
Sorafenib in Molecularly Selected Cancer Patients: Final Analysis of the MOST-Plus Sorafenib Cohort

 ,
,  ,
,  ,
,  , ,
, ,  ,
,  , , , ,
, , , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Study Procedures and Assessments
2.3. Endpoints and Statistics
3. Results
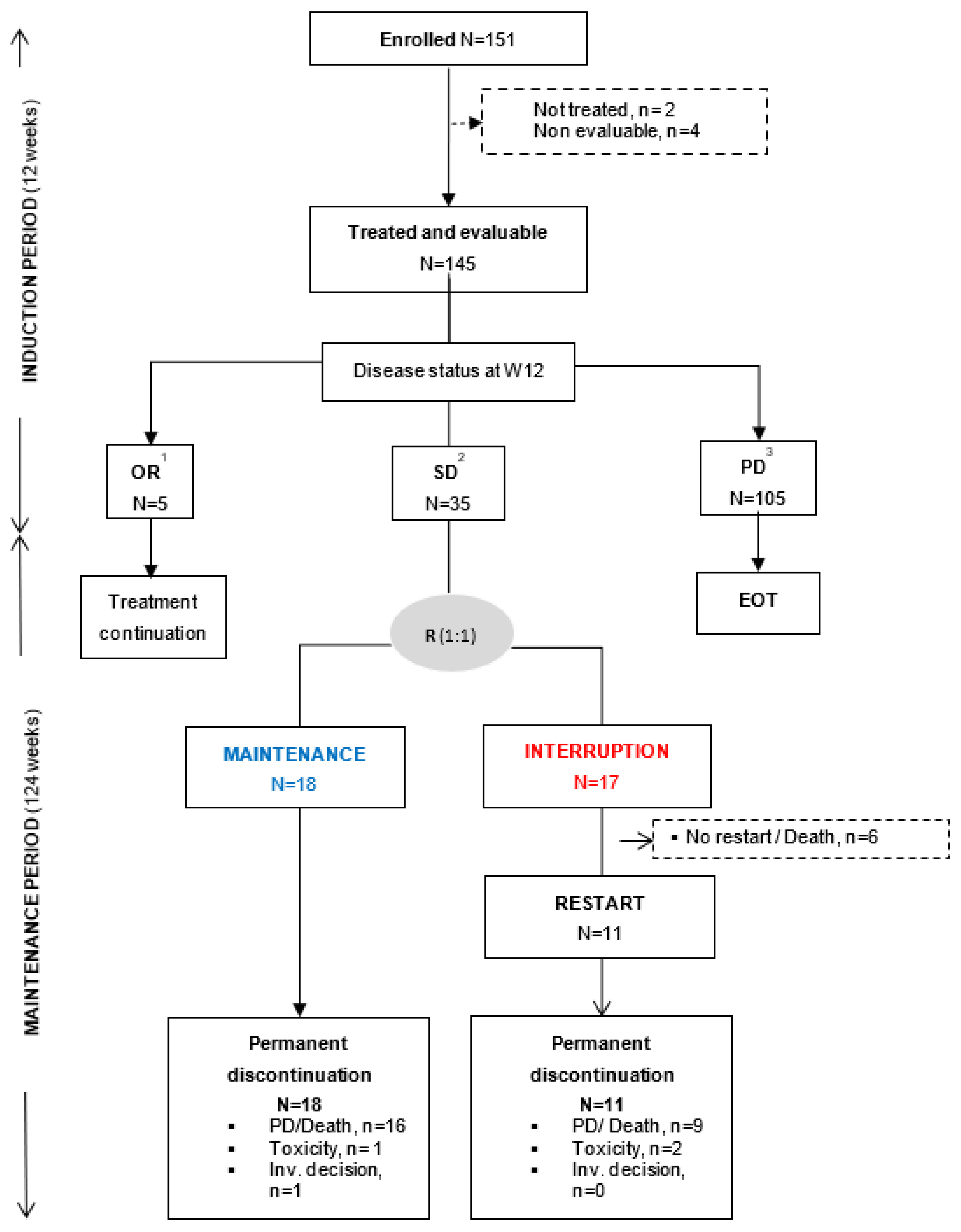
3.1. Patient Characteristics and Trial Profile
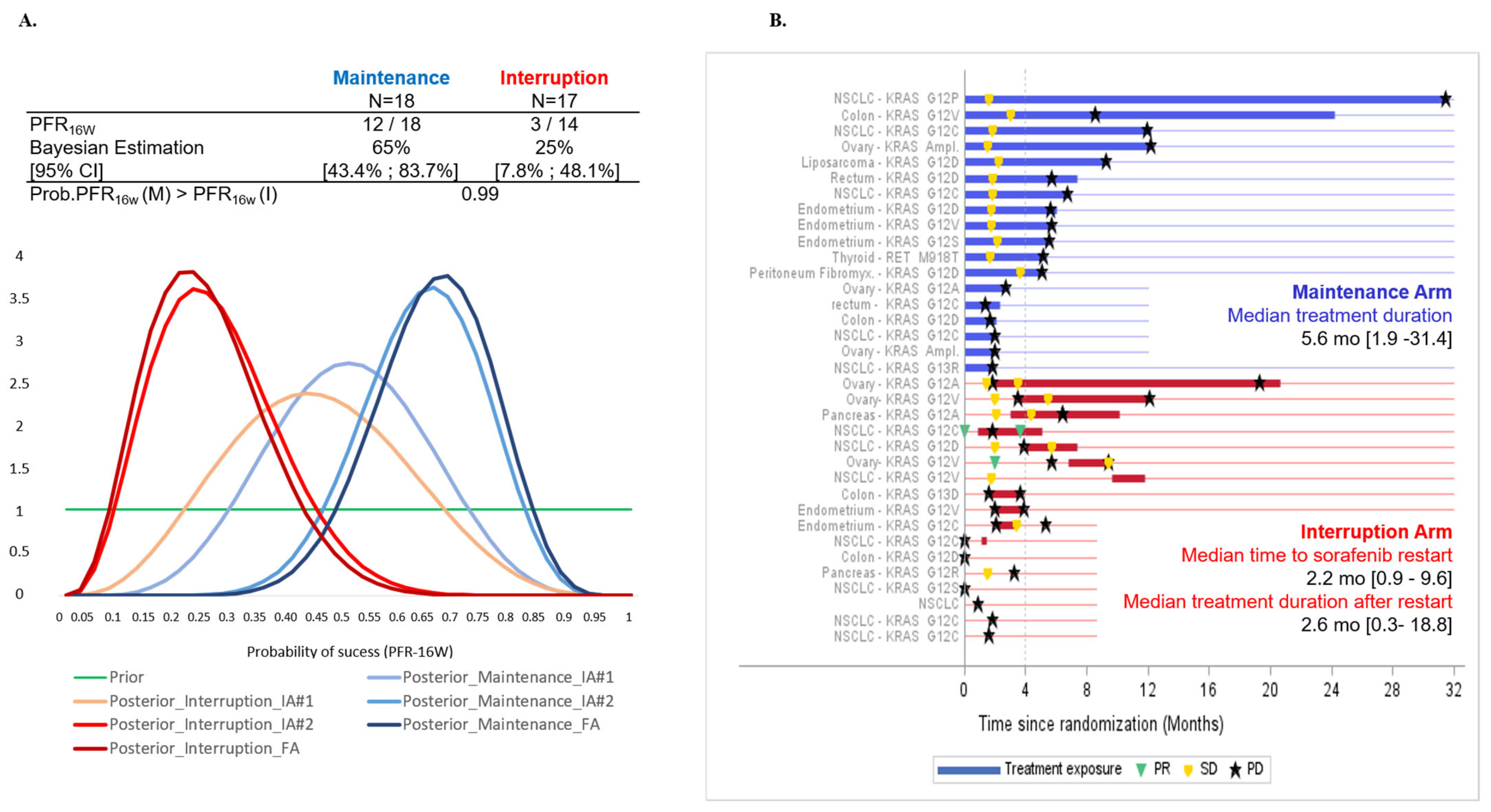
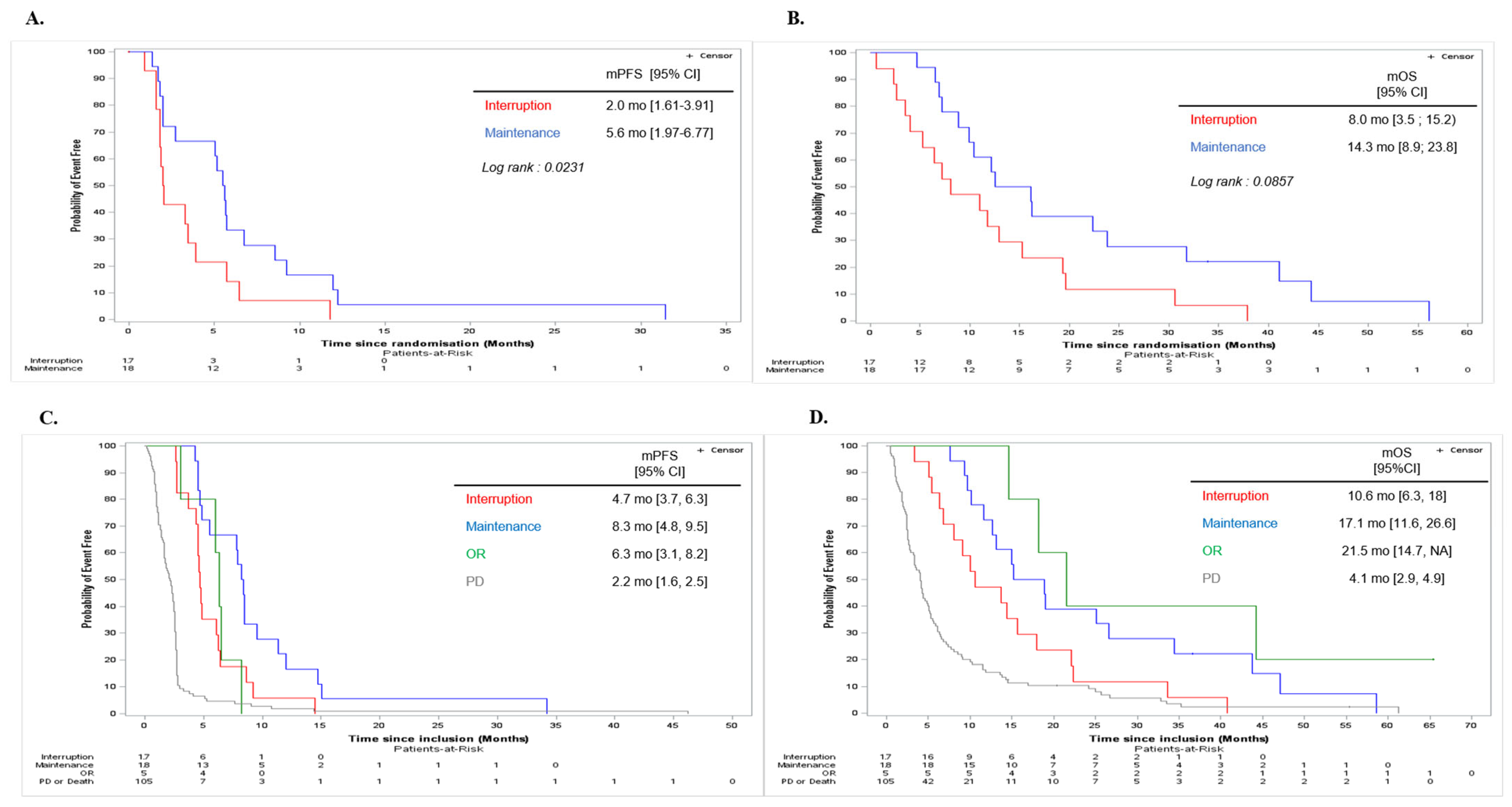
3.2. Primary Efficacy Endpoint
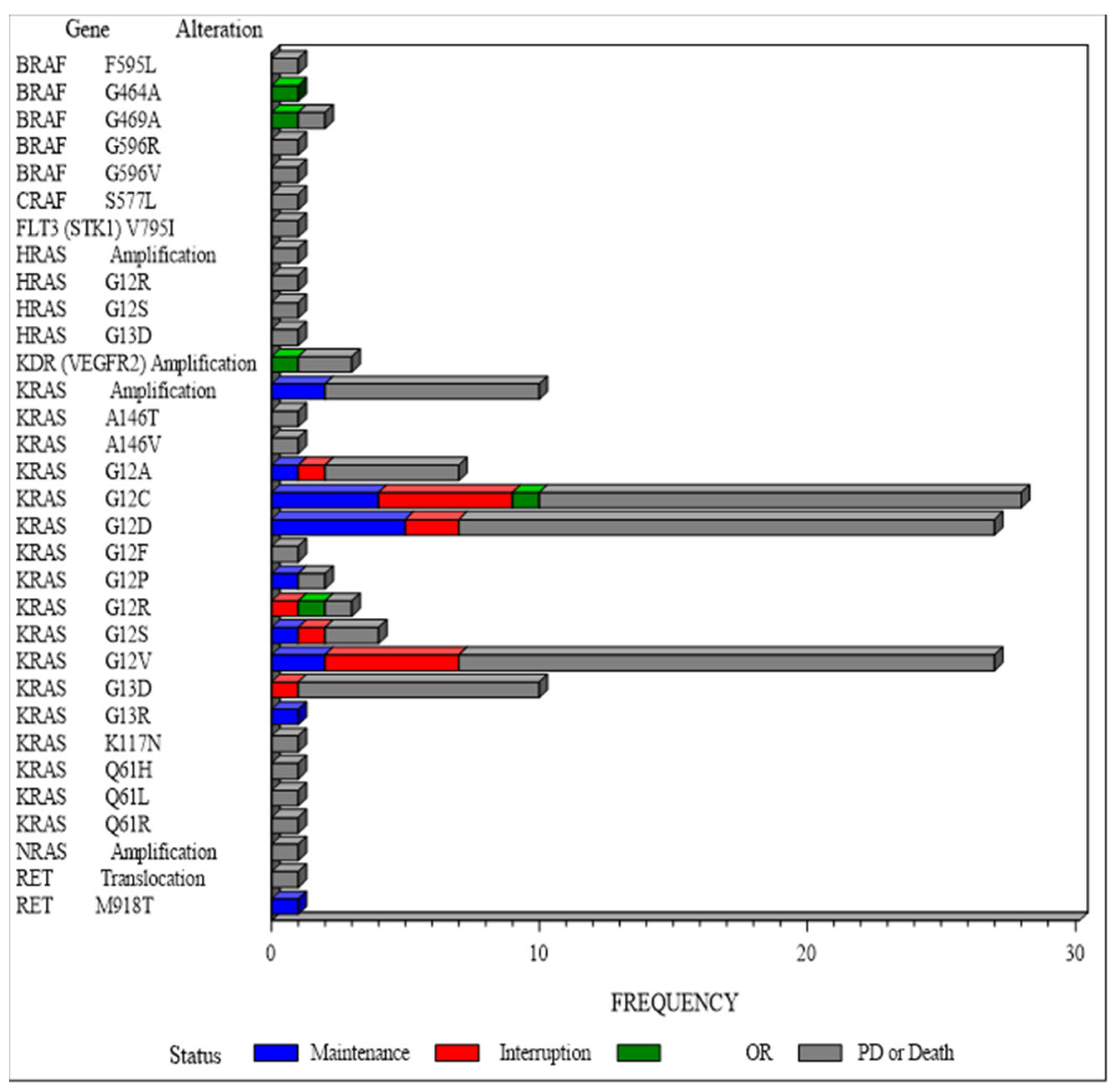
3.3. Secondary Efficacy Endpoints
3.4. Safety Endpoints
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dancey, J.E.; Bedard, P.L.; Onetto, N.; Hudson, T.J. The genetic basis for cancer treatment decisions. Cell 2012, 148, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer—Three Strikes and You’re Out. N. Engl. J. Med. 2015, 373, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Knepper, T.C.; Bell, G.C.; Hicks, J.K.; Padron, E.; Teer, J.K.; Vo, T.T.; Gillis, N.K.; Mason, N.T.; McLeod, H.L.; Walko, C.M.; et al. Key Lessons Learned from Moffitt’s Molecular Tumor Board: The Clinical Genomics Action Committee Experience. Oncologist 2017, 22, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Fu, S.; Piha-Paul, S.A.; Naing, A.; Falchook, G.S.; Janku, F.; Luthra, R.; et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Gay, L.; Otto, G.A.; White, E.; Iwanik, K.; Palmer, G.; Yelensky, R.; Lipson, D.M.; Chmielecki, J.; et al. Comprehensive Genomic Profiling of Carcinoma of Unknown Primary Site: New Routes to Targeted Therapies. JAMA Oncol. 2015, 1, 40–49. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Wen, S.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Aldape, K.; et al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: Validation and landmark analyses. Clin. Cancer Res. 2014, 20, 4827–4836. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Delord, J.P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Jiang, X.; Pissaloux, D.; De La Fouchardiere, C.; Desseigne, F.; Wang, Q.; Attignon, V.; Fondrevelle, M.E.; De La Fouchardiere, A.; Perol, M.; Cassier, P.; et al. The sum of gains and losses of genes encoding the protein tyrosine kinase targets predicts response to multi-kinase inhibitor treatment: Characterization, validation, and prognostic value. Oncotarget 2015, 6, 26388–26399. [Google Scholar] [CrossRef]
- André, F.; Bachelot, T.; Commo, F.; Campone, M.; Arnedos, M.; Dieras, V.; Lacroix-Triki, M.; Lacroix, L.; Cohen, P.; Gentien, D.; et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: A multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol. 2014, 15, 267–274. [Google Scholar] [CrossRef]
- Bernichon, E.; Vallard, A.; Wang, Q.; Attignon, V.; Pissaloux, D.; Bachelot, T.; Heudel, P.E.; Ray-Coquard, I.; Bonnet, E.; de la Fouchardière, A.; et al. Genomic alterations and radioresistance in breast cancer: An analysis of the ProfiLER protocol. Ann. Oncol. 2017, 28, 2773–2779. [Google Scholar] [CrossRef] [PubMed]
- Ratain, M.J.; Eisen, T.I.M.; Stadler, W.M.; Flaherty, K.T.; Kaye, S.B.; Rosner, G.L.; Gore, M.; Desai, A.A.; Patnaik, A.; Xiong, H.Q.; et al. Phase II Placebo-Controlled Randomized Discontinuation Trial of Sorafenib in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2006, 24, 2505-12. [Google Scholar] [CrossRef] [PubMed]
- Saville, B.R.; Berry, S.M. Efficiencies of platform clinical trials: A vision of the future. Clin. Trials 2016, 13, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Stadler, W. Other paradigms: Randomized discontinuation trial design. Cancer J. 2009, 15, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, F.; Anaganti, S.; Guida, T.; Salvatore, G.; Troncone, G.; Wilhelm, S.M.; Santoro, M. BAY 43-9006 inhibition of oncogenic RET mutants. J. Natl. Cancer Inst. 2006, 98, 326–334. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099-09. [Google Scholar] [CrossRef]
- National Cancer Institute: FDA Approval for Sorafenib Tosylate. 2015. Available online: http://www.cancer.gov/about-cancer/treatment/drugs/fda-sorafenib-tosylate (accessed on 8 June 2015).
- Llovet, J.M. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Farrell, A.T.; Saber, H.; Tang, S.; Williams, G.; Jee, J.M.; Liang, C.; Booth, B.; Chidambaram, N.; Morse, D.; et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 2006, 12, 7271–7278. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Berry, D.A. Bayesian clinical trials. Nature Review. Drug Discov. 2006, 5, 27–36. [Google Scholar] [CrossRef]
- Zohar, S.; Teramukai, S.; Zhou, Y. Bayesian design and conduct of phase II single-arm clinical trials with binary outcomes: A tutorial. Contemp Clin. Trials 2008, 29, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.; Bedard, P.; Tortora, G.; Douillard, J.-Y.; et al. A framework to rank genomic alterations as targets for cancer precision medicine: The ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Samalin, E.; de La Fouchardière, C.; Thèzenas, S.; Boige, V.; Senellart, H.; Guimbaud, R.; Taïeb, J.; François, E.; Galais, M.P.; Lièvre, A.; et al. Sorafenib Plus Irinotecan Combination in Patients with RAS-mutated Metastatic Colorectal Cancer Refractory to Standard Combined Chemotherapies: A Multicenter, Randomized Phase 2 Trial (NEXIRI-2/PRODIGE 27). Clin. Color. Cancer 2020, 19, 301–310. [Google Scholar] [CrossRef]
- Cook, J.H.; Melloni, G.E.M.; Gulhan, D.C.; Park, P.J.; Haigis, K.M. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat. Commun. 2021, 12, 1808. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-J.; Zheng, B.; Wang, H.-Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharm. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Status at End of Induction Period | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OR | PD | SD | ||||||||
| Maintenance | Interruption | |||||||||
| n = 5 | n = 105 | n = 18 | n = 17 | n = 145 | ||||||
| Age (years) | ||||||||||
| Median (IQR) | 66.0 (61.0−70.0) | 63.0 (56.0−68.0) | 60.0 (55.0−65.0) | 65.0 (57.0−68.0) | 63.0 (56.0−68.0) | |||||
| Sex | ||||||||||
| M | 3 | (60.0) | 49 | (46.7%) | 8 | (44.4%) | 6 | (35.3%) | 66 | (45.5%) |
| F | 2 | (40.0) | 56 | (53.3%) | 10 | (55.6%) | 11 | (64.7%) | 79 | (54.5%) |
| PS ECOG, n (%) | ||||||||||
| 0 | 3 | (60.0) | 28 | (26.7%) | 10 | (55.6%) | 6 | (35.3%) | 47 | (32.4%) |
| 1 | 2 | (40.0) | 63 | (60.0%) | 7 | (38.9%) | 9 | (52.9%) | 81 | (55.9%) |
| 2 | 0 | (0.0%) | 14 | (13.3%) | 1 | (5.6%) | 2 | (11.8%) | 17 | (11.7%) |
| Main primary tumor site (≥10% in at least one subgroup), n (%) | ||||||||||
| CRC | 0 | (0.0%) | 31 | (29.5%) | 4 | (22.2%) | 2 | (11.8%) | 37 | (25.5%) |
| H & N | 1 | (20.0%) | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (0.7%) |
| Retroperitoneal | 0 | (0.0%) | 0 | (0.0%) | 2 | (11.1%) | 0 | (0.0%) | 2 | (1.4%) |
| Gynecological. | 0 | (0.0%) | 18 | (17.1%) | 6 | (33.3%) | 5 | (29.4%) | 29 | (20.0%) |
| NSCLC | 3 | (60.0%) | 22 | (21.0%) | 5 | (27.8%) | 8 | (47.1%) | 38 | (26.2%) |
| Prostate | 1 | (20.0%) | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (0.7%) |
| Pancreas | 0 | (0.0%) | 16 | (15.2%) | 0 | (0.0%) | 2 | (11.8%) | 18 | (12.4%) |
| Prior number of lines in advanced/metastatic stage | ||||||||||
| Median (IQR) | 5.0 (3.0−6.0) | 3.0 (2.0−4.0) | 2.5 (2.0−4.0) | 3.0 (2.0−3.0) | 3.0 (2.0−4.0) | |||||
| 1 L or 2 L | 1 (20%) | 50 (47.6%) | 9 (50.0%) | 8 (47.1%) | 68 (46.9%) | |||||
| 3 L to 5 L | 2 (40%) | 43 (41.0%) | 8 (44.4%) | 8 (47.1%) | 61 (42.1%) | |||||
| ≥6 L | 2 (40%) | 12 (11.4%) | 1 (5.6%) | 1 (5.8%) | 16 (11.0%) | |||||
| Type of prior line before inclusion, n (%) | ||||||||||
| CT | 3 | (60.0) | 68 | (64.8%) | 7 | (38.9%) | 12 | (70.6%) | 90 | (62.1%) |
| CT + Anti-angiogenic | 0 | (0.0%) | 27 | (25.7%) | 5 | (27.8%) | 2 | (11.8%) | 34 | (23.4%) |
| Targeted therapy | 1 | (20%) | 7 | (6.6%) | 5 | (27.8%) | 2 | (11.8%) | 15 | (10.3%) |
| Immunotherapy | 0 | (0.0%) | 2 | (1.9%) | 1 | (5.6%) | 0 | (0.0%) | 3 | (2.1%) |
| Hormonotherapy | 0 | (0.0%) | 1 | (1.0%) | 0 | (0.0%) | 1 | (5.9%) | 2 | (1.4%) |
| Best response to prior line according to RECIST V1.1, n (%) | ||||||||||
| CR | 0 | (0.0%) | 2 | (1.9%) | 0 | (0.0%) | 0 | (0.0%) | 2 | (1.4%) |
| PR | 1 | (20.0) | 8 | (7.6%) | 0 | (0.0%) | 2 | (11.8%) | 11 | (7.6%) |
| SD | 0 | (0.0%) | 36 | (34.3%) | 7 | (38.9%) | 12 | (70.6%) | 55 | (37.9%) |
| PD | 2 | (40.0) | 56 | (53.3%) | 10 | (55.6%) | 2 | (11.8%) | 70 | (48.3%) |
| NE | 1 | (20.0) | 3 | (2.9%) | 1 | (5.6%) | 1 | (5.9%) | 6 | (4.1%) |
| Disease Status at End of Induction Period | TOTAL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OR | PD | SD | ||||||||
| Maintenance | Interruption | |||||||||
| n = 5 | n = 105 | n = 18 | n = 17 | n = 145 | ||||||
| Number of patients with at least, n (%) | ||||||||||
| One AE (all grades) | 5 | (100.0%) | 105 | (100.0%) | 18 | (100.0%) | 17 | (100.0%) | 145 | (100.0%) |
| One sorafenib-related AE (all grades) | 5 | (100.0%) | 86 | (81.9%) | 18 | (100.0%) | 17 | (100.0%) | 126 | (86.9%) |
| One Grade ≥ 3 AE | 1 | (20.0%) | 84 | (80.0%) | 13 | (72.2%) | 14 | (82.4%) | 112 | (77.2%) |
| One Grade ≥ 3 sorafenib-related AE | 1 | (20.0%) | 46 | (43.8%) | 12 | (66.7%) | 8 | (47.1%) | 67 | (46.2%) |
| One related SAE | 0 | (0.0%) | 33 | (31.4%) | 6 | (33.3%) | 4 | (23.5%) | 43 | (29.7%) |
| One SUSAR | 0 | (0.0%) | 10 | (9.5%) | 0 | (0.0%) | 1 | (5.9%) | 11 | (7.6%) |
| Main (≥ 5%) Grade ≥ 3 related AE, n (%) | ||||||||||
| Abdominal pain | 0 | (0.0%) | 1 | (1.0%) | 1 | (5.6%) | 0 | (0.0%) | 2 | (1.4%) |
| Intestinal perforation | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.6%) | 0 | (0.0%) | 1 | (0.7%) |
| Vomiting | 0 | (0.0%) | 4 | (3.8%) | 5 | (27.8%) | 0 | (0.0%) | 9 | (6.2%) |
| Fatigue | 0 | (0.0%) | 9 | (8.6%) | 2 | (11.1%) | 0 | (0.0%) | 11 | (7.6%) |
| QT prolonged | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.9%) | 1 | (0.7%) |
| GGT increased | 0 | (0.0%) | 3 | (2.9%) | 1 | (5.6%) | 0 | (0.0%) | 4 | (2.8%) |
| Weight decreased | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.6%) | 0 | (0.0%) | 1 | (0.7%) |
| WBC decreased | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.9%) | 1 | (0.7%) |
| Hypocalcemia | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.6%) | 0 | (0.0%) | 1 | (0.7%) |
| Dyspnea | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.9%) | 1 | (0.7%) |
| Pulmonary embolism | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.9%) | 1 | (0.7%) |
| Hand and Foot syndrome | 0 | (0.0%) | 4 | (3.8%) | 4 | (22.2%) | 1 | (5.9%) | 9 | (6.2%) |
| Rash | 0 | (0.0%) | 2 | (1.9%) | 0 | (0.0%) | 1 | (5.9%) | 3 | (2.1%) |
| Hypertension | 1 | (20.0%) | 10 | (9.5%) | 3 | (16.7%) | 4 | (23.5%) | 18 | (12.4%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trédan, O.; Toulmonde, M.; Le Tourneau, C.; Montane, L.; Italiano, A.; Ray-Coquard, I.; De La Fouchardière, C.; Bertucci, F.; Gonçalves, A.; Gomez-Roca, C.; et al. Sorafenib in Molecularly Selected Cancer Patients: Final Analysis of the MOST-Plus Sorafenib Cohort. Cancers 2023, 15, 3441. https://doi.org/10.3390/cancers15133441
Trédan O, Toulmonde M, Le Tourneau C, Montane L, Italiano A, Ray-Coquard I, De La Fouchardière C, Bertucci F, Gonçalves A, Gomez-Roca C, et al. Sorafenib in Molecularly Selected Cancer Patients: Final Analysis of the MOST-Plus Sorafenib Cohort. Cancers. 2023; 15(13):3441. https://doi.org/10.3390/cancers15133441
Chicago/Turabian StyleTrédan, Olivier, Maud Toulmonde, Christophe Le Tourneau, Laure Montane, Antoine Italiano, Isabelle Ray-Coquard, Christelle De La Fouchardière, François Bertucci, Anthony Gonçalves, Carlos Gomez-Roca, and et al. 2023. "Sorafenib in Molecularly Selected Cancer Patients: Final Analysis of the MOST-Plus Sorafenib Cohort" Cancers 15, no. 13: 3441. https://doi.org/10.3390/cancers15133441
APA StyleTrédan, O., Toulmonde, M., Le Tourneau, C., Montane, L., Italiano, A., Ray-Coquard, I., De La Fouchardière, C., Bertucci, F., Gonçalves, A., Gomez-Roca, C., You, B., Attignon, V., Boyault, S., Cassier, P. A., Dufresne, A., Tabone-Eglinger, S., Viari, A., Sohier, E., Kamal, M., ... Pérol, D. (2023). Sorafenib in Molecularly Selected Cancer Patients: Final Analysis of the MOST-Plus Sorafenib Cohort. Cancers, 15(13), 3441. https://doi.org/10.3390/cancers15133441