in HPV-16-infected cells | [165] | |
| Paba et al. | X | HPV/HSV-2 coinfected cells overexpressed cellular protein survivin | [166] | |
| Hara et al. | X | HSV-1 DNA polymerase could facilitate HPV-18 DNA replication | [167] | |
| Koanga et al. | X | HSV-2 infection may induce cervical inflammation, potentially acting as a co-factor in cervical cancer formation | [168] | |
| Okoye et al. | X | Immunocompromised status may facilitate viral-mediated oncogenesis | [169] | |
| Cao et al. | X | Current evidence does not support the idea that HSV-2 plays a role in cervical cancer development | [174] | |
| Lehtinen et al. | X | HSV-2 did not contribute to cervical cancer pathogenesis in Nordic countries | [175] | |
| Ahmadi et al. | X | Only one instance of HSV-2 infection found out of 45 patients with cervical cancer | [14] | |
| Joharinia et al. | X | No detection of HSV genomes in any patients diagnosed with low-grade squamous intraepithelial lesions, high-grade squamous intraepithelial lesions, squamous cell carcinoma or adenocarcinoma of the cervix | [176] |
| Drug | Mechanism of Action | References |
|---|---|---|
| Pembrolizumab | PD-1 inhibitor | [200,201,202,203] |
| Nivolumab | PD-1 inhibitor | [204,205,206,213] |
| Apatinib + camrelizumab | Apatinib: Tyrosine kinase inhibitor of VEGFR2 Camrelizumab: PD-1 inhibitor | [208] |
| Ipilimumab | CTLA-4 inhibitor | [211,212,213] |
| Nivolumab + ipilimumab | Nivolumab: PD-1 inhibitor Ipilimumab: CTLA-4 inhibitor | [213] |
| Tisotumab vedotin | Antibody/drug conjugate targeting tissue factor | [215,216] |
| Bevacizumab | Angiogenesis inhibitor | [217,218] |
| Brachytherapy | Radiation | [223,224] |
| SCRT v. CCRT v. RT | N/a | [225] |
| Oncolytic viruses | Cytopathic and cytotoxic killing of tumor cells | [227,228,229,230] |
| Vaccine therapies | Targeting E6/E7 | [81,231,232,233,234] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sausen, D.G.; Shechter, O.; Gallo, E.S.; Dahari, H.; Borenstein, R. Herpes Simplex Virus, Human Papillomavirus, and Cervical Cancer: Overview, Relationship, and Treatment Implications. Cancers 2023, 15, 3692. https://doi.org/10.3390/cancers15143692
Sausen DG, Shechter O, Gallo ES, Dahari H, Borenstein R. Herpes Simplex Virus, Human Papillomavirus, and Cervical Cancer: Overview, Relationship, and Treatment Implications. Cancers. 2023; 15(14):3692. https://doi.org/10.3390/cancers15143692
Chicago/Turabian StyleSausen, Daniel G., Oren Shechter, Elisa S. Gallo, Harel Dahari, and Ronen Borenstein. 2023. "Herpes Simplex Virus, Human Papillomavirus, and Cervical Cancer: Overview, Relationship, and Treatment Implications" Cancers 15, no. 14: 3692. https://doi.org/10.3390/cancers15143692
APA StyleSausen, D. G., Shechter, O., Gallo, E. S., Dahari, H., & Borenstein, R. (2023). Herpes Simplex Virus, Human Papillomavirus, and Cervical Cancer: Overview, Relationship, and Treatment Implications. Cancers, 15(14), 3692. https://doi.org/10.3390/cancers15143692