
Co-Targeting FASN and mTOR Suppresses Uveal Melanoma Growth

 , , , , , , , , ,
, , , , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Generation of Cell Lines
2.2. Single-Cell RNA Analysis
2.3. Isolation of Human Choroidal Melanocytes
2.4. Analysis of Mass Spectrum Data
2.5. Tissue Specimens
2.6. Inhibitors
2.7. Short-Interfering RNA (siRNA) Transfection
2.8. Cell Growth Assay
2.9. Reverse Phase Protein Array (RPPA)
2.10. Annexin V/Propidium Iodide (PI) Apoptosis Assay
2.11. EdU Cell S Phase Entry Assay
2.12. Tumor 3D Spheroid Formation
2.13. Immunohistochemistry (IHC) and Scoring
2.14. Western Blotting
2.15. Metabolomics and Lipidomics
2.16. Statistical Analysis
3. Results
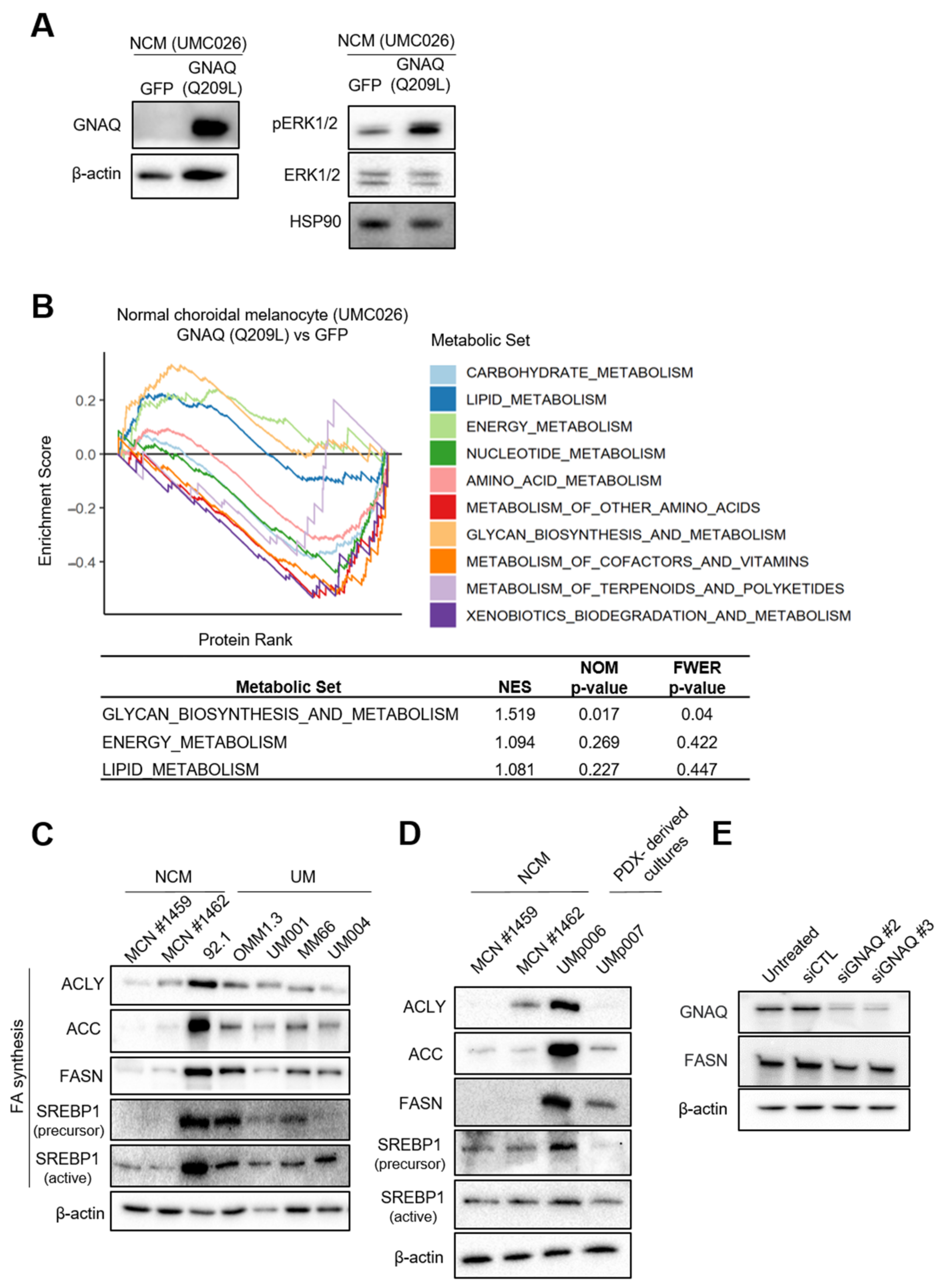
3.1. UM Cells Have Elevated Lipogenic Enzyme Expression
3.2. Co-Targeting FASN and mTOR Represses the Growth of UM Cells
3.3. Inhibition of FASN and mTOR Alters Cell Cycle Regulator Expression in UM Cells
3.4. Inhibition of FASN and mTOR Reduce Cell Growth through Cell Cycle Arrest in UM Cells
3.5. FASN and mTOR Inhibitors Diminish Fatty Acid Biosynthesis in UM Cells
3.6. Suppression of FASN and mTOR Impairs Glucose Utilization and TCA Cycle Metabolism in UM Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Schulze, A. Lipid desaturation—The next step in targeting lipogenesis in cancer? FEBS J. 2016, 283, 2767–2778. [Google Scholar] [CrossRef] [PubMed]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar]
- Ray, U.; Roy, S.S. Aberrant lipid metabolism in cancer cells—The role of oncolipid-activated signaling. FEBS J. 2018, 285, 432–443. [Google Scholar] [CrossRef]
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef]
- Szutowicz, A.; Kwiatkowski, J.; Angielski, S. Lipogenetic and glycolytic enzyme activities in carcinoma and nonmalignant diseases of the human breast. Br. J. Cancer 1979, 39, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.M.; Yeung, S.C.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gomez, H.; Kellum, J.A.; Ronco, C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat. Rev. Nephrol. 2017, 13, 143–151. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef]
- Guo, D.; Bell, E.H.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Sharen, G.; Yuan, F.; Peng, Y.; Chen, R.; Zhou, X.; Wei, H.; Li, B.; Jing, W.; Zhao, J. TIP30 regulates lipid metabolism in hepatocellular carcinoma by regulating SREBP1 through the Akt/mTOR signaling pathway. Oncogenesis 2017, 6, e347. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Oh, Y.-T.; Yue, P.; Khuri, F.R.; Sun, S.-Y. Inhibition of mTOR complex 2 induces GSK3/FBXW7-dependent degradation of sterol regulatory element-binding protein 1 (SREBP1) and suppresses lipogenesis in cancer cells. Oncogene 2016, 35, 642. [Google Scholar] [CrossRef]
- Krantz, B.A.; Dave, N.; Komatsubara, K.M.; Marr, B.P.; Carvajal, R.D. Uveal melanoma: Epidemiology, etiology, and treatment of primary disease. Clin. Ophthalmol. 2017, 11, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.D.; Bergman, L.; Seregard, S. Uveal melanoma: Epidemiologic aspects. Ophthalmol. Clin. N. Am. 2005, 18, 75–84. [Google Scholar] [CrossRef]
- Souto, E.B.; Zielinska, A.; Luis, M.; Carbone, C.; Martins-Gomes, C.; Souto, S.B.; Silva, A.M. Uveal melanoma: Physiopathology and new in situ-specific therapies. Cancer Chemother. Pharmacol. 2019, 84, 15–32. [Google Scholar] [CrossRef]
- Chua, V.; Aplin, A.E. Novel therapeutic strategies and targets in advanced uveal melanoma. Curr. Opin. Oncol. 2018, 30, 134–141. [Google Scholar] [CrossRef]
- Johansson, P.A.; Brooks, K.; Newell, F.; Palmer, J.M.; Wilmott, J.S.; Pritchard, A.L.; Broit, N.; Wood, S.; Carlino, M.S.; Leonard, C.; et al. Whole genome landscapes of uveal melanoma show an ultraviolet radiation signature in iris tumours. Nat. Commun. 2020, 11, 2408. [Google Scholar] [CrossRef]
- Karlsson, J.; Nilsson, L.M.; Mitra, S.; Alsen, S.; Shelke, G.V.; Sah, V.R.; Forsberg, E.M.V.; Stierner, U.; All-Eriksson, C.; Einarsdottir, B.; et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat. Commun. 2020, 11, 1894. [Google Scholar] [CrossRef]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef]
- Caksa, S.; Baqai, U.; Aplin, A.E. The future of targeted kinase inhibitors in melanoma. Pharmacol. Ther. 2022, 239, 108200. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination with Dacarbazine in Patients with Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Heppt, M.V.; Amaral, T.; Kahler, K.C.; Heinzerling, L.; Hassel, J.C.; Meissner, M.; Kreuzberg, N.; Loquai, C.; Reinhardt, L.; Utikal, J.; et al. Combined immune checkpoint blockade for metastatic uveal melanoma: A retrospective, multi-center study. J. Immunother. Cancer 2019, 7, 299. [Google Scholar] [CrossRef]
- Xu, B.; Ma, R.; Ren, H.; Qian, J. Genome-Wide Analysis of Uveal Melanoma Metastasis-Associated LncRNAs and Their Functional Network. DNA Cell. Biol. 2018, 37, 99–108. [Google Scholar] [CrossRef]
- Ness, C.; Garred, Ø.; Eide, N.A.; Kumar, T.; Olstad, O.K.; Bærland, T.P.; Petrovski, G.; Moe, M.C.; Noer, A. Multicellular tumor spheroids of human uveal melanoma induce genes associated with anoikis resistance, lipogenesis, and SSXs. Mol. Vis. 2017, 23, 680–694. [Google Scholar]
- Han, A.; Schug, Z.T.; Aplin, A.E. Metabolic Alterations and Therapeutic Opportunities in Rare Forms of Melanoma. Trends Cancer 2021, 7, 671–681. [Google Scholar] [CrossRef]
- Chattopadhyay, C.; Oba, J.; Roszik, J.; Marszalek, J.R.; Chen, K.; Qi, Y.; Eterovic, K.; Robertson, A.G.; Burks, J.K.; McCannel, T.A.; et al. Elevated Endogenous SDHA Drives Pathological Metabolism in Highly Metastatic Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4187–4195. [Google Scholar] [CrossRef] [PubMed]
- Chua, V.; Han, A.; Bechtel, N.; Purwin, T.J.; Hunter, E.; Liao, C.; Harbour, J.W.; Aplin, A.E. The AMP-dependent kinase pathway is upregulated in BAP1 mutant uveal melanoma. Pigment Cell Melanoma Res. 2021, 35, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Han, A.; Chua, V.; Baqai, U.; Purwin, T.J.; Bechtel, N.; Hunter, E.; Tiago, M.; Seifert, E.; Speicher, D.W.; Schug, Z.T.; et al. Pyruvate dehydrogenase inactivation causes glycolytic phenotype in BAP1 mutant uveal melanoma. Oncogene 2022, 41, 1129–1139. [Google Scholar] [CrossRef]
- Han, A.; Purwin, T.J.; Aplin, A.E. Roles of the BAP1 Tumor Suppressor in Cell Metabolism. Cancer Res. 2021, 81, 2807–2814. [Google Scholar] [CrossRef]
- Han, A.; Purwin, T.J.; Bechtel, N.; Liao, C.; Chua, V.; Seifert, E.; Sato, T.; Schug, Z.T.; Speicher, D.W.; William Harbour, J.; et al. BAP1 mutant uveal melanoma is stratified by metabolic phenotypes with distinct vulnerability to metabolic inhibitors. Oncogene 2021, 40, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Chua, V.; Liao, C.; Purwin, T.J.; Terai, M.; Kageyama, K.; Davies, M.A.; Sato, T.; Aplin, A.E. Co-targeting HGF/cMET Signaling with MEK Inhibitors in Metastatic Uveal Melanoma. Mol. Cancer Ther. 2017, 16, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Raimondi, F.; Kadji, F.M.N.; Singh, G.; Kishi, T.; Uwamizu, A.; Ono, Y.; Shinjo, Y.; Ishida, S.; Arang, N.; et al. Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 2019, 177, 1933–1947.e1925. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck III, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Djigo, A.D.; Berube, J.; Landreville, S.; Proulx, S. Characterization of a tissue-engineered choroid. Acta Biomater. 2019, 84, 305–316. [Google Scholar] [CrossRef]
- Hu, D.N.; McCormick, S.A.; Ritch, R.; Pelton-Henrion, K. Studies of human uveal melanocytes in vitro: Isolation, purification and cultivation of human uveal melanocytes. Investig. Ophthalmol. Vis. Sci. 1993, 34, 2210–2219. [Google Scholar]
- Kaler, C.J.; Dollar, J.J.; Cruz, A.M.; Kuznetsoff, J.N.; Sanchez, M.I.; Decatur, C.L.; Licht, J.D.; Smalley, K.S.M.; Correa, Z.M.; Kurtenbach, S.; et al. BAP1 Loss Promotes Suppressive Tumor Immune Microenvironment via Upregulation of PROS1 in Class 2 Uveal Melanomas. Cancers 2022, 14, 3678. [Google Scholar] [CrossRef]
- Kuznetsov, J.N.; Aguero, T.H.; Owens, D.A.; Kurtenbach, S.; Field, M.G.; Durante, M.A.; Rodriguez, D.A.; King, M.L.; Harbour, J.W. BAP1 regulates epigenetic switch from pluripotency to differentiation in developmental lineages giving rise to BAP1-mutant cancers. Sci. Adv. 2019, 5, eaax1738. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Tibes, R.; Qiu, Y.; Lu, Y.; Hennessy, B.; Andreeff, M.; Mills, G.B.; Kornblau, S.M. Reverse phase protein array: Validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther. 2006, 5, 2512–2521. [Google Scholar] [CrossRef]
- Sandri, S.; Faiao-Flores, F.; Tiago, M.; Pennacchi, P.C.; Massaro, R.R.; Alves-Fernandes, D.K.; Berardinelli, G.N.; Evangelista, A.F.; de Lima Vazquez, V.; Reis, R.M.; et al. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol. Res. 2016, 111, 523–533. [Google Scholar] [CrossRef]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef]
- Melamud, E.; Vastag, L.; Rabinowitz, J.D. Metabolomic analysis and visualization engine for LC-MS data. Anal. Chem. 2010, 82, 9818–9826. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, Q.; Tan, L.; Porter, D.; Jager, M.J.; Emery, C.; Bastian, B.C. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 2014, 33, 4724–4734. [Google Scholar] [CrossRef] [PubMed]
- Phelps, G.B.; Hagen, H.R.; Amsterdam, A.; Lees, J.A. MITF deficiency accelerates GNAQ-driven uveal melanoma. Proc. Natl. Acad. Sci. USA 2022, 119, e2107006119. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef]
- Larribere, L.; Utikal, J. Update on GNA Alterations in Cancer: Implications for Uveal Melanoma Treatment. Cancers 2020, 12, 1524. [Google Scholar] [CrossRef] [PubMed]
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W. Fasnall, a selective FASN inhibitor, shows potent anti-tumor activity in the MMTV-Neu model of HER2+ breast cancer. Cell Chem. Biol. 2016, 23, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, Y.; Miyazaki, H.; Vaidyan, L.K.; Kagawa, Y.; Ebrahimi, M.; Yamamoto, Y.; Ogata, M.; Katsuyama, Y.; Sadahiro, H.; Suzuki, M. Inhibition of fatty acid synthase decreases expression of stemness markers in glioma stem cells. PLoS ONE 2016, 11, e0147717. [Google Scholar] [CrossRef]
- Oh, J.E.; Jung, B.H.; Park, J.; Kang, S.; Lee, H. Deciphering Fatty Acid Synthase Inhibition-Triggered Metabolic Flexibility in Prostate Cancer Cells through Untargeted Metabolomics. Cells 2020, 9, 2447. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Ricoult, S.J.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef]
- Bueno, M.J.; Jimenez-Renard, V.; Samino, S.; Capellades, J.; Junza, A.; Lopez-Rodriguez, M.L.; Garcia-Carceles, J.; Lopez-Fabuel, I.; Bolanos, J.P.; Chandel, N.S.; et al. Essentiality of fatty acid synthase in the 2D to anchorage-independent growth transition in transforming cells. Nat. Commun. 2019, 10, 5011. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Harris, J.W.; Mitov, M.I.; Kim, J.T.; Butterfield, D.A.; Lee, E.Y.; Weiss, H.L.; Gao, T.; Evers, B.M. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget 2015, 6, 18891–18904. [Google Scholar] [CrossRef]
- Mignard, C.; Deschamps Huvier, A.; Gillibert, A.; Duval Modeste, A.B.; Dutriaux, C.; Khammari, A.; Avril, M.F.; Kramkimel, N.; Mortier, L.; Marcant, P.; et al. Efficacy of Immunotherapy in Patients with Metastatic Mucosal or Uveal Melanoma. J. Oncol. 2018, 2018, 1908065. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Rychahou, P.G.; Le, A.-T.; Scott, T.L.; Flight, R.M.; Kim, J.T.; Harris, J.; Liu, J.; Wang, C.; Morris, A.J. Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer. Oncotarget 2018, 9, 24787. [Google Scholar] [CrossRef] [PubMed]
- Nanda, C.S.; Venkateswaran, S.V.; Patani, N.; Yuneva, M. Defining a metabolic landscape of tumours: Genome meets metabolism. Br. J. Cancer 2020, 122, 136–149. [Google Scholar] [CrossRef]
- Rabbie, R.; Ferguson, P.; Molina-Aguilar, C.; Adams, D.J.; Robles-Espinoza, C.D. Melanoma subtypes: Genomic profiles, prognostic molecular markers and therapeutic possibilities. J. Pathol. 2019, 247, 539–551. [Google Scholar] [CrossRef]
- Zukotynski, K.; Yap, J.T.; Giobbie-Hurder, A.; Weber, J.; Gonzalez, R.; Gajewski, T.F.; O’Day, S.; Kim, K.; Hodi, F.S.; Van den Abbeele, A.D. Metabolic response by FDG-PET to imatinib correlates with exon 11 KIT mutation and predicts outcome in patients with mucosal melanoma. Cancer Imaging 2014, 14, 30. [Google Scholar] [CrossRef]
- An, J.; Wan, H.; Zhou, X.; Hu, D.-N.; Wang, L.; Hao, L.; Yan, D.; Shi, F.; Zhou, Z.; Wang, J. A comparative transcriptomic analysis of uveal melanoma and normal uveal melanocyte. PLoS ONE 2011, 6, e16516. [Google Scholar] [CrossRef]
- Giallongo, S.; Di Rosa, M.; Caltabiano, R.; Longhitano, L.; Reibaldi, M.; Distefano, A.; Lo Re, O.; Amorini, A.M.; Puzzo, L.; Salvatorelli, L.; et al. Loss of macroH2A1 decreases mitochondrial metabolism and reduces the aggressiveness of uveal melanoma cells. Aging 2020, 12, 9745–9760. [Google Scholar] [CrossRef] [PubMed]
- Angi, M.; Kalirai, H.; Prendergast, S.; Simpson, D.; Hammond, D.E.; Madigan, M.C.; Beynon, R.J.; Coupland, S.E. In-depth proteomic profiling of the uveal melanoma secretome. Oncotarget 2016, 7, 49623–49635. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, A.; Brożyna, A.A.; Podgórska, E.; Elas, M.; Urbańska, K.; Jetten, A.M.; Slominski, A.T.; Jóźwicki, W.; Orłowska-Heitzman, J.; Dyduch, G. Vitamin D receptors (VDR), hydroxylases CYP27B1 and CYP24A1 and retinoid-related orphan receptors (ROR) level in human uveal tract and ocular melanoma with different melanization levels. Sci. Rep. 2019, 9, 9142. [Google Scholar] [CrossRef]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671. [Google Scholar] [CrossRef]
- Amirouchene-Angelozzi, N.; Nemati, F.; Gentien, D.; Nicolas, A.; Dumont, A.; Carita, G.; Camonis, J.; Desjardins, L.; Cassoux, N.; Piperno-Neumann, S. Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol. Oncol. 2014, 8, 1508–1520. [Google Scholar] [CrossRef] [PubMed]
- Amirouchene-Angelozzi, N.; Frisch-Dit-Leitz, E.; Carita, G.; Dahmani, A.; Raymondie, C.; Liot, G.; Gentien, D.; Némati, F.; Decaudin, D.; Roman-Roman, S. The mTOR inhibitor Everolimus synergizes with the PI3K inhibitor GDC0941 to enhance anti-tumor efficacy in uveal melanoma. Oncotarget 2016, 7, 23633. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ho, A.L.; Musi, E.; Ambrosini, G.; Nair, J.S.; Vasudeva, S.D.; de Stanchina, E.; Schwartz, G.K. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PLoS ONE 2012, 7, e40439. [Google Scholar] [CrossRef]
- Guichard, S.M.; Curwen, J.; Bihani, T.; D’Cruz, C.M.; Yates, J.W.; Grondine, M.; Howard, Z.; Davies, B.R.; Bigley, G.; Klinowska, T.; et al. AZD2014, an Inhibitor of mTORC1 and mTORC2, Is Highly Effective in ER+ Breast Cancer When Administered Using Intermittent or Continuous Schedules. Mol. Cancer Ther. 2015, 14, 2508–2518. [Google Scholar] [CrossRef]
- Bakan, I.; Laplante, M. Connecting mTORC1 signaling to SREBP-1 activation. Curr. Opin. Lipidol. 2012, 23, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, A.; Mukha, D.; Chua, V.; Purwin, T.J.; Tiago, M.; Modasia, B.; Baqai, U.; Aumiller, J.L.; Bechtel, N.; Hunter, E.; et al. Co-Targeting FASN and mTOR Suppresses Uveal Melanoma Growth. Cancers 2023, 15, 3451. https://doi.org/10.3390/cancers15133451
Han A, Mukha D, Chua V, Purwin TJ, Tiago M, Modasia B, Baqai U, Aumiller JL, Bechtel N, Hunter E, et al. Co-Targeting FASN and mTOR Suppresses Uveal Melanoma Growth. Cancers. 2023; 15(13):3451. https://doi.org/10.3390/cancers15133451
Chicago/Turabian StyleHan, Anna, Dzmitry Mukha, Vivian Chua, Timothy J. Purwin, Manoela Tiago, Bhavik Modasia, Usman Baqai, Jenna L. Aumiller, Nelisa Bechtel, Emily Hunter, and et al. 2023. "Co-Targeting FASN and mTOR Suppresses Uveal Melanoma Growth" Cancers 15, no. 13: 3451. https://doi.org/10.3390/cancers15133451
APA StyleHan, A., Mukha, D., Chua, V., Purwin, T. J., Tiago, M., Modasia, B., Baqai, U., Aumiller, J. L., Bechtel, N., Hunter, E., Danielson, M., Terai, M., Wedegaertner, P. B., Sato, T., Landreville, S., Davies, M. A., Kurtenbach, S., Harbour, J. W., Schug, Z. T., & Aplin, A. E. (2023). Co-Targeting FASN and mTOR Suppresses Uveal Melanoma Growth. Cancers, 15(13), 3451. https://doi.org/10.3390/cancers15133451