
Overview of the Genetic Causes of Hereditary Breast and Ovarian Cancer Syndrome in a Large French Patient Cohort

 ,
,  ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. NGS Analysis
2.3. Variant Interpretation
2.4. Additional Analyzes
3. Results
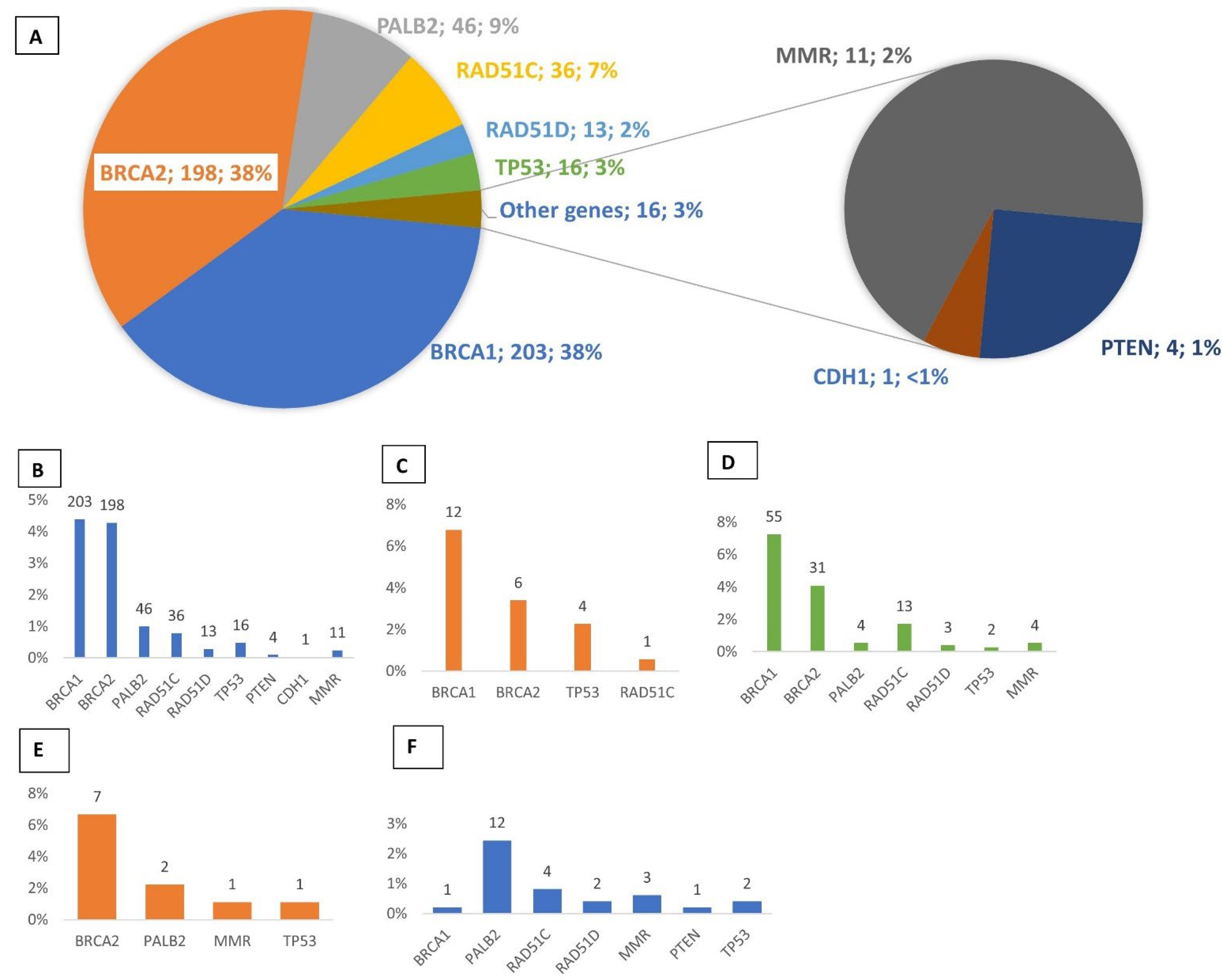
3.1. Multigene Panel Screening
3.2. Novel Mutations
3.3. Recurrent Mutations
3.4. Identification of Double Mutations
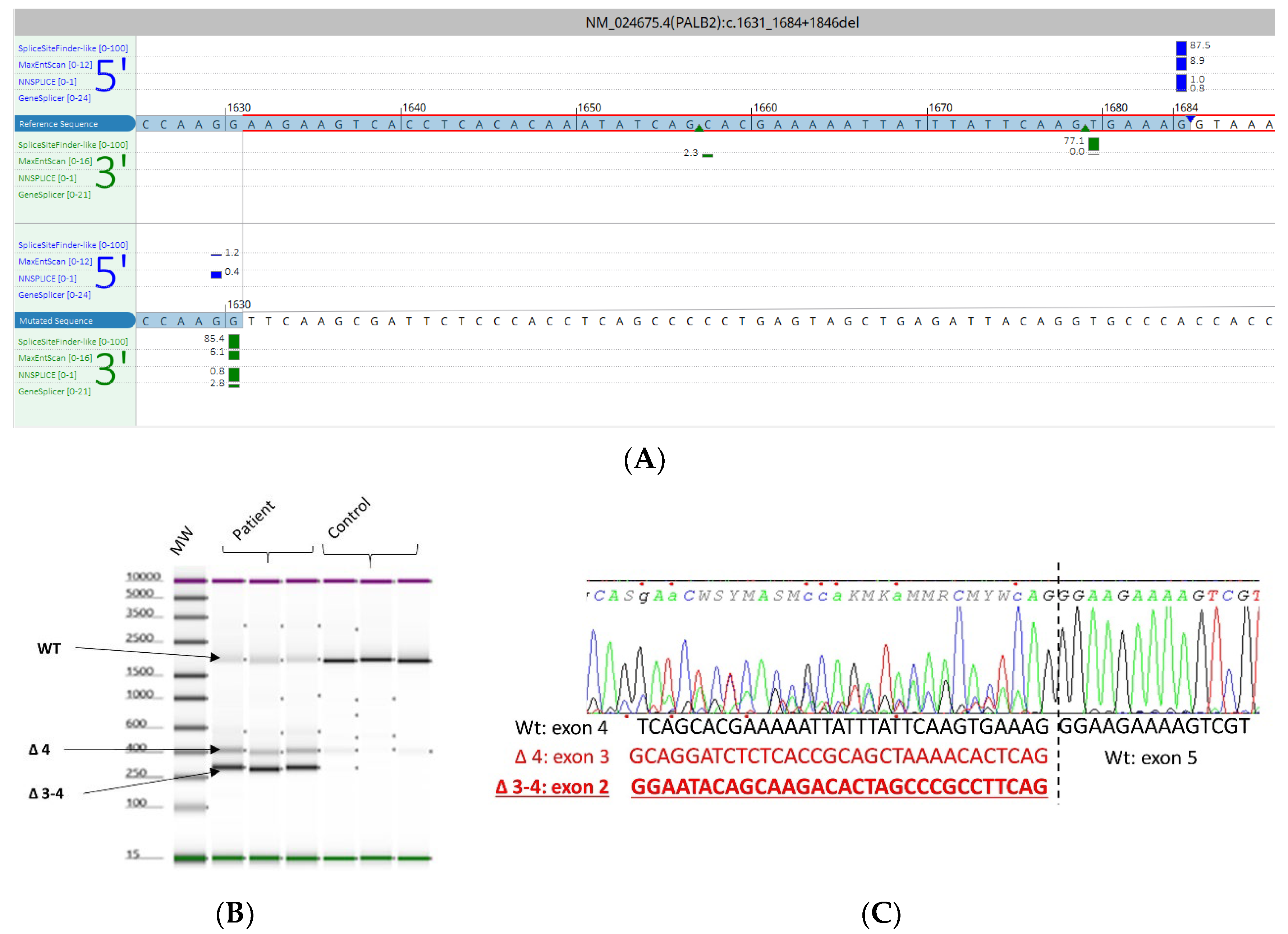
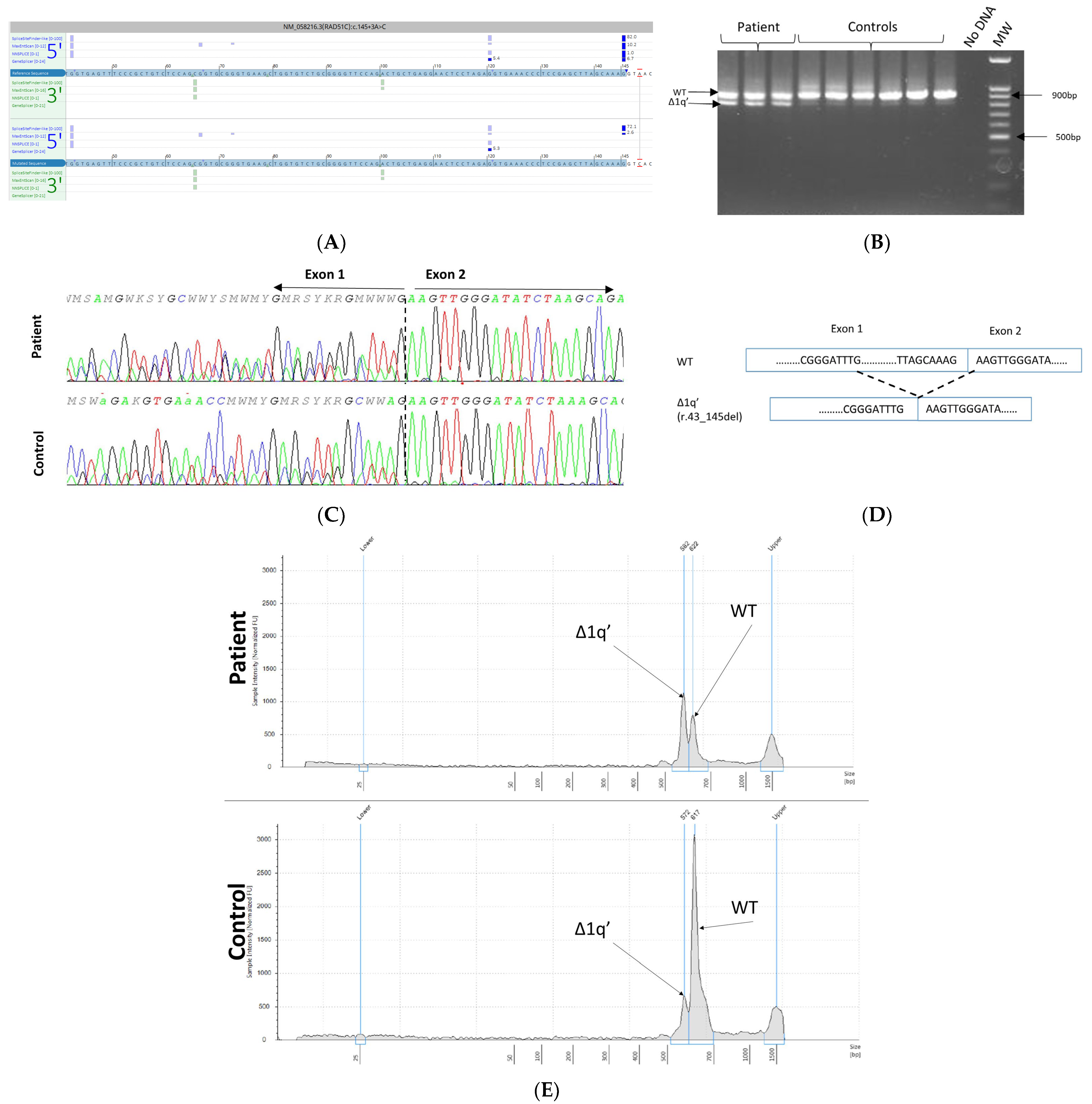
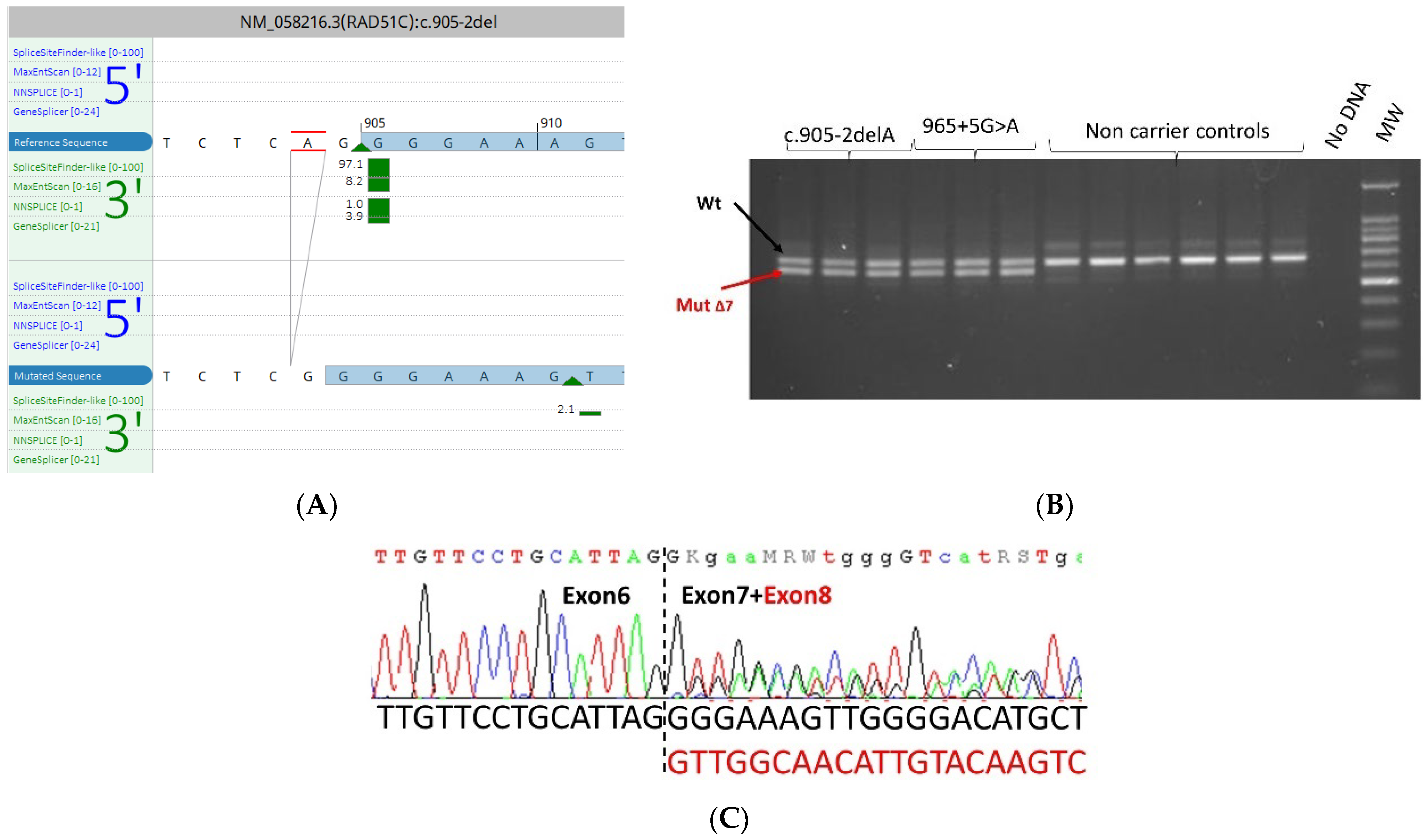
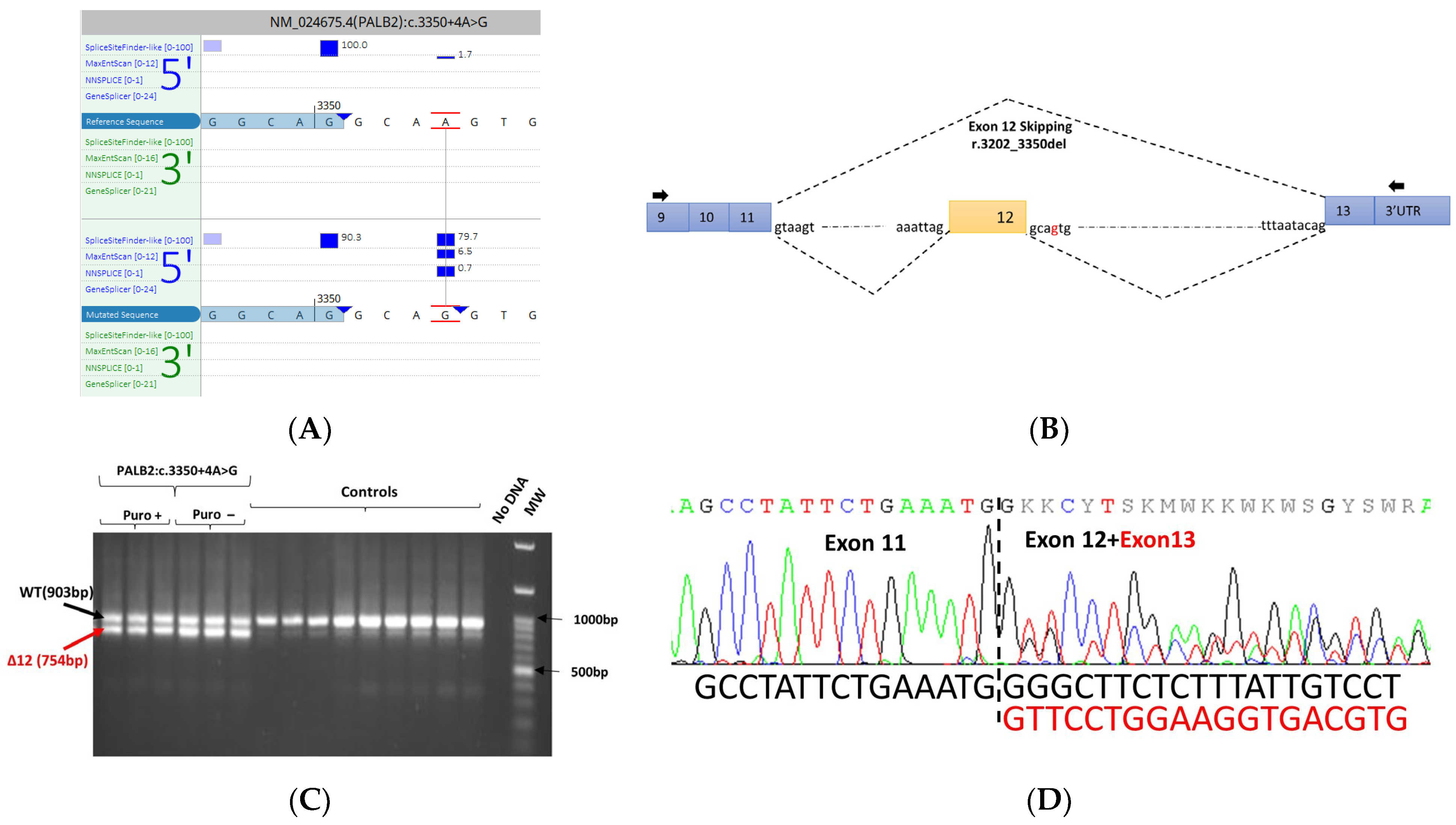
3.5. mRNA Transcript Analysis of Patients with P/LP Splice Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoshida, R. Hereditary Breast and Ovarian Cancer (HBOC): Review of Its Molecular Characteristics, Screening, Treatment, and Prognosis. Breast Cancer 2021, 28, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Ohno, S.; Sasaki, Y.; Matsuura, M. Hereditary Breast and Ovarian Cancer Susceptibility Genes (Review). Oncol. Rep. 2013, 30, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Castéra, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of Pathogenic Variations in a Panel of 34 Genes and Cancer Risk Estimation from 5131 HBOC Families. Genet. Med. 2018, 20, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A Study of over 35,000 Women with Breast Cancer Tested with a 25-Gene Panel of Hereditary Cancer Genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef]
- Moretta, J.; Berthet, P.; Bonadona, V.; Caron, O.; Cohen-Haguenauer, O.; Colas, C.; Corsini, C.; Cusin, V.; De Pauw, A.; Delnatte, C.; et al. The French Genetic and Cancer Consortium guidelines for multigene panel analysis in hereditary breast and ovarian cancer predisposition. Bull. Cancer 2018, 105, 907–917. [Google Scholar] [CrossRef]
- Aloraifi, F.; Boland, M.R.; Green, A.J.; Geraghty, J.G. Gene Analysis Techniques and Susceptibility Gene Discovery in Non-BRCA1/BRCA2 Familial Breast Cancer. Surg. Oncol. 2015, 24, 100–109. [Google Scholar] [CrossRef]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D Mutations Are Associated with High Susceptibility to Ovarian Cancer: Mutation Prevalence and Precise Risk Estimates Based on a Pooled Analysis of ~30,000 Cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef]
- Eisinger, F.; Bressac, B.; Castaigne, D.; Cottu, P.-H.; Lansac, J.; Lefranc, J.-P.; Lesur, A.; Noguès, C.; Pierret, J.; Puy-Pernias, S.; et al. Identification and management of hereditary predisposition to cancer of the breast and the ovary (update 2004). Bull. Cancer 2004, 91, 219–237. [Google Scholar]
- Caputo, S.M.; Golmard, L.; Léone, M.; Damiola, F.; Guillaud-Bataille, M.; Revillion, F.; Rouleau, E.; Derive, N.; Buisson, A.; Basset, N.; et al. Classification of 101 BRCA1 and BRCA2 Variants of Uncertain Significance by Cosegregation Study: A Powerful Approach. Am. J. Hum. Genet. 2021, 108, 1907–1923. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Byrnes, G.B.; Goldgar, D.E.; Thomas, A. Classification of Rare Missense Substitutions, Using Risk Surfaces, with Genetic- and Molecular-Epidemiology Applications. Hum. Mutat. 2008, 29, 1342–1354. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Leman, R.; Parfait, B.; Vidaud, D.; Girodon, E.; Pacot, L.; Le Gac, G.; Ka, C.; Ferec, C.; Fichou, Y.; Quesnelle, C.; et al. SPiP: Splicing Prediction Pipeline, a Machine Learning Tool for Massive Detection of Exonic and Intronic Variant Effects on MRNA Splicing. Hum. Mutat. 2022, 43, 2308–2323. [Google Scholar] [CrossRef]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Houdayer, C.; Caux-Moncoutier, V.; Krieger, S.; Barrois, M.; Bonnet, F.; Bourdon, V.; Bronner, M.; Buisson, M.; Coulet, F.; Gaildrat, P.; et al. Guidelines for Splicing Analysis in Molecular Diagnosis Derived from a Set of 327 Combined in Silico/in Vitro Studies on BRCA1 and BRCA2 Variants. Hum. Mutat. 2012, 33, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Davy, G.; Rousselin, A.; Goardon, N.; Castéra, L.; Harter, V.; Legros, A.; Muller, E.; Fouillet, R.; Brault, B.; Smirnova, A.S.; et al. Detecting Splicing Patterns in Genes Involved in Hereditary Breast and Ovarian Cancer. Eur. J. Hum. Genet. 2017, 25, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Coulet, F.; Fajac, A.; Colas, C.; Eyries, M.; Dion-Minière, A.; Rouzier, R.; Uzan, S.; Lefranc, J.-P.; Carbonnel, M.; Cornelis, F.; et al. Germline RAD51C Mutations in Ovarian Cancer Susceptibility. Clin. Genet. 2013, 83, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Palomo, A.; Bueno-Martínez, E.; Sanoguera-Miralles, L.; Lorca, V.; Fraile-Bethencourt, E.; Esteban-Sánchez, A.; Gómez-Barrero, S.; Carvalho, S.; Allen, J.; García-Álvarez, A.; et al. Splicing Predictions, Minigene Analyses, and ACMG-AMP Clinical Classification of 42 Germline PALB2 Splice-Site Variants. J. Pathol. 2022, 256, 321–334. [Google Scholar] [CrossRef]
- Feliubadaló, L.; López-Fernández, A.; Pineda, M.; Díez, O.; del Valle, J.; Gutiérrez-Enríquez, S.; Teulé, A.; González, S.; Stjepanovic, N.; Salinas, M.; et al. Opportunistic Testing of BRCA1, BRCA2 and Mismatch Repair Genes Improves the Yield of Phenotype Driven Hereditary Cancer Gene Panels. Int. J. Cancer 2019, 145, 2682–2691. [Google Scholar] [CrossRef]
- Cavaillé, M.; Uhrhammer, N.; Privat, M.; Ponelle-Chachuat, F.; Gay-Bellile, M.; Lepage, M.; Viala, S.; Bidet, Y.; Bignon, Y.-J. Feedback of Extended Panel Sequencing in 1530 Patients Referred for Suspicion of Hereditary Predisposition to Adult Cancers. Clin. Genet. 2021, 99, 166–175. [Google Scholar] [CrossRef]
- Walsh, T.; Lee, M.K.; Casadei, S.; Thornton, A.M.; Stray, S.M.; Pennil, C.; Nord, A.S.; Mandell, J.B.; Swisher, E.M.; King, M.-C. Detection of Inherited Mutations for Breast and Ovarian Cancer Using Genomic Capture and Massively Parallel Sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 12629–12633. [Google Scholar] [CrossRef]
- Gustafson, S.; Zbuk, K.M.; Scacheri, C.; Eng, C. Cowden Syndrome. Semin. Oncol. 2007, 34, 428–434. [Google Scholar] [CrossRef]
- Olivier, M.; Goldgar, D.E.; Sodha, N.; Ohgaki, H.; Kleihues, P.; Hainaut, P.; Eeles, R.A. Li-Fraumeni and Related Syndromes: Correlation between Tumor Type, Family Structure, and TP53 Genotype. Cancer Res. 2003, 63, 6643–6650. [Google Scholar]
- Bakhuizen, J.J.; Hogervorst, F.B.; Velthuizen, M.E.; Ruijs, M.W.; van Engelen, K.; van Os, T.A.; Gille, J.J.; Collée, M.; van den Ouweland, A.M.; van Asperen, C.J.; et al. TP53 Germline Mutation Testing in Early-Onset Breast Cancer: Findings from a Nationwide Cohort. Fam. Cancer 2019, 18, 273–280. [Google Scholar] [CrossRef]
- Garrett, A.; Talukdar, S.; Izatt, L.; Brady, A.F.; Whyte, S.; Josephs, K.S.; Shanmugasundaram, M.; Guillemot, L.S.; Vakili, D.; Ey, S.; et al. Results from London Regional Clinical Genetics Services over a 5-Year Period on Germline TP53 Testing in Women Diagnosed with Breast Cancer at <30 Years. J. Med. Genet. 2022, 59, 554–558. [Google Scholar] [CrossRef]
- Ferrarini, A.; Auteri-Kaczmarek, A.; Pica, A.; Boesch, N.; Heinimann, K.; Schäfer, S.C.; Vesnaver-Megalo, S.; Cina, V.; Beckmann, J.S.; Monnerat, C. Early Occurrence of Lung Adenocarcinoma and Breast Cancer after Radiotherapy of a Chest Wall Sarcoma in a Patient with a de Novo Germline Mutation in TP53. Fam. Cancer 2011, 10, 187–192. [Google Scholar] [CrossRef]
- Limacher, J.M.; Frebourg, T.; Natarajan-Ame, S.; Bergerat, J.P. Two Metachronous Tumors in the Radiotherapy Fields of a Patient with Li-Fraumeni Syndrome. Int. J. Cancer 2001, 96, 238–242. [Google Scholar] [CrossRef]
- Heymann, S.; Delaloge, S.; Rahal, A.; Caron, O.; Frebourg, T.; Barreau, L.; Pachet, C.; Mathieu, M.-C.; Marsiglia, H.; Bourgier, C. Radio-Induced Malignancies after Breast Cancer Postoperative Radiotherapy in Patients with Li-Fraumeni Syndrome. Radiat. Oncol. 2010, 5, 104. [Google Scholar] [CrossRef]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. JCO 2015, 33, 2901–2907. [Google Scholar] [CrossRef]
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.-J.; Caron, O.; et al. Cancer Risks Associated with Germline Mutations in MLH1, MSH2, and MSH6 Genes in Lynch Syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Taieb, J.; Svrcek, M.; Cohen, R.; Basile, D.; Tougeron, D.; Phelip, J.-M. Deficient Mismatch Repair/Microsatellite Unstable Colorectal Cancer: Diagnosis, Prognosis and Treatment. Eur. J. Cancer 2022, 175, 136–157. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.C.; Steele, L.; Kuan, C.-J.; Greilac, S.; Neuhausen, S.L. Mutations in BRCA2 and PALB2 in Male Breast Cancer Cases from the United States. Breast Cancer Res. Treat. 2011, 126, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Pensabene, M.; Von Arx, C.; De Laurentiis, M. Male Breast Cancer: From Molecular Genetics to Clinical Management. Cancers 2022, 14, 2006. [Google Scholar] [CrossRef]
- Chamseddine, R.S.; Wang, C.; Yin, K.; Wang, J.; Singh, P.; Zhou, J.; Robson, M.E.; Braun, D.; Hughes, K.S. Penetrance of Male Breast Cancer Susceptibility Genes: A Systematic Review. Breast Cancer Res. Treat. 2022, 191, 31–38. [Google Scholar] [CrossRef]
- Weitzel, J.N.; Neuhausen, S.L.; Adamson, A.; Tao, S.; Ricker, C.; Maoz, A.; Rosenblatt, M.; Nehoray, B.; Sand, S.; Steele, L.; et al. Pathogenic and Likely Pathogenic Variants in PALB2, CHEK2, and Other Known Breast Cancer Susceptibility Genes among 1054 BRCA-Negative Hispanics with Breast Cancer. Cancer 2019, 125, 2829–2836. [Google Scholar] [CrossRef]
- Gonzalez, A.; Del Greco, F.; Vargas-Roig, L.; Brun, B.; Tabares, G.; Mampel, A.; Montes, C.; Martin, C.; Lopez, M.; Rossi, N.; et al. PALB2 Germline Mutations in a Multi-Gene Panel Testing Cohort of 1905 Breast-Ovarian Cancer Patients in Argentina. Breast Cancer Res. Treat. 2022, 194, 403–412. [Google Scholar] [CrossRef]
- Lerner-Ellis, J.; Sopik, V.; Wong, A.; Lázaro, C.; Narod, S.A.; Charames, G.S. Retesting of Women Who Are Negative for a BRCA1 and BRCA2 Mutation Using a 20-Gene Panel. J. Med. Genet. 2020, 57, 380–384. [Google Scholar] [CrossRef]
- Dragoo, D.D.; Taher, A.; Wong, V.K.; Elsaiey, A.; Consul, N.; Mahmoud, H.S.; Mujtaba, B.; Stanietzky, N.; Elsayes, K.M. PTEN Hamartoma Tumor Syndrome/Cowden Syndrome: Genomics, Oncogenesis, and Imaging Review for Associated Lesions and Malignancy. Cancers 2021, 13, 3120. [Google Scholar] [CrossRef]
- Bleuyard, J.-Y.; Buisson, R.; Masson, J.-Y.; Esashi, F. ChAM, a Novel Motif That Mediates PALB2 Intrinsic Chromatin Binding and Facilitates DNA Repair. EMBO Rep. 2012, 13, 135–141. [Google Scholar] [CrossRef]
- Sanoguera-Miralles, L.; Bueno-Martínez, E.; Valenzuela-Palomo, A.; Esteban-Sánchez, A.; Llinares-Burguet, I.; Pérez-Segura, P.; García-Álvarez, A.; de la Hoya, M.; Velasco-Sampedro, E.A. Minigene Splicing Assays Identify 20 Spliceogenic Variants of the Breast/Ovarian Cancer Susceptibility Gene RAD51C. Cancers 2022, 14, 2960. [Google Scholar] [CrossRef]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural Basis for Recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef]
- Montalban, G.; Fraile-Bethencourt, E.; López-Perolio, I.; Pérez-Segura, P.; Infante, M.; Durán, M.; Alonso-Cerezo, M.C.; López-Fernández, A.; Diez, O.; de la Hoya, M.; et al. Characterization of Spliceogenic Variants Located in Regions Linked to High Levels of Alternative Splicing: BRCA2 c. 7976+5G > T as a Case Study. Hum. Mutat. 2018, 39, 1155–1160. [Google Scholar] [CrossRef]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic Mutations in PALB2 Cause Fanconi Anemia Subtype FA-N and Predispose to Childhood Cancer. Nat. Genet. 2007, 39, 162–164. [Google Scholar] [CrossRef]
- Southey, M.C.; Goldgar, D.E.; Winqvist, R.; Pylkäs, K.; Couch, F.; Tischkowitz, M.; Foulkes, W.D.; Dennis, J.; Michailidou, K.; van Rensburg, E.J.; et al. PALB2, CHEK2 and ATM Rare Variants and Cancer Risk: Data from COGS. J. Med. Genet. 2016, 53, 800–811. [Google Scholar] [CrossRef]
- Lesueur, F.; Eon-Marchais, S.; Bonnet-Boissinot, S.; Beauvallet, J.; Dondon, M.-G.; Golmard, L.; Rouleau, E.; Garrec, C.; Martinez, M.; Toulas, C.; et al. TUMOSPEC: A Nation-Wide Study of Hereditary Breast and Ovarian Cancer Families with a Predicted Pathogenic Variant Identified through Multigene Panel Testing. Cancers 2021, 13, 3659. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Transcript | Gene | Transcript |
|---|---|---|---|
| BRCA1 | NM_007294.3 | CDH1 | NM_004360.4 |
| BRCA2 | NM_000059.3 | MLH1 | NM_000249.3 |
| PALB2 | NM_024675.3 | MSH2 | NM_000251.2 |
| RAD51C | NM_058216.2 | MSH6 | NM_000179.2 |
| RAD51D | NM_002878.3 | PMS2 * | NM_000535.6 |
| TP53 | NM_000546.5 | EPCAM ** | NM_002354.2 |
| PTEN | NM_000314.6 |
| Personal and Familial Criteria | Total | Detection Rate of P/LP Variants, n (%) |
|---|---|---|
| Breast carcinoma ≤ 31 years | 177 | 23 (13%) |
| Breast carcinoma ≤ 36 years | 564 | 23 (14%) |
| Male breast carcinoma | 91 | 11 (12%) |
| Ovary adenocarcinoma | 759 | 112 (15%) |
| Bilateral breast carcinoma | 244 | 33 (14%) |
| Free of cancer index case with a strong familial history of HBOC | 108 | 9 (8.3%) |
| Index case with both breast and ovarian cancer | 74 | 9 (17.6%) |
| Patients previously tested negative for BRCA pathogenic variants retested using the panel of 13 HBOC genes | 492 | 25 (5%) |
| Genes | cDNA Position | Protein | Frequency | % |
|---|---|---|---|---|
| BRCA1 | c.5266dup | p.(Gln1756Profs*14) | 15 | 7.4 |
| c.3481_3491del | p.(Glu1161Phefs*3) | 10 | 5 | |
| c.1115G>A | p.(Trp372*) | 9 | 4 | |
| c.4391del | p.(Pro1464Leufs*2) | 5 | 2 | |
| c.4327C>T | p.(Arg1443*) | 4 | 2 | |
| c.3756_3759delGTCT | p.(Ser1253Argfs*10) | 4 | 2 | |
| c.191G>A | p.(Cys64Tyr) | 4 | 2 | |
| BRCA2 | c.4889C>G | p.(Ser1630*) | 11 | 5 |
| c.3847_3848delGT | p.Val1283Lysfs*2 | 7 | 4 | |
| c.9294C>A | p.(Tyr3098*) | 6 | 3 | |
| c.1813dupA | p.(Ile605Asnfs*11) | 6 | 3 | |
| c.1310_1313delAAGA | p.(Lys437Ilefs*22) | 6 | 3 | |
| c.7680dup | p.(Gln2561Serfs*5) | 5 | 3 | |
| c.8364G>A | p.Trp2788* | 4 | 2 | |
| c.2612C>A | p.(Ser871*) | 4 | 2 | |
| c.5909C>A | p.(Ser1970*) | 4 | 2 | |
| RAD51C | c.1026+5_1026+7delGTA | p.(Arg322Serfs*22) | 9 | 25 |
| c.965+5G>A | p.(Glu303Trpfs*41) | 7 | 19 | |
| RAD51D | c.803G>A | p.(Trp268*) | 5 | 40 |
| c.170del | p.(Leu57Argfs*10) | 4 | 30 | |
| PALB2 | c.2257C>T | p.(Arg753*) | 3 | 7 |
| c.1915G>T | p.(Glu639*) | 3 | 7 |
| Gene | Diagnosis | Variant | Protein Effect | Class |
|---|---|---|---|---|
| Patient 1 | ||||
| BRCA1 | Uterus cancer at 30 years. Triple-negative breast carcinoma at 33 years | c.5309G>T | p.(Gly1770Val) | 5 |
| BRCA2 | c.7234_7235insG | p.(Thr2412Serfs*2) | 5 | |
| Patient 2 | ||||
| BRCA1 | Breast carcinoma at 36 years, contralateral triple-negative breast carcinoma at 62 years | c.212+3A>G | p.Cys64fs* | 5 |
| RAD51C | c.1026+5_1026+7delGTA | p.(Arg322Serfs*22) | 5 | |
| Patient 3 | ||||
| BRCA2 | Breast carcinoma at 55 years | c.9097dupA | p.(Thr3033fs) | 5 |
| RAD51C | c.905-2del | p.(Glu303Trpfs*41) | 5 | |
| Patient 4 | ||||
| BRCA2 | Ovarian cancer | c.1842dupT | p.(Asn615*) | 5 |
| RAD51C | c.773G>A | p.(Arg258His) | 5 | |
| Patient 5 | ||||
| BRCA2 | Epithelial thyroid cancer at 29 years Breast carcinoma at 37 years | c.3645_3646delGTinsTAAAAAG | p.(Phe1216Lysfs*14) | 5 |
| PTEN | c.1003C>T | p.(Arg335*) | 5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouras, A.; Guidara, S.; Leone, M.; Buisson, A.; Martin-Denavit, T.; Dussart, S.; Lasset, C.; Giraud, S.; Bonnet-Dupeyron, M.-N.; Kherraf, Z.-E.; et al. Overview of the Genetic Causes of Hereditary Breast and Ovarian Cancer Syndrome in a Large French Patient Cohort. Cancers 2023, 15, 3420. https://doi.org/10.3390/cancers15133420
Bouras A, Guidara S, Leone M, Buisson A, Martin-Denavit T, Dussart S, Lasset C, Giraud S, Bonnet-Dupeyron M-N, Kherraf Z-E, et al. Overview of the Genetic Causes of Hereditary Breast and Ovarian Cancer Syndrome in a Large French Patient Cohort. Cancers. 2023; 15(13):3420. https://doi.org/10.3390/cancers15133420
Chicago/Turabian StyleBouras, Ahmed, Souhir Guidara, Mélanie Leone, Adrien Buisson, Tanguy Martin-Denavit, Sophie Dussart, Christine Lasset, Sophie Giraud, Marie-Noëlle Bonnet-Dupeyron, Zine-Eddine Kherraf, and et al. 2023. "Overview of the Genetic Causes of Hereditary Breast and Ovarian Cancer Syndrome in a Large French Patient Cohort" Cancers 15, no. 13: 3420. https://doi.org/10.3390/cancers15133420
APA StyleBouras, A., Guidara, S., Leone, M., Buisson, A., Martin-Denavit, T., Dussart, S., Lasset, C., Giraud, S., Bonnet-Dupeyron, M.-N., Kherraf, Z.-E., Sanlaville, D., Fert-Ferrer, S., Lebrun, M., Bonadona, V., Calender, A., & Boutry-Kryza, N. (2023). Overview of the Genetic Causes of Hereditary Breast and Ovarian Cancer Syndrome in a Large French Patient Cohort. Cancers, 15(13), 3420. https://doi.org/10.3390/cancers15133420